Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues


 , ,
, , 
Abstract
1. Introduction
2. Results
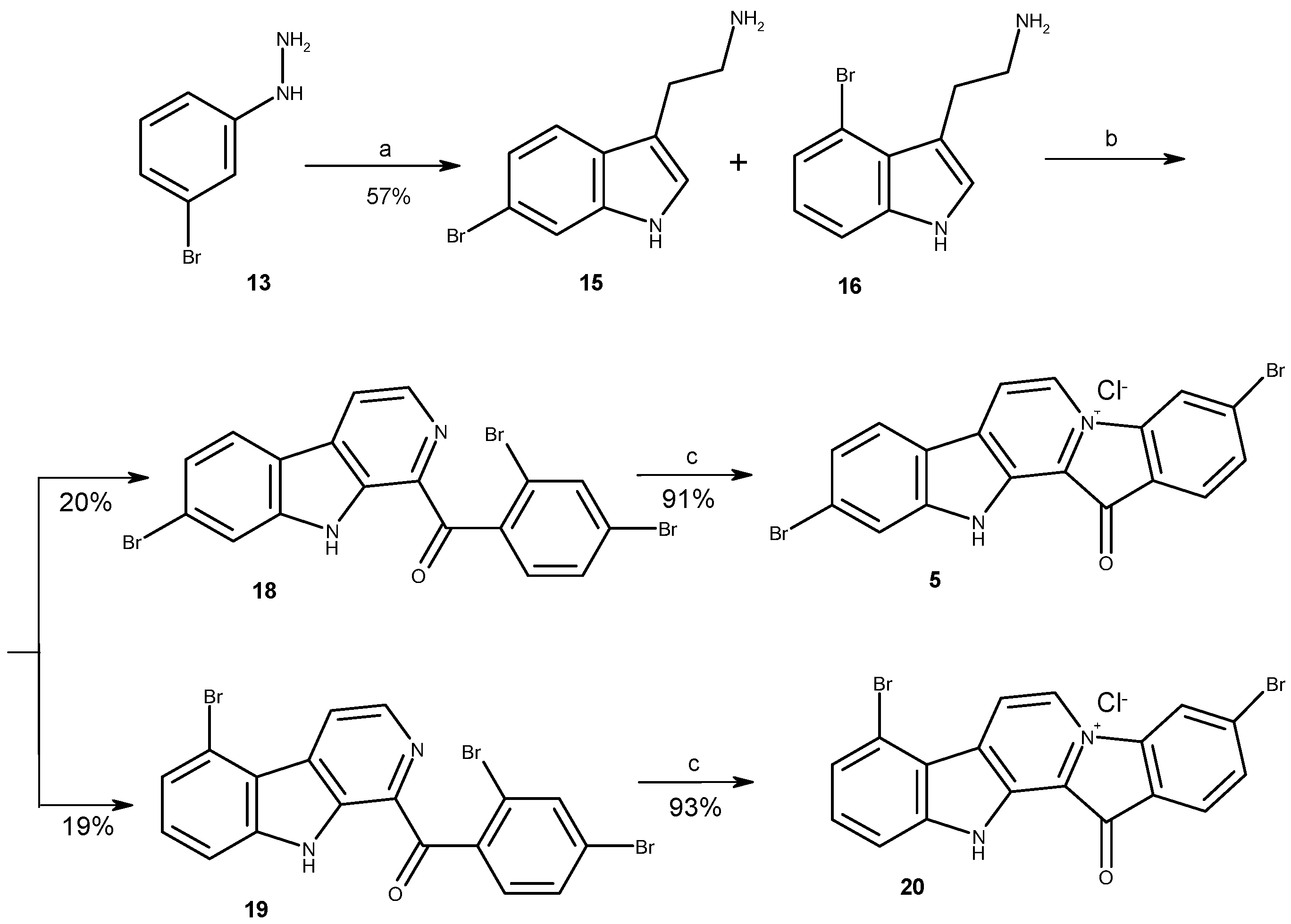
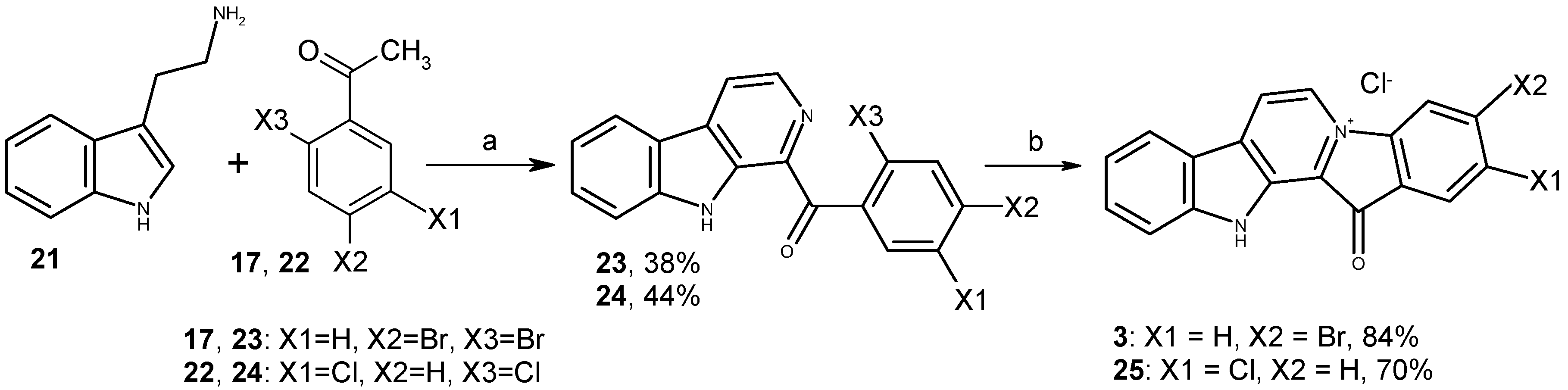
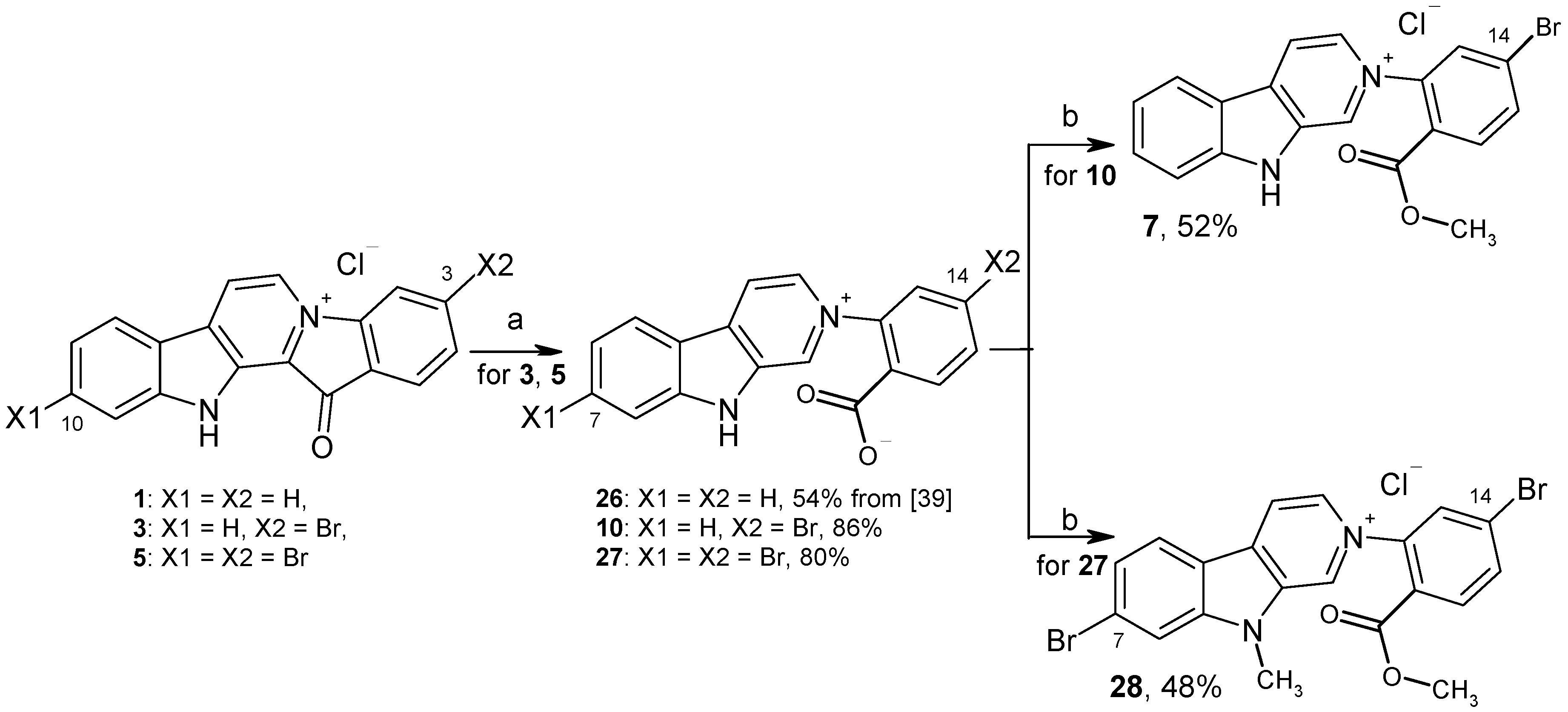
2.1. Chemistry
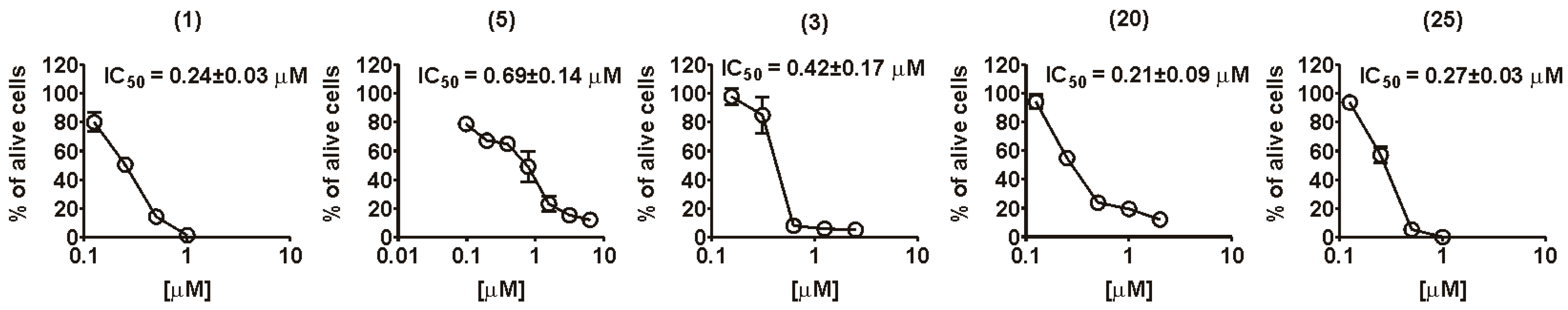
2.2. Biology
3. Materials and Methods
3.1. Chemistry
3.1.1. Preparation of Mixture of Tryptamines 15 and 16
3.1.2. Preparation of Substituted 1-Benzoyl-β-Carbolines 18, 19, 23, 24
3.1.3. Preparation of Fascaplysin Derivatives
3.1.4. Preparation of Compounds 10, 27
3.1.5. Preparation of 14-Bromoreticulatine (7) and Compound 28
3.2. Biological Evaluation
3.2.1. Cell Lines and Culture Conditions
3.2.2. Cytotoxicity Assays
3.2.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-derived CDK 4 Inhibitor. Mini Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an Unusual Antimicrobial Pigment from the Marine Sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Jimenez, C.; Quinoa, E.; Adamczeski, M.; Hunter, L.M.; Crews, P. Novel Sponge-Derived Amino Acids. 12. Tryptophan-Derived Pigments and Accompanying Sesterterpenes from Fascaplysinopis reticulata. J. Org. Chem. 1991, 56, 3403–3410. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Faulkner, D.J. Palauolol, a New Anti-inflammatory Sesterterpene from the Sponge Fascaplysinopsis sp. from Palau. Tetrahedron Lett. 1996, 37, 3951–3954. [Google Scholar] [CrossRef]
- Kirsch, G.; Konig, G.M.; Wright, A.D.; Kaminsky, R. A New Bioactive Sesterterpene and Antiplasmodial Alkaloids from the Marine Sponge Hyrtios cf. erecta. J. Nat. Prod. 2000, 63, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Charan, R.D.; McKee, T.C.; Gustafson, K.R.; Pannell, L.K.; Boyd, M.R. Cytotoxic Alkaloids from the Marine Sponge Thorectandra sp. Tetrahedron Lett. 2002, 43, 5201–5204. [Google Scholar] [CrossRef]
