Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products


Abstract
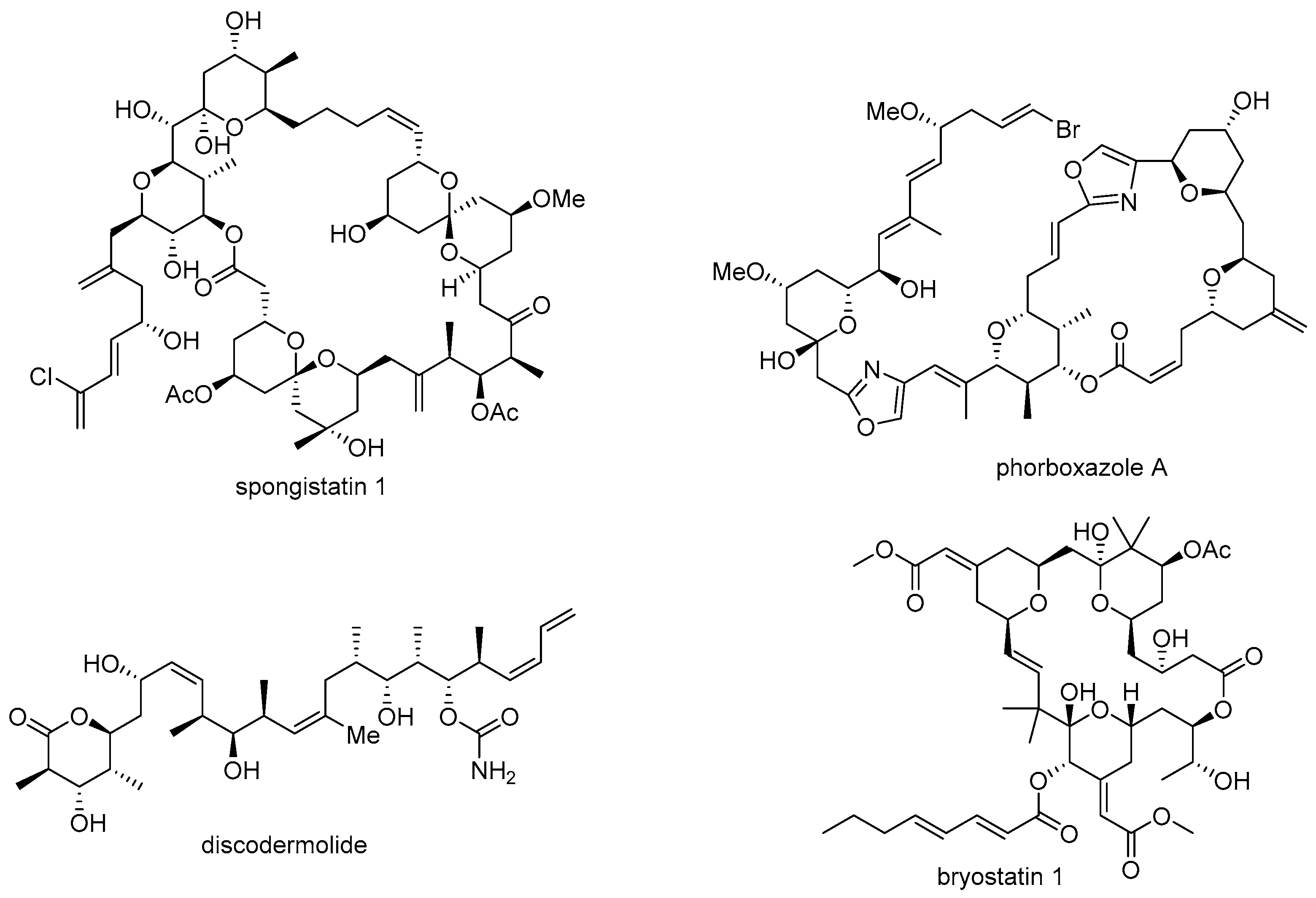
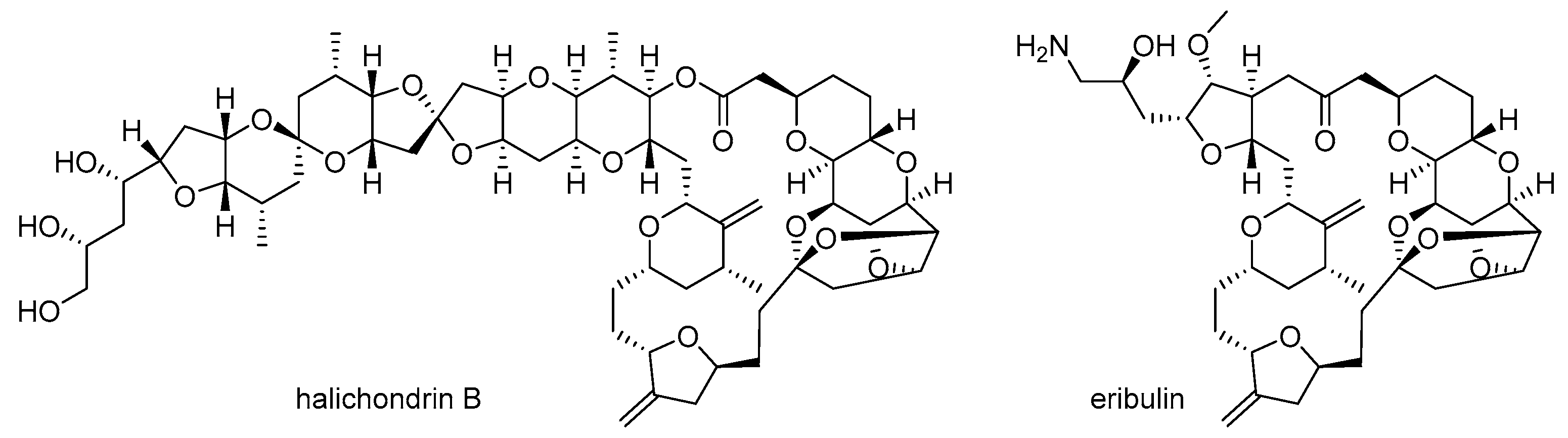
1. Introduction
2. Results
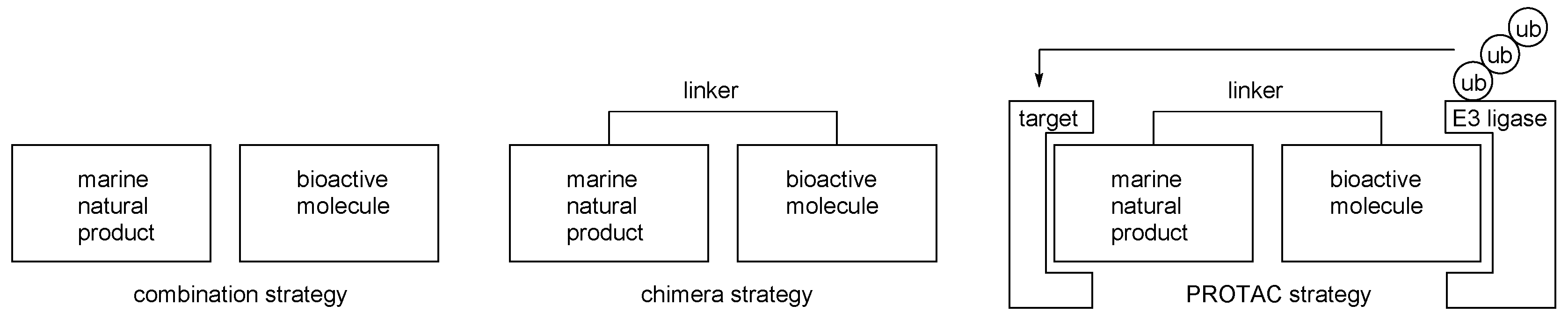
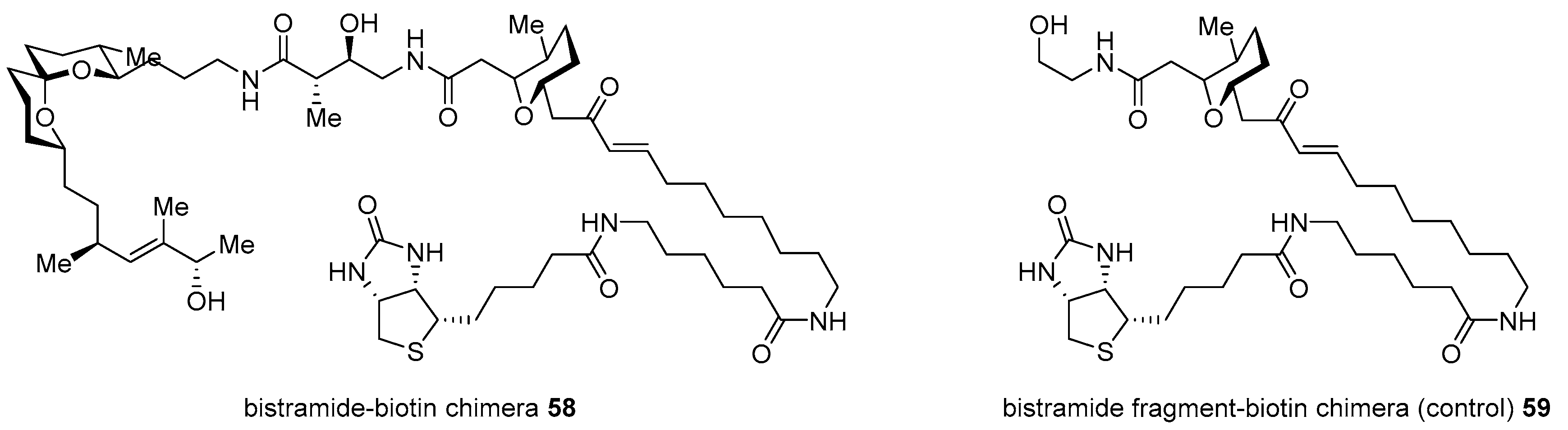
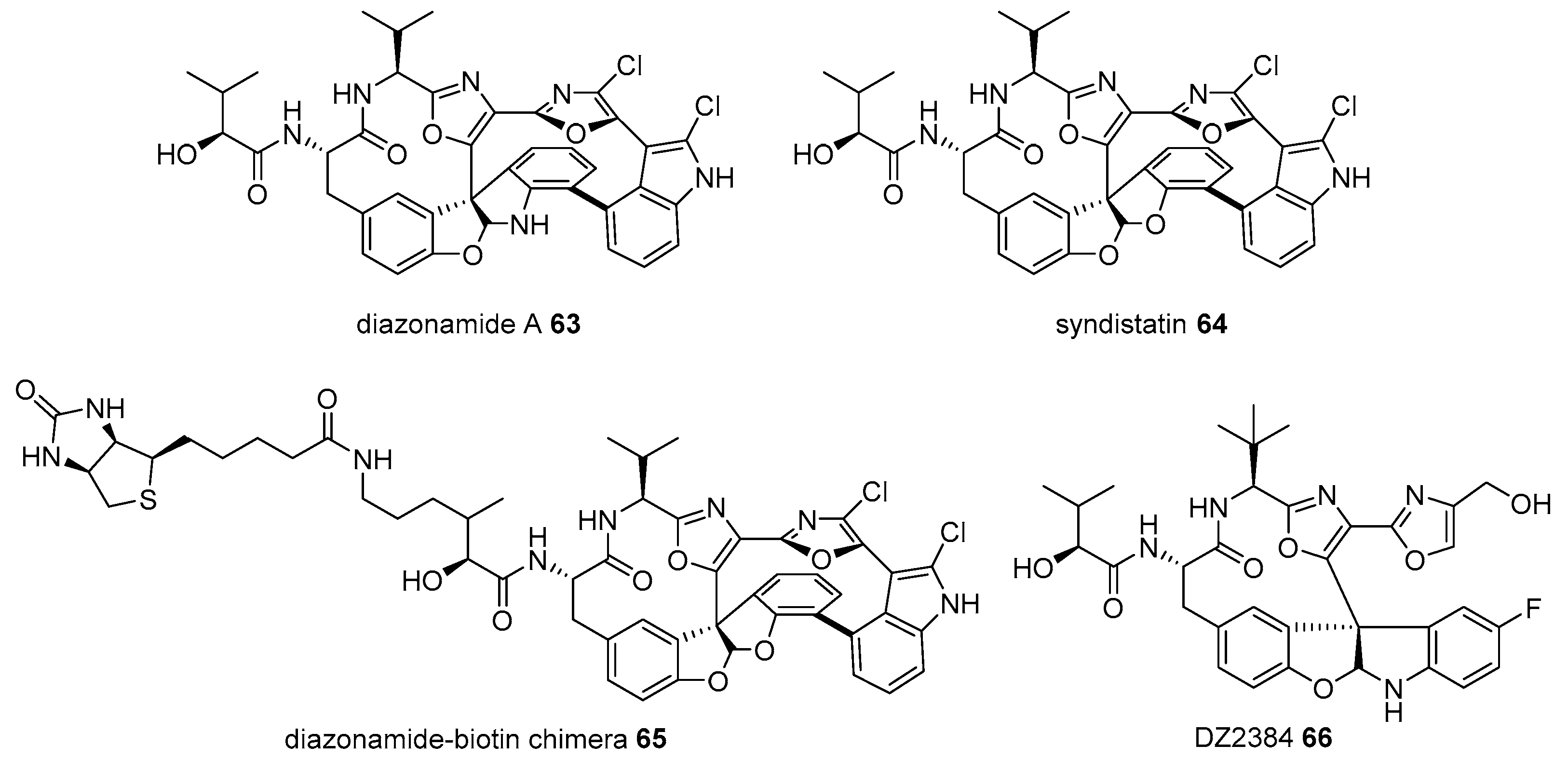
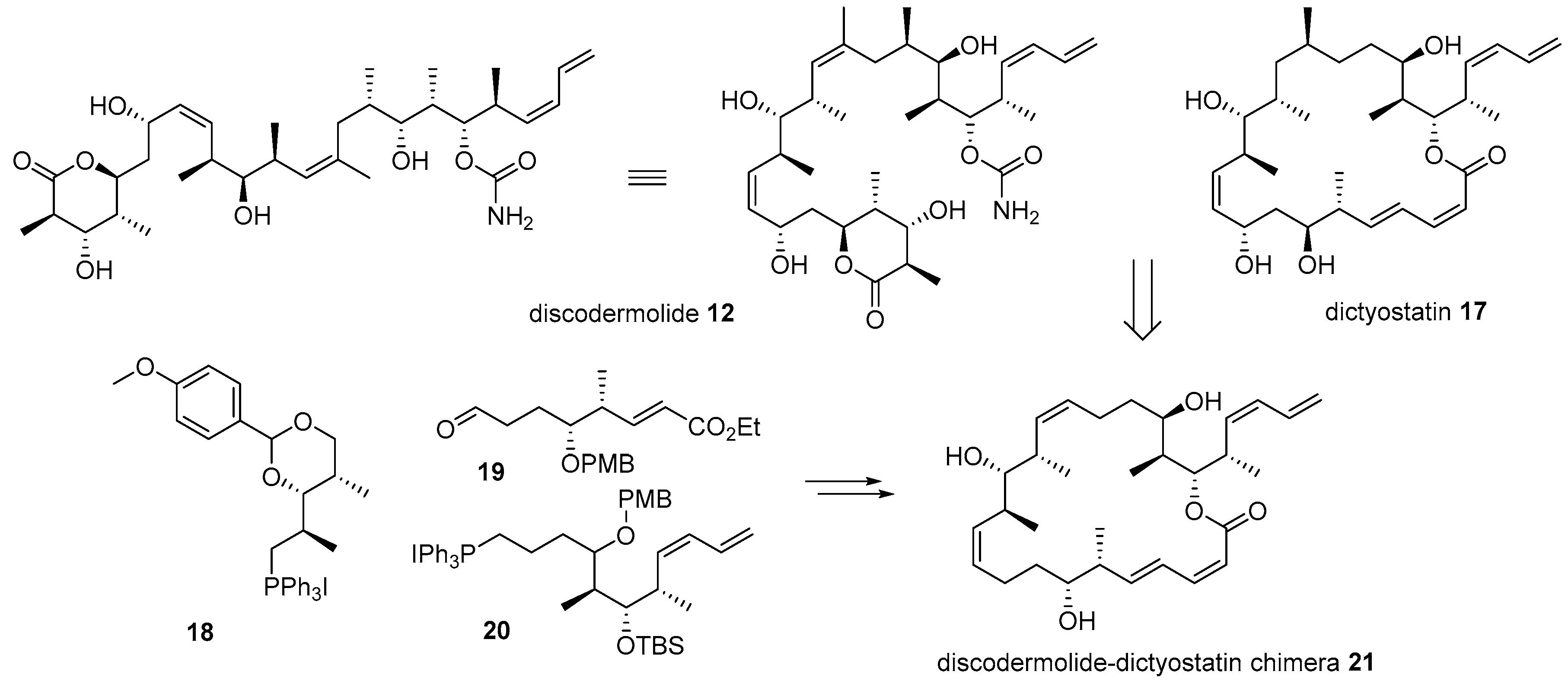
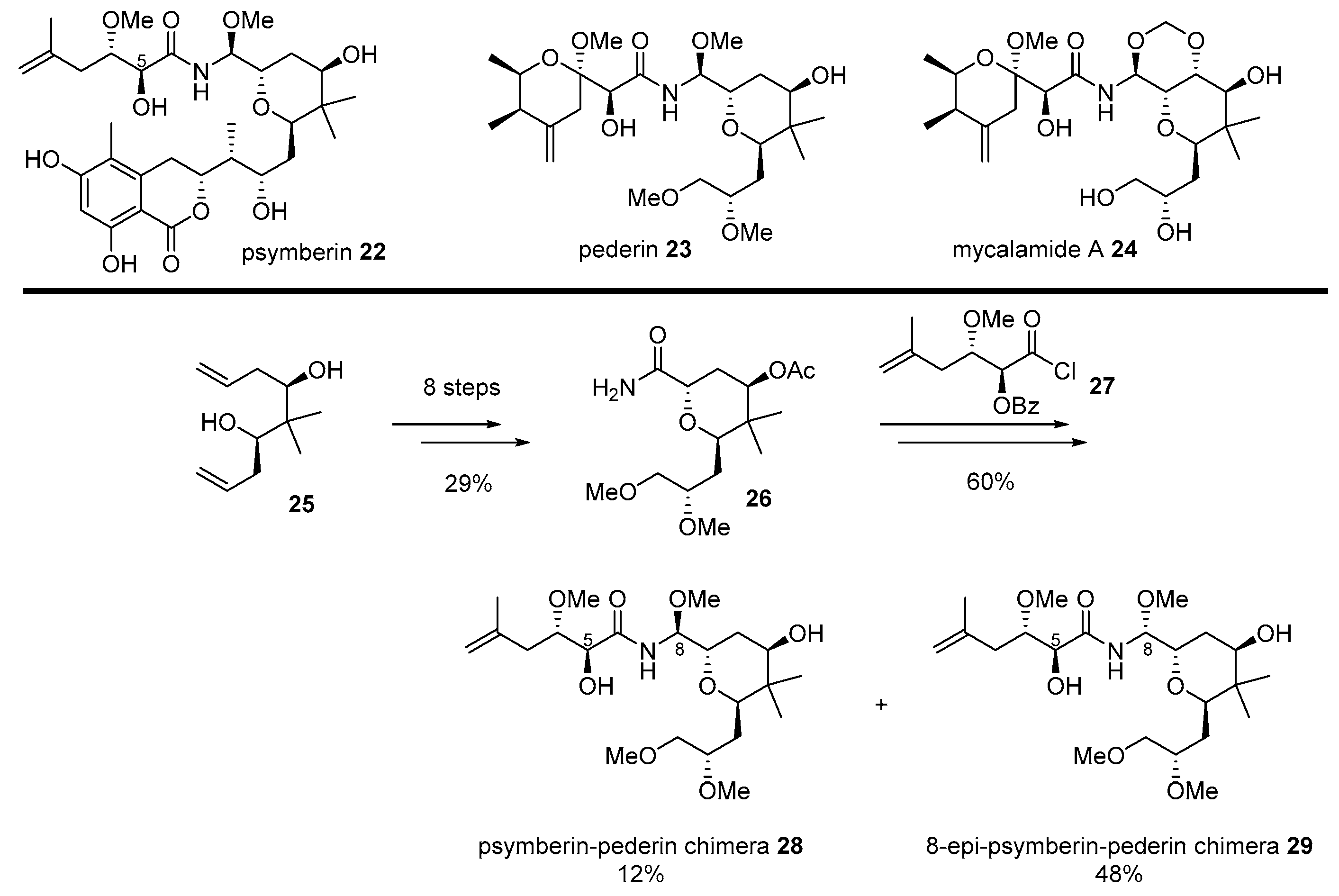
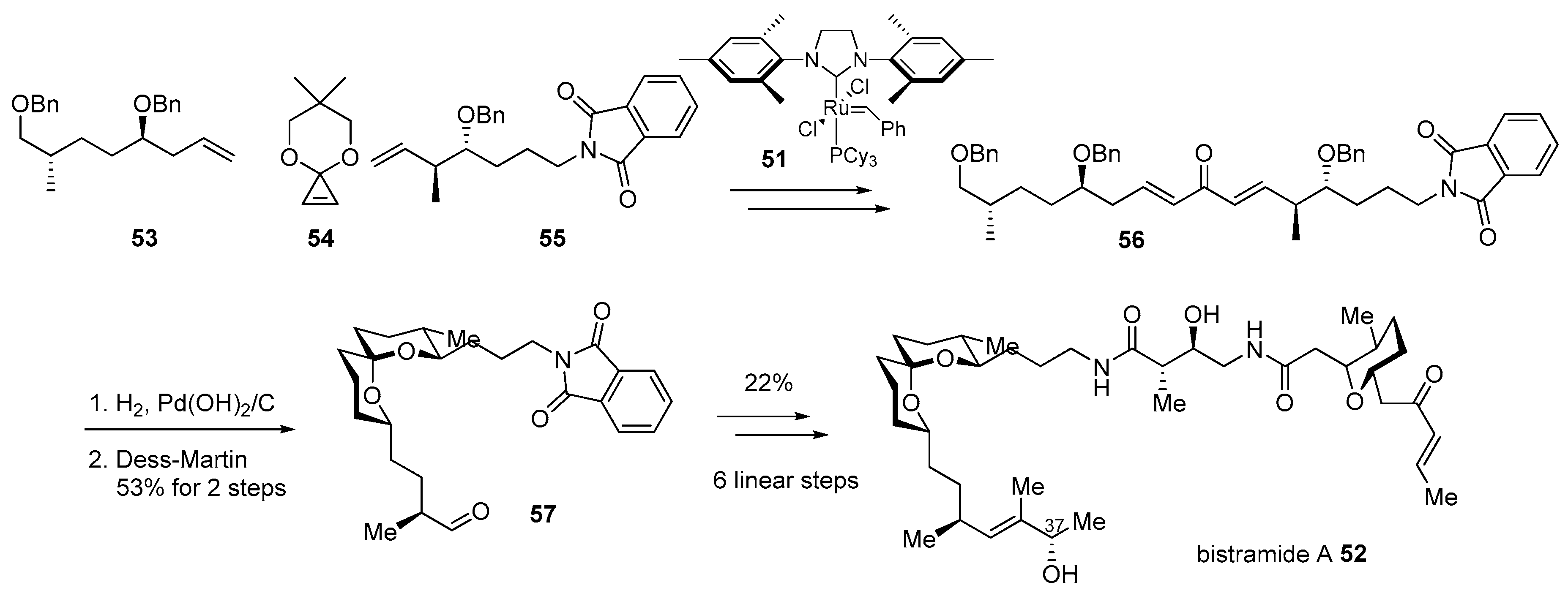
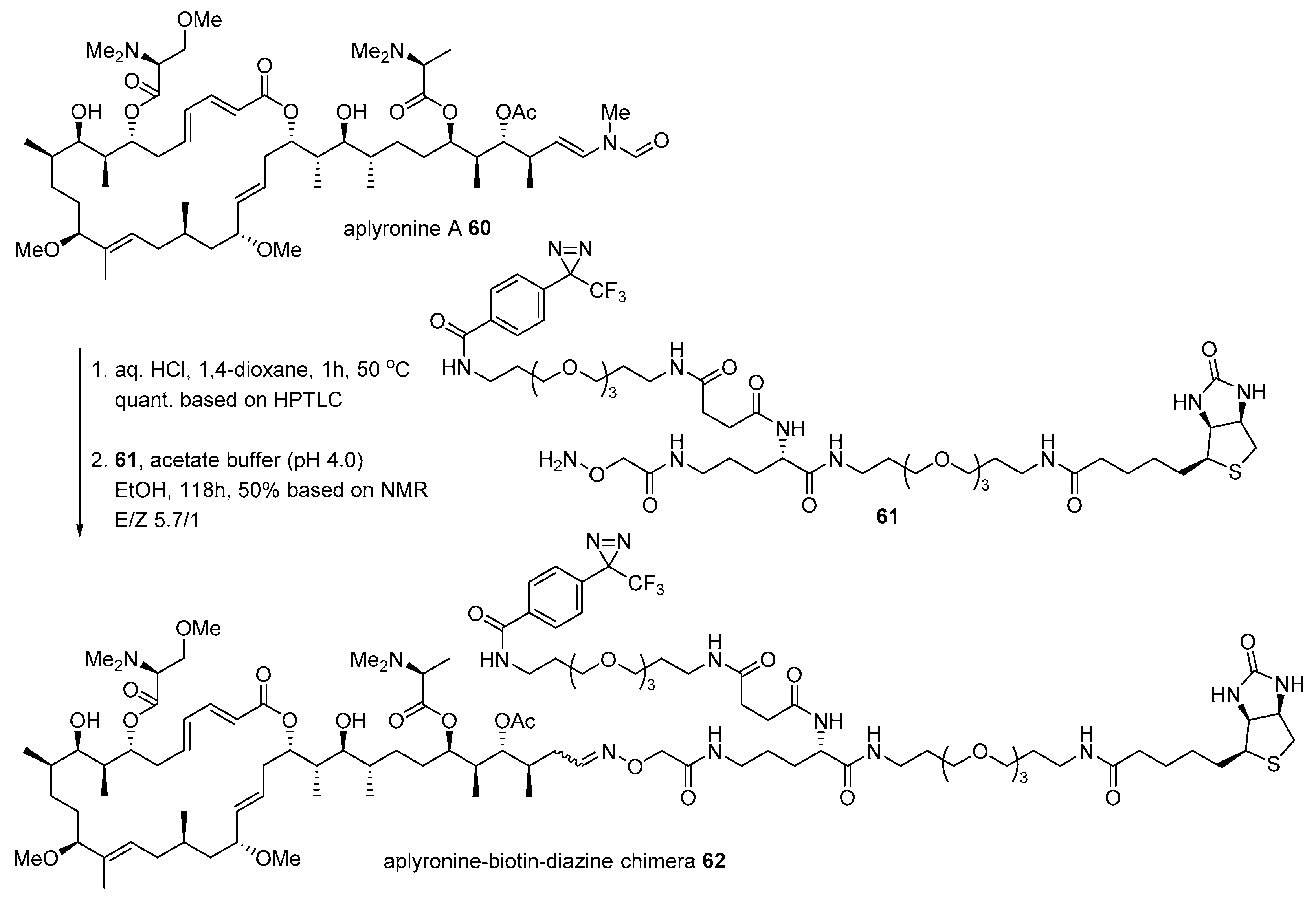
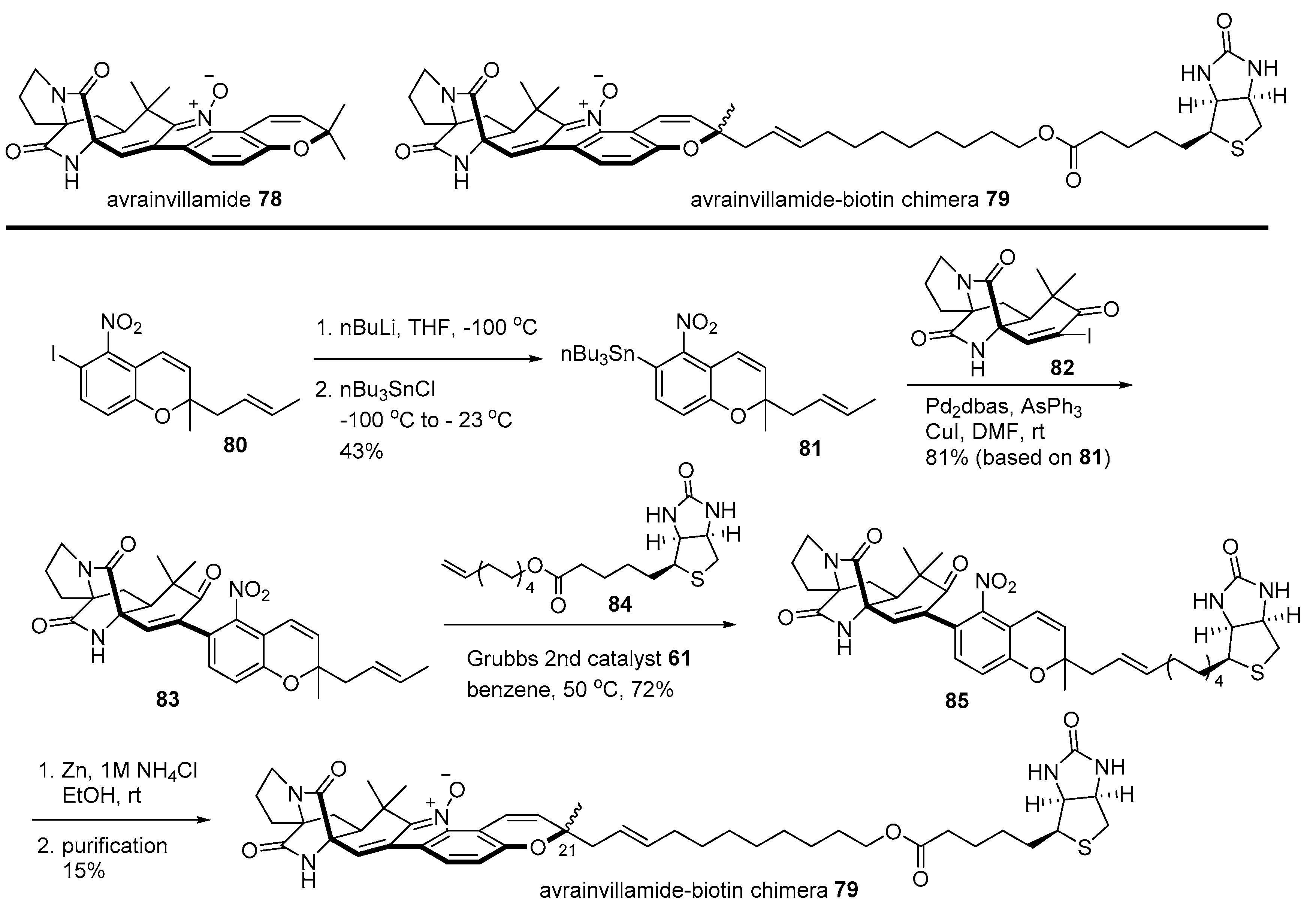
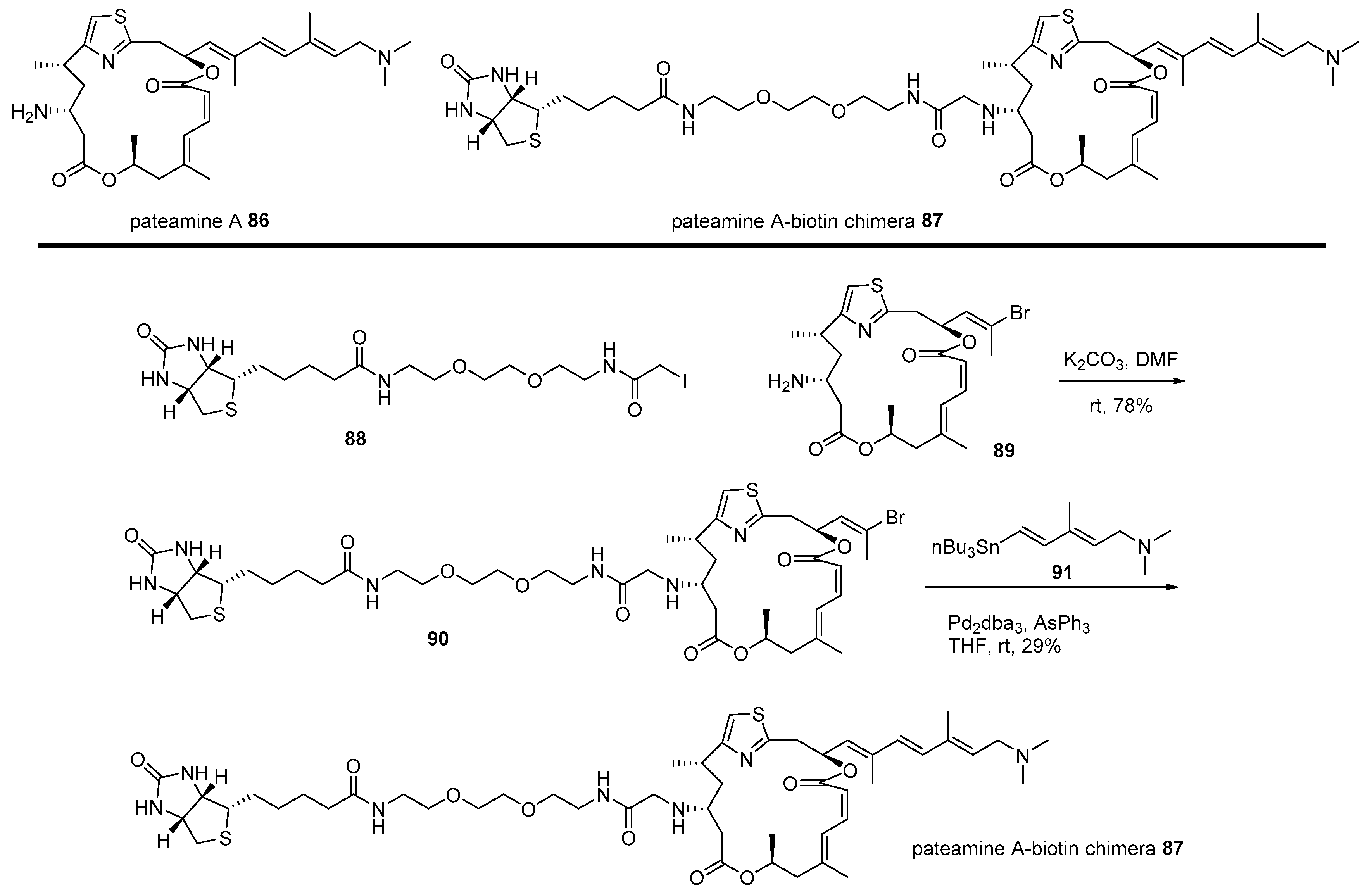
2.1. Conjugation with Other Active Molecules
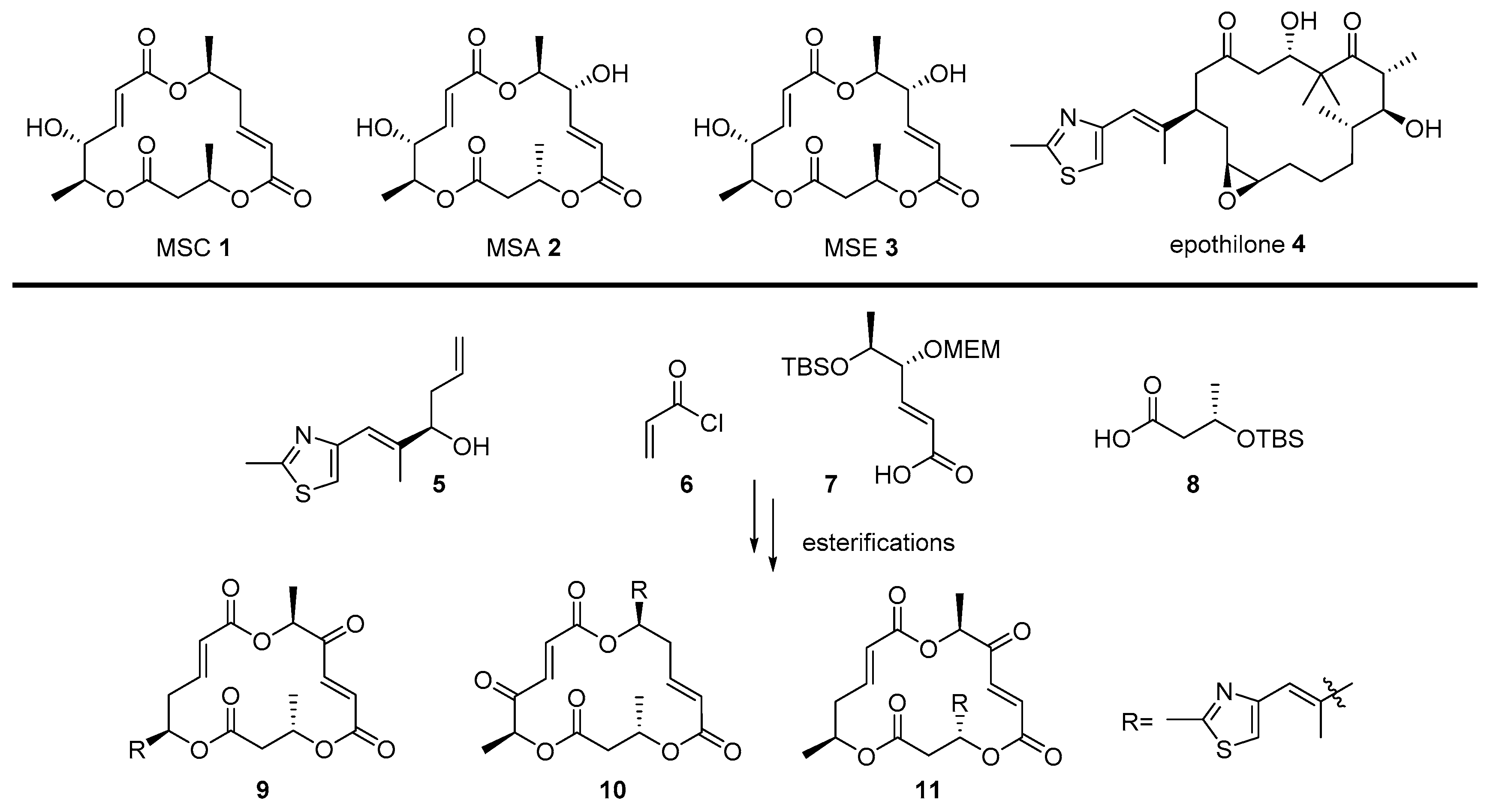
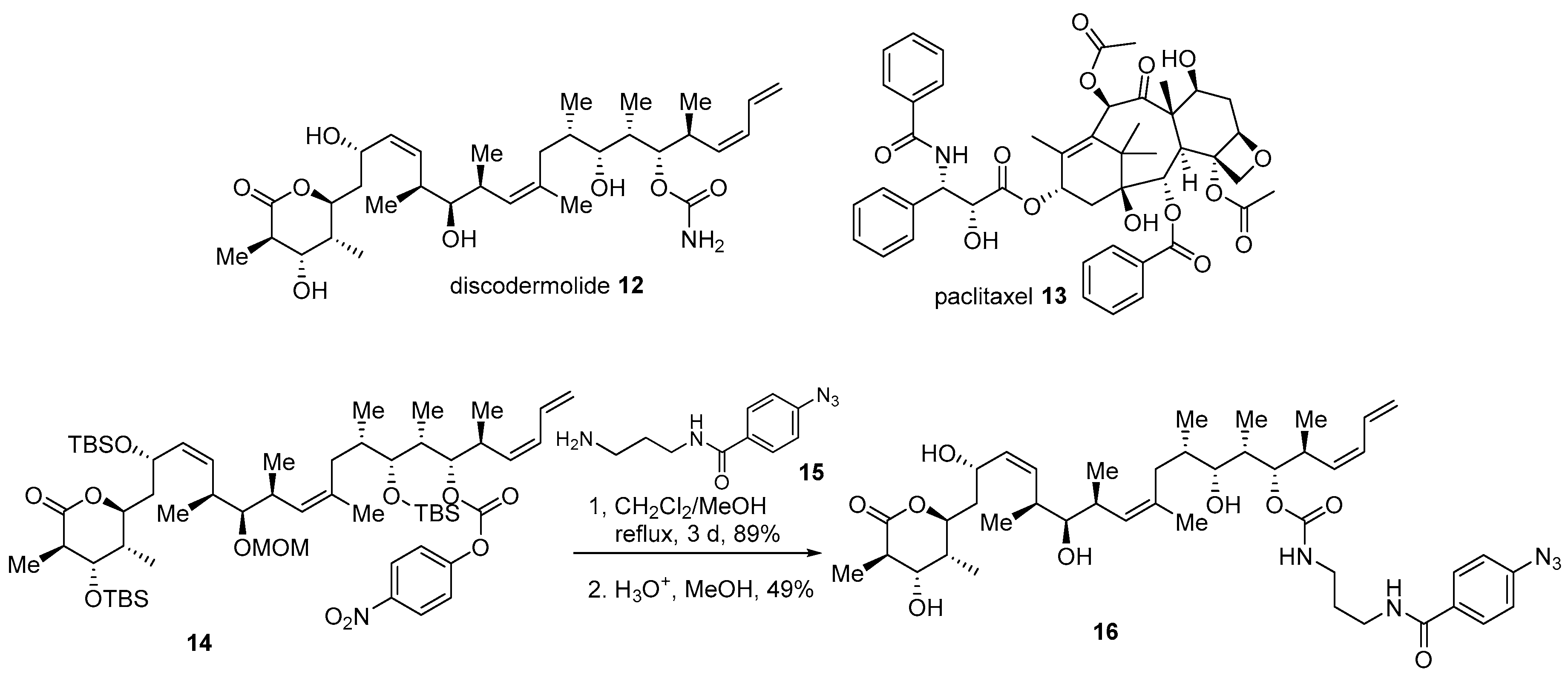
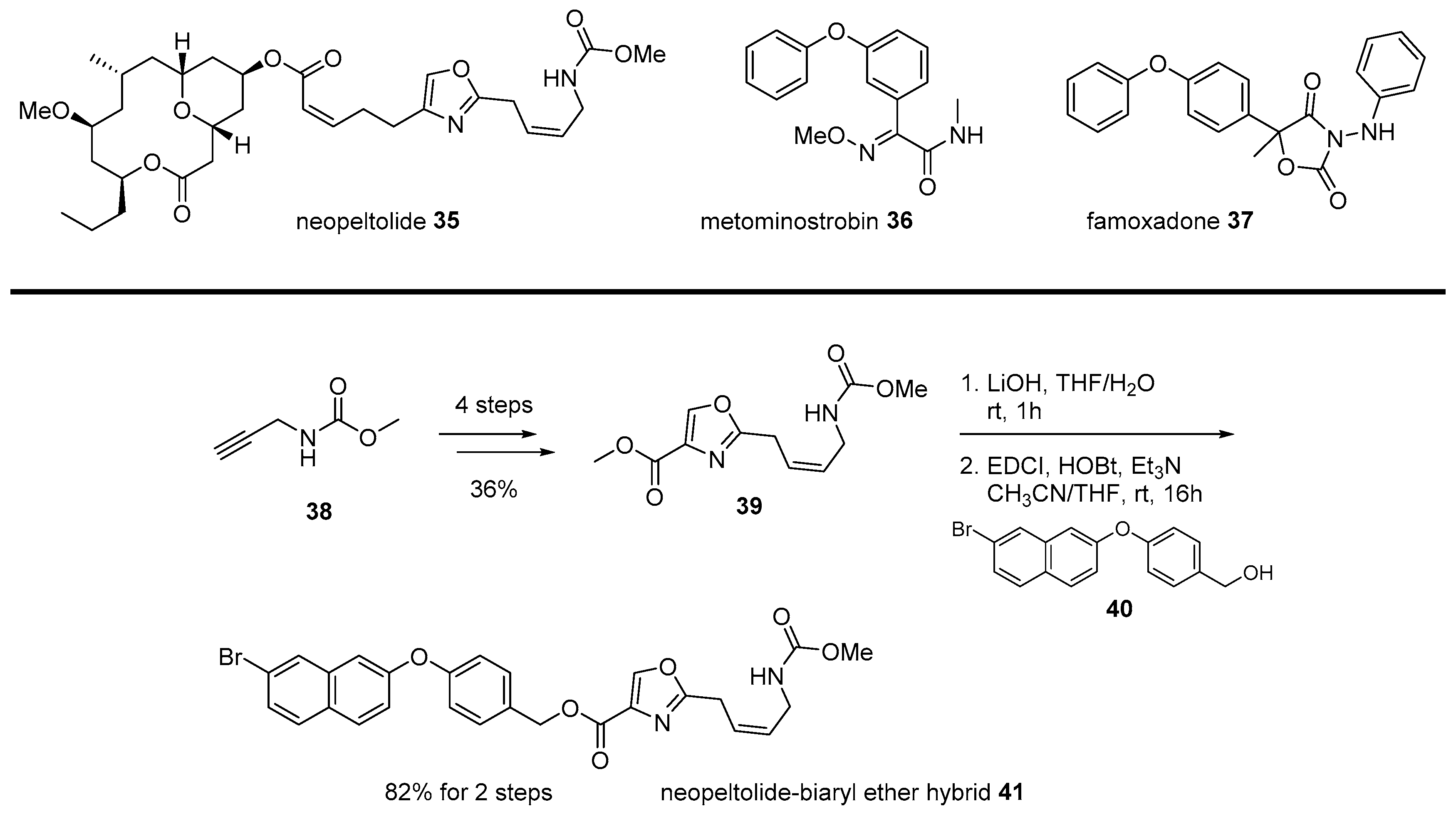
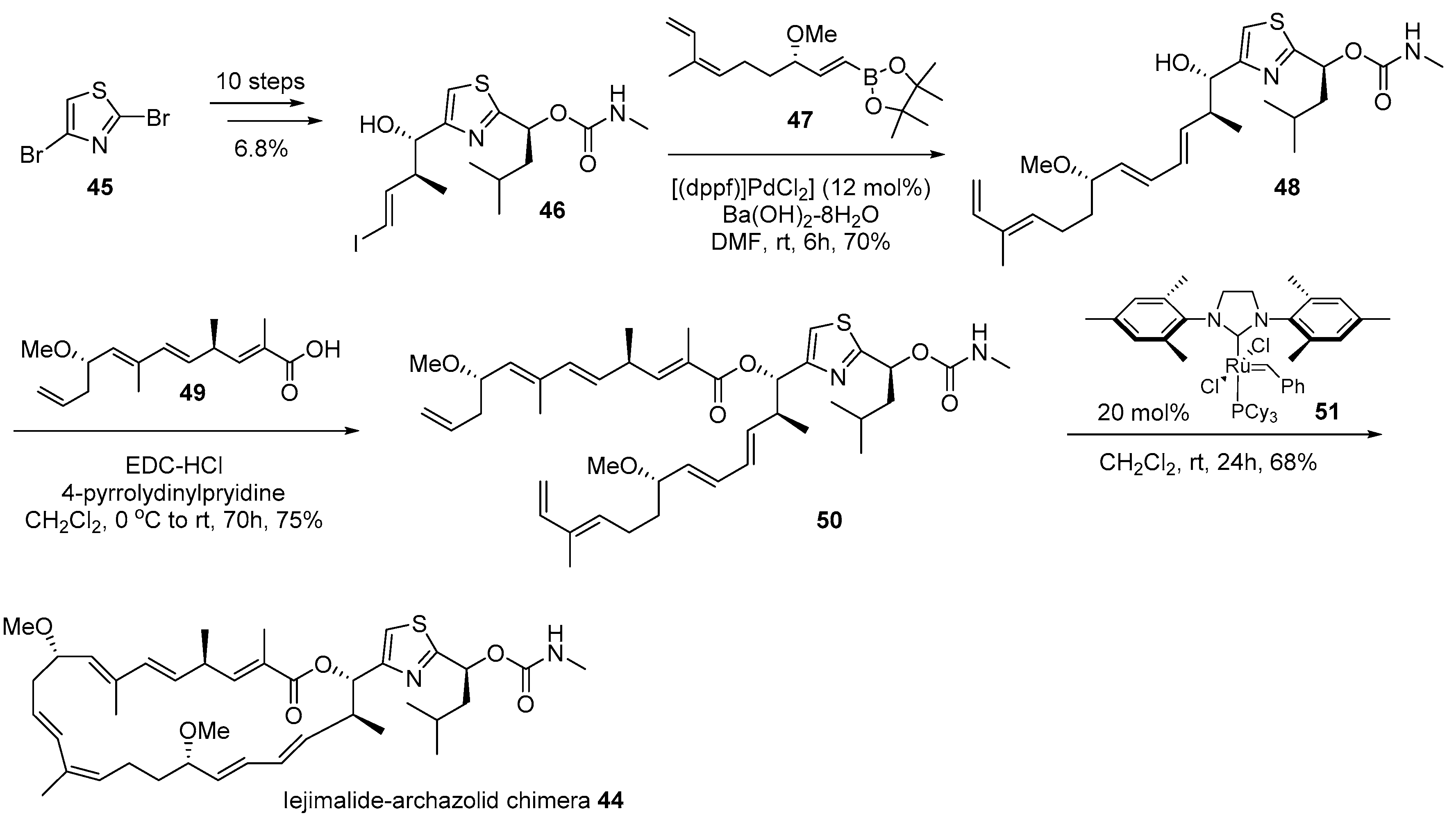
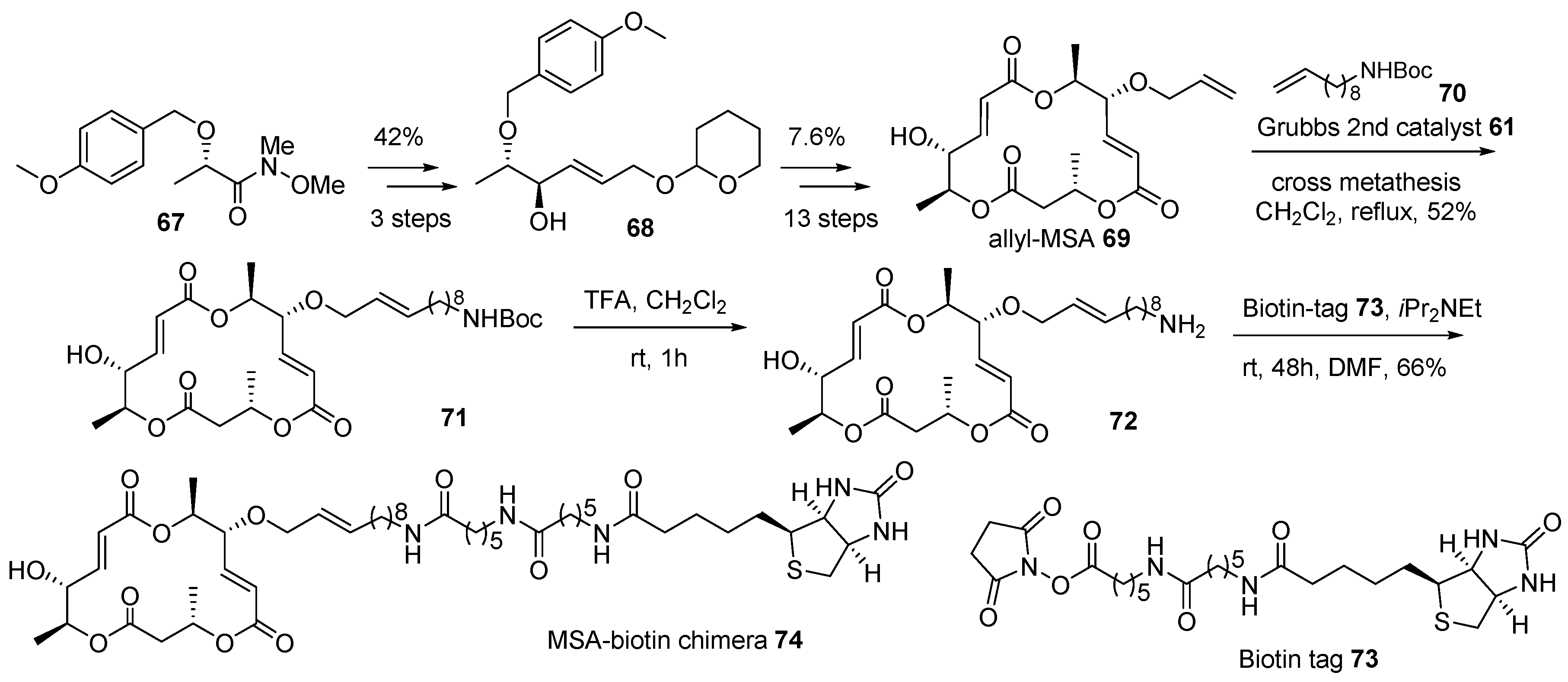
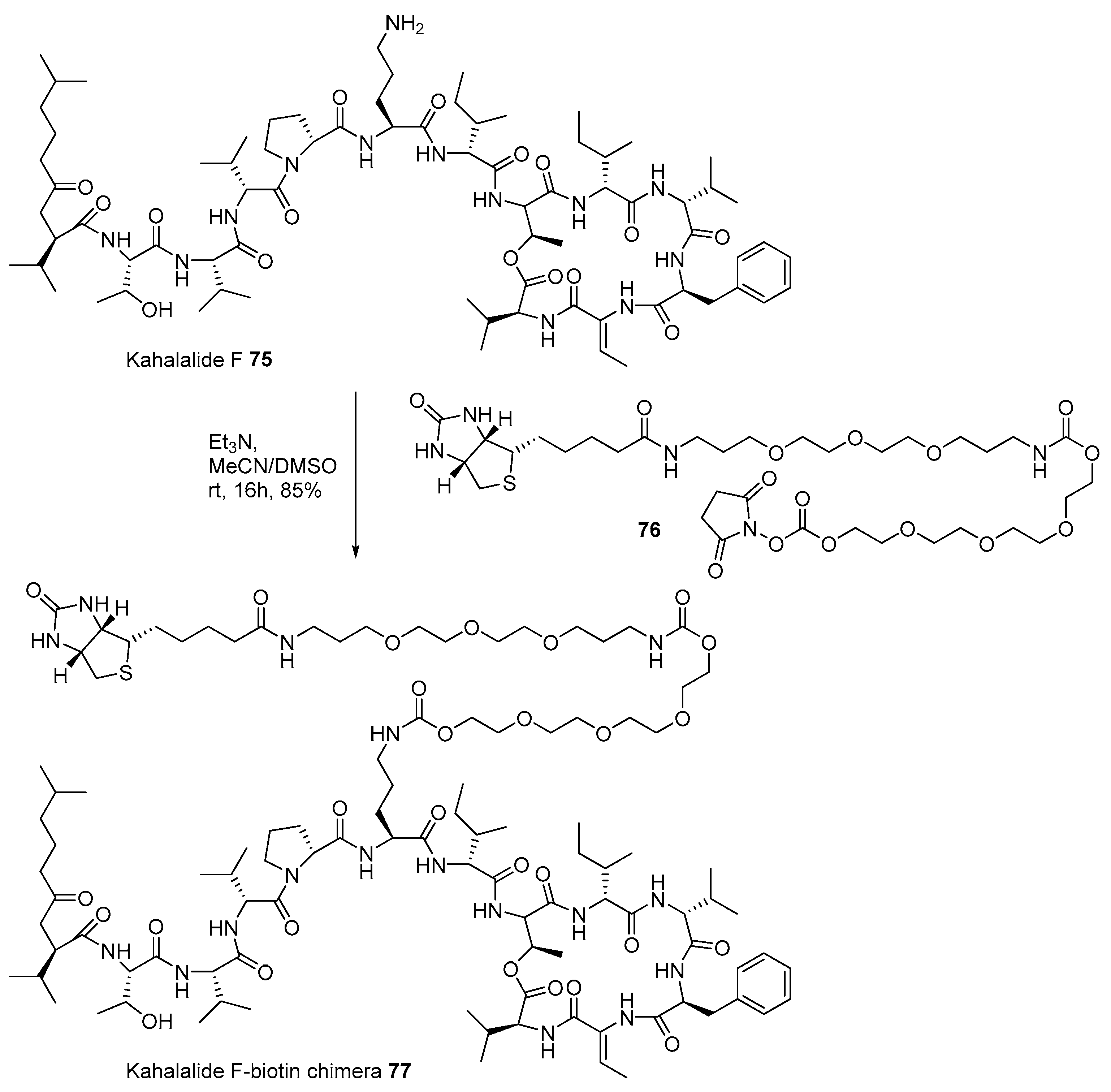
2.2. Conjugation with Other Functional Compounds
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.M.; Goncalves, B.D.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, P.; D’Auria, M.V.; Muller, C.D.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring marine resources for bioactive compounds. Planta Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lou, H.-X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Chicacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N. Antineoplastic agents. 257. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Smith, A.B.; Razler, T.M.; Meis, R.M.; Pettit, G.R. Synthesis and biological evaluation of phorboxazole congeners leading to the discovery and preparative-scale synthesis of (+)-chlorophorboxazole a possessing picomolar human solid tumor cell growth inhibitory activity. J. Org. Chem. 2008, 73, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Schaufelberger, D.E.; Koleck, M.P.; Beutler, J.A.; Vatakis, A.M.; Alvarado, A.B.; Andrews, P.; Marzo, L.; Muschik, G.; Roach, J.; Ross, J.T. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991, 54, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B., III; Tomioka, T.; Risatti, C.A.; Sperry, J.B.; Sfouggatakis, C. Gram-scale synthesis of (+)-spongistatin 1: Development of an improved, scalable synthesis of the F-ring subunit, fragment union, and final elaboration. Org. Lett. 2008, 10, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Beauchamp, T.J.; LaMarche, M.J.; Kaufman, M.D.; Qiu, Y.; Arimoto, H.; Jones, D.R.; Kobayashi, K. Evolution of a gram-scale synthesis of (+)-discodermolide. J. Am. Chem. Soc. 2000, 122, 8654–8664. [Google Scholar] [CrossRef]