- Popov, A.M.; Stonik, V.A. Physiological activity of fascaplisine—An unusual pigment from tropical sea fishes. Antibiot. Chemoter. 1991, 36, 12–14. [Google Scholar]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer Activity of Fascaplysin against Lung Cancer Cell and Small Cell Lung Cancer Circulating Tumor Cell Lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zunstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of Cyclin-Dependent Kinase 4 (Cdk4) by Fascaplysin, a Marine Natural Product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, A.; Chaudhuri, B.; Fretz, H. DNA Binding Properties of the Marine Sponge Pigment Fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Lu, X.L.; Zheng, Y.L.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Anti-proliferation of human cervical cancer HeLa cell line by fascaplysin through apoptosis induction. Acta Pharm. Sin. 2009, 44, 980–986. [Google Scholar]
- Wang, F.; Chen, H.; Yan, X.; Zheng, Y. Fascaplysin sensitizes cells to TRAIL-induced apoptosis through upregulating DR5 expression. Chin. J. Oceanol. Limnol. 2013, 31, 560–569. [Google Scholar] [CrossRef]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk Between Apoptosis and Autophagy Through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, Y.M.; Nam, T.J.; Ko, Y.S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, J.H.; Kim, S.; Nam, T.J.; Kim, Y.S.; Kim, B.M.; Yim, W.J.; Lim, J.H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules 2018, 23, 42. [Google Scholar] [CrossRef]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A. Discovery of a marine-derived bis-indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure-activity relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Johnson, T.A.; Milan-Lobo, L.; Che, T.; Ferwerda, M.; Lambu, E.; McIntosh, N.L.; Li, F.; He, L.; Lorig-Roach, N.; Crews, P. Identification of the First Marine-Derived Opioid Receptor “balanced” Agonist with a Signaling Profile That Resembles the Endorphins. ACS Chem. Neurosci. 