- Mickel, S.J.; Niederer, D.; Daeffler, R.; Osmani, A.; Kuesters, E.; Schmid, E.; Schaer, K.; Gamboni, R.; Chen, W.; Loeser, E. Large-scale synthesis of the anti-cancer marine natural product (+)-Discodermolide. Part 5: Linkage of fragments C1-6 and C7-24 and finale. Org. Process. Res. Dev. 2004, 8, 122–130. [Google Scholar] [CrossRef]
- Choudhary, A.; Naughton, L.; Montánchez, I.; Dobson, A.; Rai, D. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Lear, M.J.; Hirai, K.; Ogawa, K.; Yamashita, S.; Hirama, M. A convergent total synthesis of the kedarcidin chromophore: 20-years in the making. J. Antibiot. 2019, 72, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.-H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- McBride, A.; Butler, S.K. Eribulin mesylate: A novel halichondrin B analogue for the treatment of metastatic breast cancer. Am. J. Health Syst. Pharm. 2012, 69, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561. [Google Scholar] [CrossRef] [PubMed]
- Bebbington, M.W. Natural product analogues: Towards a blueprint for analogue-focused synthesis. Chem. Soc. Rev. 2017, 46, 5059–5109. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Edit. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Edit. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Mehta, G.; Singh, V. Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101. [Google Scholar] [CrossRef]
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Olaáh, J.; Ovaádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2017, 61, 482–491. [Google Scholar] [CrossRef]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar] [CrossRef]
- Paek, S. Synthetic advances in macrosphelides: Natural anticancer agents. Molecules 2014, 19, 15982–16000. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysia sea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, Y.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Sunazuka, T.; Hirose, T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Ōmura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell—Cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar] [CrossRef]
- Paek, S. Development of advanced macrosphelides: Potent anticancer agents. Molecules 2015, 20, 4430–4449. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.; Kondo, T.; Nemoto, H. Synthesis of macrosphelides with a thiazole side chain: New antitumor candidates having apoptosis-inducing property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar] [CrossRef]
- Meng, D.; Bertinato, P.; Balog, A.; Su, D.; Kamenecka, T.; Sorensen, E.J.; Danishefsky, S.J. Total syntheses of epothilones A and B. J. Am. Chem. Soc. 1997, 119, 10073–10092. [Google Scholar] [CrossRef]
- Xia, S.; Kenesky, C.S.; Rucker, P.V.; Smith, A.B.; Orr, G.A.; Horwitz, S.B. A photoaffinity analogue of discodermolide specifically labels a peptide in β-tubulin. Biochemistry 2006, 45, 11762–11775. [Google Scholar] [CrossRef]
- Smith, A.B., III; Sugasawa, K.; Atasoylu, O.; Yang, C.H.; Horwitz, S.B. Design and synthesis of (+)-discodermolide–paclitaxel hybrids leading to enhanced biological activity. J. Med. Chem. 2011, 54, 6319–6327. [Google Scholar] [CrossRef]
- Shin, Y.; Choy, N.; Balachandran, R.; Madiraju, C.; Day, B.W.; Curran, D.P. Discodermolide/dictyostatin hybrids: Synthesis and biological evaluation. Org. Lett. 2002, 4, 4443–4446. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Schmidt, J.M. Isolation and structure of the cancer cell growth inhibitor dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994, 1111–1112. [Google Scholar] [CrossRef]
- Isbrucker, R.A.; Cummins, J.; Pomponi, S.A.; Longley, R.E.; Wright, A.E. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem. Pharmacol. 2003, 66, 75–82. [Google Scholar] [CrossRef]
- Zanato, C.; Pignataro, L.; Hao, Z.; Gennari, C. A practical synthesis of the C1–C9 fragment of dictyostatin. Synthesis 2008, 2008, 2158–2162. [Google Scholar]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef]
- Clark, W.D.; Corbett, T.; Valeriote, F.; Crews, P. Cyclocinamide A. An unusual cytotoxic halogenated hexapeptide from the marine sponge Psammocinia. J. Am. Chem. Soc. 1997, 119, 9285–9286. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tanaka, J.I.; Katori, T.; Matsuura, M.; Kitagawa, I. Structure of swinholide A, a potent cytotoxic macrolide from the Okinawan marine sponge Tehonella Swinhoei. Tetrahedron Lett. 1989, 30, 2963–2966. [Google Scholar] [CrossRef]
- Faulkner, D.J. Variabilin, an antibiotic from the sponge, Ircinia variabilis. Tetrahedron Lett. 1973, 39, 3821–3822. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.; Chapuis, J.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge Ircinia r amosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef]
- Jiang, X.; García-Fortanet, J.; De Brabander, J.K. Synthesis and complete stereochemical assignment of psymberin/irciniastatin A. J. Am. Chem. Soc. 2005, 127, 11254–11255. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Jiang, X.; Williams, N.; De Brabander, J.K. Synthesis of psymberin analogues: Probing a functional correlation with the pederin/mycalamide family of natural products. Org. Lett. 2007, 9, 227–230. [Google Scholar] [CrossRef]