2017, 8, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, F.; Sang, J.; Lin, M.; Ma, J.; Xiao, X.; Yan, S.; Naman, C.B.; Wang, N.; He, S.; et al. 9-Methylfascaplysin Is a More Potent Aβ Aggregation Inhibitor than the Marine-Derived Alkaloid, Fascaplysin, and Produces Nanomolar Neuroprotective Effects in SH-SY5Y Cells. Mar. Drugs 2019, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Biol. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.-I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3- and 10-bromofascaplysins is mediated by caspase-8, -9, -3-dependent apoptosis. Bioorg. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef] [PubMed]
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor Activity of Fascaplysin Derivatives on Glioblastoma Model In Vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Baranova, O.V.; Balaneva, N.N.; Fedorov, S.N.; Radchenko, O.S.; Dubovitskii, S.V. The first syntheses of 3-bromofascaplysin, 10-bromofascaplysin and 3,10-dibromofascaplysin—Marine alkaloids from Fascaplysinopsis reticulata and Didemnum sp. by application of a simple and effective approach to the pyrido [1,2-a:3,4-b′] diindole system. Tetrahedron Lett. 2007, 48, 7998–8000. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of Fascaplysin and Related Alkaloids: A Study of Structures, Cytotoxicities, and Sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W.; Pelcman, B. Total Syntheses of the Marine Sponge Pigments Fascaplysin and Homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Rocca, P.; Marsais, F.; Godart, A.; Quéguiner, G. A Short Synthesis of the Antimicrobial Marine Sponge Pigment Fascaplysin. Tetrahedron Lett. 1993, 34, 7917–7918. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; Garcia-Zafra, S.; Almendros, P. Iminophosphorane—Mediated Syntheses of the Fascaplysin Alkaloid of Marine Origin and Nitramarine. Tetrahedron Lett. 1994, 35, 8851–8854. [Google Scholar] [CrossRef]
- Radchenko, O.S.; Novikov, V.L.; Elyakov, G.B. A Simple and Practical Approach to the Synthesis of the Marine Sponge Pigment Fascaplysin and Related Compounds. Tetrahedron Lett. 1997, 38, 5339–5342. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittstein, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Baranova, O.V.; Kravchenko, N.S.; Dubovitskii, S.V. A new method for the synthesis of the marine alkaloid fascaplysin. Tetrahedron Lett. 2010, 51, 6498–6499. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R.A. Total synthesis and anti-cholinesterase activity of marine-derived bisindole alkaloid fascaplysin. Med. Chem. Commun. 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Liu, M.C.; Cai, Q.; Jia, F.C.; Wu, A.X. A Cascade Coupling Strategy for One-Pot Total Synthesis of β-Carboline and Isoquinoline-Containing Natural Products and Derivatives. Chem. Eur. J. 2013, 19, 10132–10137. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Sidorova, M.A.; Lyakhova, I.A. One-step transformation of the marine alkaloid fascaplysin into homofascaplysins B and B-1. The first syntheses of 3-bromohomofascaplysin B and 3–bromohomofascaplysin B-1. Tetrahedron Lett. 2018, 59, 1417–1420. [Google Scholar] [CrossRef]
- Fretz, H.; Ucci-Stoll, K.; Hug, P.; Schoepfer, J.; Lang, M. Investigations on the reactivity of fascaplysin. Part II. General stability considerations and products formed with nucleophiles. Helv. Chim. Acta 2001, 84, 867–873. [Google Scholar] [CrossRef]
- Laboratory of Microbiology. Available online: http://www.piboc.dvo.ru/en/structure/biosintez/lab6.php (accessed on 24 June 2019).