- Bartik, K.; Braekman, J.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentins, new toxic bis-indole alkaloids from the marine sponge Topsentia genitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis (indolyl) imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Gu, X.; Wan, X.; Jiang, B. Syntheses and biological activities of bis (3-indolyl) thiazoles, analogues of marine bis (indole) alkaloid nortopsentins. Bioorg. Med. Chem. Lett. 1999, 9, 569–572. [Google Scholar] [CrossRef]
- Hogan, I.; Sainsbury, M. The synthesis of dendrodoine, 5-[3-(N, N-dimethylamino-1, 2, 4-thiadiazolyl]-3-indolylmethanone, a metabolite of the marine tunicate dendroda grossular. Tetrahedron 1984, 40, 681–682. [Google Scholar] [CrossRef]
- Juneja, M.; Vanam, U.; Paranthaman, S.; Bharathan, A.; Keerthi, V.S.; Reena, J.K.; Rajaram, R.; Rajasekharan, K.N.; Karunagaran, D. 4-amino-2-arylamino-5-indoloyl/cinnamoythiazoles, analogs of topsentin-class of marine alkaloids, induce apoptosis in hela cells. Eur. J. Med. Chem. 2013, 63, 474–483. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total synthesis and structural revision of the marine macrolide neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Hines, J.; Crews, C.M.; Scheidt, K.A. Total synthesis and structure—Activity investigation of the marine natural product neopeltolide. J. Am. Chem. Soc. 2009, 131, 12406–12414. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Janjic, J.; Suzuki, M.; Sabharwal, S.S.; Schumacker, P.T.; Kron, S.J.; Kozmin, S.A. Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat. Chem. Biol. 2008, 4, 418. [Google Scholar] [CrossRef]
- Zhu, X.L.; Z’hang, R.; Wu, Q.; Song, Y.; Wang, Y.; Yang, J.; Yang, G. Natural Product Neopeltolide as A Cytochrome bc1 Complex Inhibitor: Mechanism of Action and Structural Modification. J. Agric. Food Chem. 2019, 67, 2774–2781. [Google Scholar] [CrossRef]
- Vintonyak, V.V.; Kunze, B.; Sasse, F.; Maier, M.E. Total synthesis and biological activity of neopeltolide and analogues. Chem. Eur. J. 2008, 14, 11132–11140. [Google Scholar] [CrossRef]
- Xiong, L.; Li, H.; Jiang, L.; Ge, J.; Yang, W.; Zhu, X.L.; Yang, G. Structure-based discovery of potential fungicides as succinate ubiquinone oxidoreductase inhibitors. J. Agric. Food Chem. 2017, 65, 1021–1029. [Google Scholar] [CrossRef]
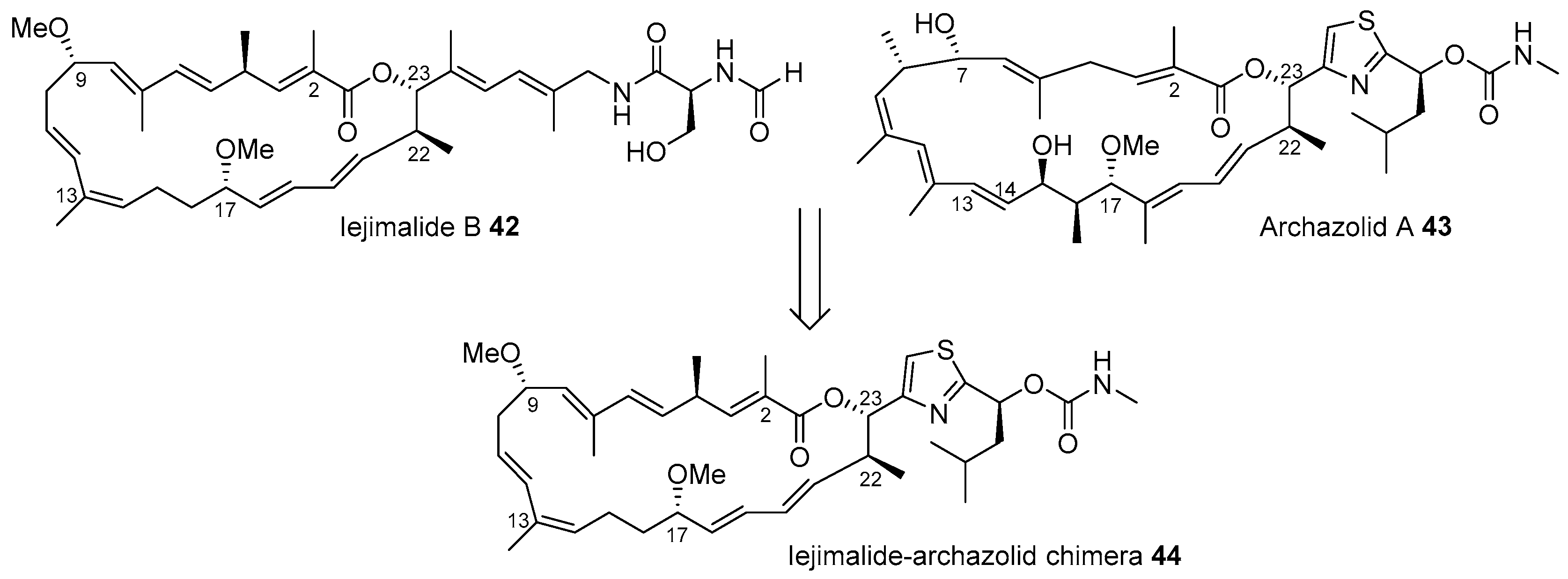
- Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.; Fürstner, A. Synthesis and evaluation of an Iejimalide-archazolid chimera. Tetrahedron 2010, 66, 6421–6428. [Google Scholar] [CrossRef]