- Bilay, T.I. Methods of Experimental Mycology; Naukova Dumka: Kiev, Ukraine, 1982; p. 550. [Google Scholar]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in Vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Madanchi, R.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Schumacher, U.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Honecker, F.; et al. The marine triterpene glycoside frondoside A induces p53-independent apoptosis and inhibits autophagy in urothelial carcinoma cells. BMC Cancer 2017, 17, 93. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibiting Concentration, (IC50), µM | IC50 (ViCell)/IC50 (MTT) for 22Rv1 Cells | |||||
|---|---|---|---|---|---|---|---|
| HT-29 a | T-47D a | SK-MEL-28 a | PC-3 b | 22Rv1 b | 22Rv1 c | ||
| Fascaplysin (1) | 2.7 ± 0.05 | 5 ± 0.2 | 1.3 ± 0.08 | 0.78 ± 0.16 | 0.24 ± 0.04 | 0.34 ± 0.11 | 1.39 |
| 3-Bromofascaplysin (3) | 3.3 ± 0.12 | >5 | 1.9 ± 0.04 | 10 ± 1.75 | 0.42 ± 0.29 | 0.24 ± 0.14 | 0.58 |
| Compound 20 | >5 | >5 | >5 | 1.39 ± 0.43 | 0.21 ± 0.04 | 0.26 ± 0.05 | 1.24 |
| Compound 25 | >5 | >5 | 1.8±0.02 | 0.91 ± 0.06 | 0.27 ± 0.01 | 0.5 ± 0.19 | 1.88 |
| 14-Bromoreticulatate (10) | >5 | >5 | >5 | n/d | n/d | n/d | n/d |
| 14-Bromoreticulatine (7) | >5 | >5 | 1.2 ± 0.1 | > 50 | 35.72 ± 10.1 | n/d | n/d |
| 3,10-Dibromofascaplysin (5) | >5 | >5 | >5 | 7.28 ± 0.73 | 0.69 ± 0.05 | 5.14 ± 1.16 | 7.45 |
| Compound | Conc., mg/disc | Zone Unit Differentials in the Disk Diffusion Soft Agar Assay a | ||||
|---|---|---|---|---|---|---|
| Bacillus subtilis (KMM 430) | Staphylococcus aureus (ATCC 21027) | Pseudomonas aeruginosa (KMM 433) | Escherichia coli (ATCC 15034) | Candida albicans (KMM 455) | ||
| 1 | 0.4 | 25 | 25 | >35 | 20 | n/a |
| 3 | 0.1 | 25 | 20 | >35 | 20 | * |
| 25 | 0.2 | 20 | 20 | >35 | 20 | n/a |
| 7 | 0.2 | 10 | n/a | >35 | 10 | n/a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhidkov, M.E.; Smirnova, P.A.; Tryapkin, O.A.; Kantemirov, A.V.; Khudyakova, Y.V.; Malyarenko, O.S.; Ermakova, S.P.; Grigorchuk, V.P.; Kaune, M.; von Amsberg, G.; et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Mar. Drugs 2019, 17, 496. https://doi.org/10.3390/md17090496
Zhidkov ME, Smirnova PA, Tryapkin OA, Kantemirov AV, Khudyakova YV, Malyarenko OS, Ermakova SP, Grigorchuk VP, Kaune M, von Amsberg G, et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Marine Drugs. 2019; 17(9):496. https://doi.org/10.3390/md17090496
Chicago/Turabian StyleZhidkov, Maxim E., Polina A. Smirnova, Oleg A. Tryapkin, Alexey V. Kantemirov, Yuliya V. Khudyakova, Olesya S. Malyarenko, Svetlana P. Ermakova, Valeria P. Grigorchuk, Moritz Kaune, Gunhild von Amsberg, and et al. 2019. "Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues" Marine Drugs 17, no. 9: 496. https://doi.org/10.3390/md17090496
APA StyleZhidkov, M. E., Smirnova, P. A., Tryapkin, O. A., Kantemirov, A. V., Khudyakova, Y. V., Malyarenko, O. S., Ermakova, S. P., Grigorchuk, V. P., Kaune, M., von Amsberg, G., & Dyshlovoy, S. A. (2019). Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Marine Drugs, 17(9), 496. https://doi.org/10.3390/md17090496