- Kobayashi, J.; Cheng, J.; Ohta, T.; Nakamura, H.; Nozoe, S.; Hirata, Y.; Ohizumi, Y.; Sasaki, T. Iejimalides A and B, novel 24-membered macrolides with potent antileukemic activity from the Okinawan tunicate Eudistoma cf. rigida. J. Org. Chem. 1988, 53, 6147–6150. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Iejimalides C and D, new antineoplastic 24-membered macrolide sulfates from the Okinawan marine tunicate Eudistoma cf. rigida. Tetrahedron Lett. 1991, 32, 797–798. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Waser, M. Total synthesis of iejimalide B. Angew. Chem. Int. Edit. 2006, 45, 5837–5842. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Waser, M.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Moulin, E.; Müller, O. Total Synthesis of Iejimalide A–D and Assessment of the Remarkable Actin-Depolymerizing Capacity of These Polyene Macrolides. J. Am. Chem. Soc. 2007, 129, 9150–9161. [Google Scholar] [CrossRef]
- Sasse, F.; Steinmetz, H.; Hoefle, G.; Reichenbach, H. Archazolids, New Cytotoxic Macrolactones from Archangium gephyra (Myxobacteria). J. Antibiot. 2003, 56, 520–525. [Google Scholar] [CrossRef]
- Corey, E.J.; Helal, C.J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Edit. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Marshall, J.A. Chiral allylic and allenic metal reagents for organic synthesis. J. Org. Chem. 2007, 72, 8153–8166. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Ogba, O.; Warner, N.; O’Leary, D.; Grubbs, R. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Nordlund, H.R.; Hytönen, V.P.; Kulomaa, M.S. Brave new (strept) avidins in biotechnology. Trends Biotechnol. 2007, 25, 269–277. [Google Scholar] [CrossRef]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin–biotin technology: Improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef]
- Kim, J.; Cantor, A.B.; Orkin, S.H.; Wang, J. Use of in vivo biotinylation to study protein–protein and protein–DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009, 4, 506–517. [Google Scholar] [CrossRef]
- Manz, B.; Heubner, A.; Kohler, I.; Grill, H.; Pollow, K. Synthesis of Biotin-Labelled Dexamethasone Derivatives: Novel Hormone-Affinity Probes. Eur. J. Biochem. 1983, 131, 333–338. [Google Scholar] [CrossRef]
- Wright, M.; Sieber, S. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2010, 6, 34–46. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Shirakawa, T.; Uesugi, M. Biochemical target isolation for novices: Affinity-based strategies. Chem. Biol. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Gouiffes, D.; Moreau, S.; Helbecque, N.; Bernier, J.; Henichart, J.; Barbin, Y.; Laurent, D.; Verbist, J. Proton nuclear magnetic study of bistramide A, a new cytotoxic drug isolated from Lissoclinum bistratum Sluiter. Tetrahedron 1988, 44, 451–459. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; McCaffrey, E.J.; Parry, D.L.; Watters, D.J. Novel cytotoxic compounds from the ascidian Lissoclinum bistratum. J. Med. Chem. 1989, 32, 1354–1359. [Google Scholar] [CrossRef]
- Biard, J.; Roussakis, C.; Kornprobst, J.; Gouiffes-Barbin, D.; Verbist, J.; Cotelle, P.; Foster, M.P.; Ireland, C.M.; Debitus, C. Bistramides A, B, C, D, and K: A new class of bioactive cyclic polyethers from Lissoclinum bistratum. J. Nat. Prod. 1994, 57, 1336–1345. [Google Scholar] [CrossRef]
- Riou, D.; Roussakis, C.; Biard, J.F.; Verbist, J.F. Comparative study of the antitumor activity of bistramides A, D and K against a non-small cell broncho-pulmonary carcinoma. Anticancer Res. 1993, 13, 2331–2334. [Google Scholar]
- Wipf, P.; Uto, Y.; Yoshimura, S. Total synthesis of a stereoisomer of bistramide C and assignment of configuration of the natural product. Chem. Eur. J. 2002, 8, 1670–1681. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Liu, D.; Kozmin, S.A. Synthesis of bistramide A. J. Am. Chem. Soc. 2004, 126, 9546–9547. [Google Scholar] [CrossRef]
- Gagne, M.R.; Grubbs, R.H.; Feldman, J.; Ziller, J.W. Catalytic activity of a well-defined binuclear ruthenium alkylidene complex. Organometallics 1992, 11, 3933–3935. [Google Scholar] [CrossRef]
- Foster, M.P.; Mayne, C.L.; Dunkel, R.; Pugmire, R.J.; Grant, D.M.; Kornprobst, J.M.; Verbist, J.F.; Biard, J.F.; Ireland, C.M. Revised structure of bistramide A (bistratene A): Application of a new program for the automated analysis of 2D INADEQUATE spectra. J. Am. Chem. Soc. 1992, 114, 1110–1111. [Google Scholar] [CrossRef]
- Johnson, W.E.B.; Watters, D.J.; Suniara, R.K.; Brown, G.; Bunce, C.M. Bistratene A induces a microtubule-dependent block in cytokinesis and altered stathmin expression in HL60 cells. Biochem. Biophys. Res. Commun. 1999, 260, 80–88. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Sun, H.; Low, K.E.; Woo, S.; Noble, R.L.; Graham, R.J.; Connaughton, S.S.; Gee, M.A.; Lee, L.G. Real-time protein kinase assay. Anal. Chem. 2005, 77, 2043–2049. [Google Scholar] [CrossRef]
- Yamada, K.; Ojika, M.; Ishigaki, T.; Yoshida, Y.; Ekimoto, H.; Arakawa, M. Aplyronine A, a potent antitumor substance and the congeners aplyronines B and C isolated from the sea hare Aplysia kurodai. J. Am. Chem. Soc. 1993, 115, 11020–11021. [Google Scholar] [CrossRef]
- Kita, M.; Hirayama, Y.; Yoneda, K.; Yamagishi, K.; Chinen, T.; Usui, T.; Sumiya, E.; Uesugi, M.; Kigoshi, H. Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J. Am. Chem. Soc. 2013, 135, 18089–18095. [Google Scholar] [CrossRef]
- Paterson, I.; Fink, S.J.; Lee, L.Y.; Atkinson, S.J.; Blakey, S.B. Total synthesis of aplyronine C. Org. Lett. 2013, 15, 3118–3121. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; Van Duyne, G.D.; Clardy, J. Isolation and structure determination of diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. [Google Scholar] [CrossRef]
- Li, J.; Jeong, S.; Esser, L.; Harran, P.G. Total synthesis of nominal Diazonamides—Part 1: Convergent preparation of the structure proposed for (−)-Diazonamide A. Angew. Chem. Int. Edit. 2001, 40, 4765–4769. [Google Scholar] [CrossRef]
- Li, J.; Burgett, A.W.; Esser, L.; Amezcua, C.; Harran, P.G. Total synthesis of nominal diazonamides—Part 2: On the true structure and origin of natural isolates. Angew. Chem. Int. Edit. 2001, 40, 4770–4773. [Google Scholar] [CrossRef]
- Nicolaou, K.; Bella, M.; Chen, D.Y.; Huang, X.; Ling, T.; Snyder, S.A. Total synthesis of diazonamide A. Angew. Chem. Int. Edit. 2002, 41, 3495–3499. [Google Scholar] [CrossRef]
- Wang, G.; Shang, L.; Burgett, A.W.; Harran, P.G.; Wang, X. Diazonamide toxins reveal an unexpected function for ornithine δ-amino transferase in mitotic cell division. Proc. Natl. Acad. Sci. USA 2007, 104, 2068–2073. [Google Scholar] [CrossRef]
- Seiler, N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Curr. Drug Targets 2000, 1, 119–154. [Google Scholar] [CrossRef]
- Wieczorek, M.; Tcherkezian, J.; Bernier, C.; Prota, A.E.; Chaaban, S.; Rolland, Y.; Godbout, C.; Hancock, M.A.; Arezzo, J.C.; Ocal, O. The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med. 2016, 8, 365ra159. [Google Scholar] [CrossRef]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.; Suh, Y. Design and synthesis of a macrosphelide A-biotin chimera. Org. Biomol. Chem. 2014, 12, 7127–7135. [Google Scholar] [CrossRef]
- Paek, S.; Seo, S.; Kim, S.; Jung, J.; Lee, Y.; Jung, J.; Suh, Y. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Faircloth, G.; Marchante, M.d.C.C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs. In Molluscs; Springer: Berlin/Heidelberg, Germany, 2006; pp. 363–379. [Google Scholar]
- Piggott, A.M.; Karuso, P. Rapid identification of a protein binding partner for the marine natural product kahalalide F by using reverse chemical proteomics. ChemBioChem 2008, 9, 524–530. [Google Scholar] [CrossRef]
- Wulff, J.E.; Siegrist, R.; Myers, A.G. The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J. Am. Chem. Soc. 2007, 129, 14444–14451. [Google Scholar] [CrossRef]
- Sugie, Y.; Hirai, H.; Inagaki, T.; Ishiguro, M.; Kim, Y.; Kojima, Y.; Sakakibara, T.; Sakemi, S.; Sugiura, A.; Suzuki, Y. A new antibiotic CJ-17, 665 from Aspergillus ochraceus. J. Antibiot. 2001, 54, 911–916. [Google Scholar] [CrossRef]
- Herzon, S.B.; Myers, A.G. Enantioselective synthesis of stephacidin B. J. Am. Chem. Soc. 2005, 127, 5342–5344. [Google Scholar] [CrossRef]
- Wulff, J.E.; Herzon, S.B.; Siegrist, R.; Myers, A.G. Evidence for the rapid conversion of stephacidin B into the electrophilic monomer avrainvillamide in cell culture. J. Am. Chem. Soc. 2007, 129, 4898–4899. [Google Scholar] [CrossRef]
- Chan, W.Y.; Liu, Q.R.; Borjigin, J.; Busch, H.; Rennert, O.M.; Tease, L.A.; Chan, P.K. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28, 1033–1039. [Google Scholar] [CrossRef]
- Myers, A.G.; Herzon, S.B.; Wulff, J.E.; Siegrist, R.; Svenda, J.; Zajac, M.A. Synthesis of Avrainvillamide, Stephacidin B, and Analogues Thereof. U.S. Patent 20110166170A1, 1 July 2011. [Google Scholar]
- Northcote, P.T.; Blunt, J.W.; Munro, M.H. Pateamine: A potent cytotoxin from the New Zealand marine sponge, Mycale sp. Tetrahedron Lett. 1991, 32, 6411–6414. [Google Scholar] [CrossRef]
- Romo, D.; Rzasa, R.M.; Shea, H.A.; Park, K.; Langenhan, J.M.; Sun, L.; Akhiezer, A.; Liu, J.O. Total synthesis and immunosuppressive activity of (−)-pateamine A and related compounds: Implementation of a β-lactam-based macrocyclization. J. Am. Chem. Soc. 1998, 120, 12237–12254. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Rzasa, R.M.; Shea, H.A.; Li, S.; Park, K.; Ma, G. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product Pateamine A. Meth. Enzymol. 2007, 431, 303–324. [Google Scholar]
- Low, W.; Dang, Y.; Bhat, S.; Romo, D.; Liu, J.O. Substrate-dependent targeting of eukaryotic translation initiation factor 4A by pateamine A: Negation of domain-linker regulation of activity. Chem. Biol. 2007, 14, 715–727. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, M.W.; Song, B.R.; Chung, H.J.; Paek, S.-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Mar. Drugs 2019, 17, 500. https://doi.org/10.3390/md17090500
Ha MW, Song BR, Chung HJ, Paek S-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Marine Drugs. 2019; 17(9):500. https://doi.org/10.3390/md17090500
Chicago/Turabian StyleHa, Min Woo, Bo Reum Song, Hye Jin Chung, and Seung-Mann Paek. 2019. "Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products" Marine Drugs 17, no. 9: 500. https://doi.org/10.3390/md17090500
APA StyleHa, M. W., Song, B. R., Chung, H. J., & Paek, S.-M. (2019). Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Marine Drugs, 17(9), 500. https://doi.org/10.3390/md17090500