Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons



Abstract
1. Introduction
2. Results and Discussion
2.1. Lipid Classes
2.2. Fatty Acid Analyses
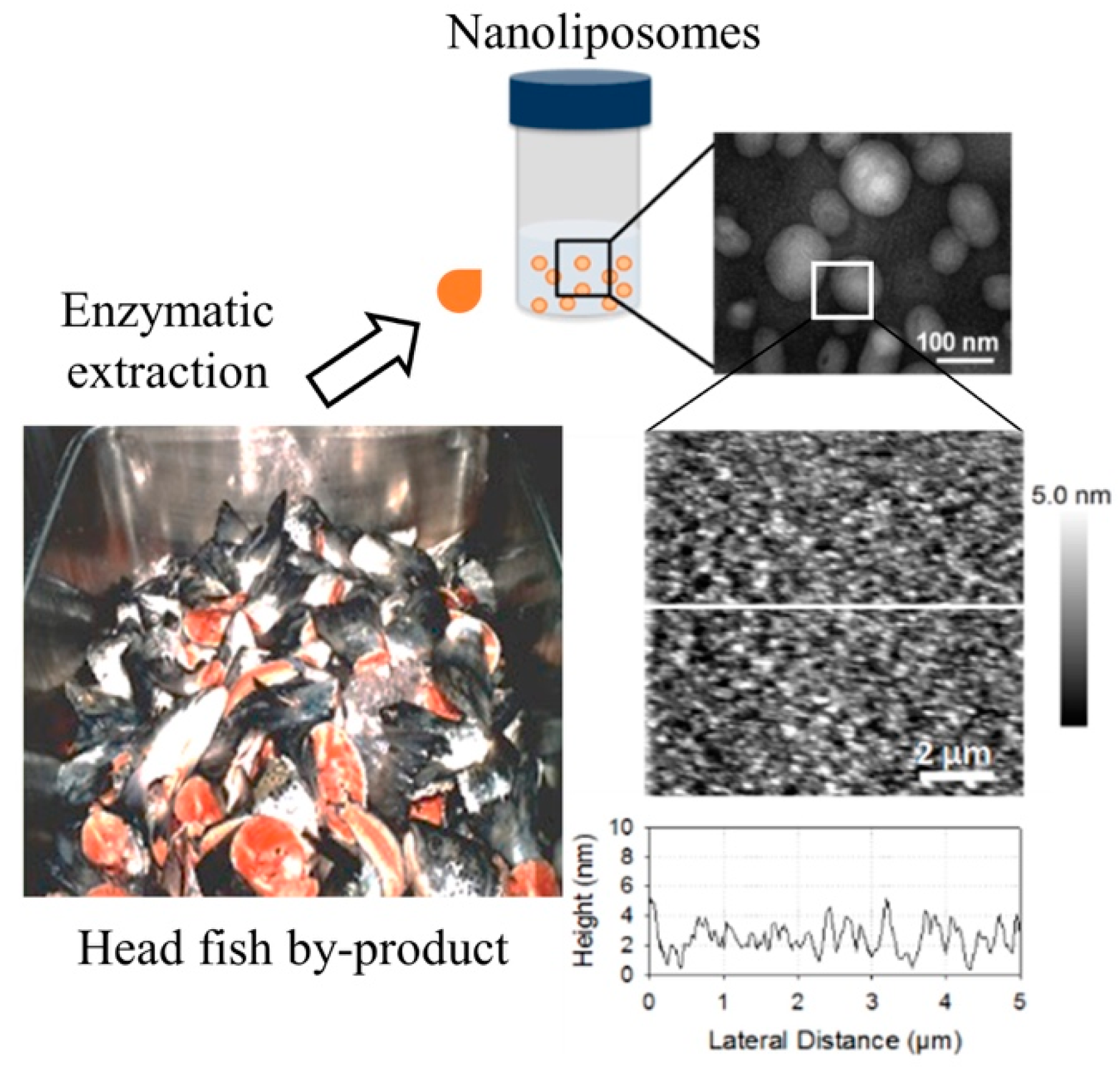
2.3. Liposome Size and Electrophoretic Mobility Measurements and Morphological Properties
2.4. Membrane Fluidity
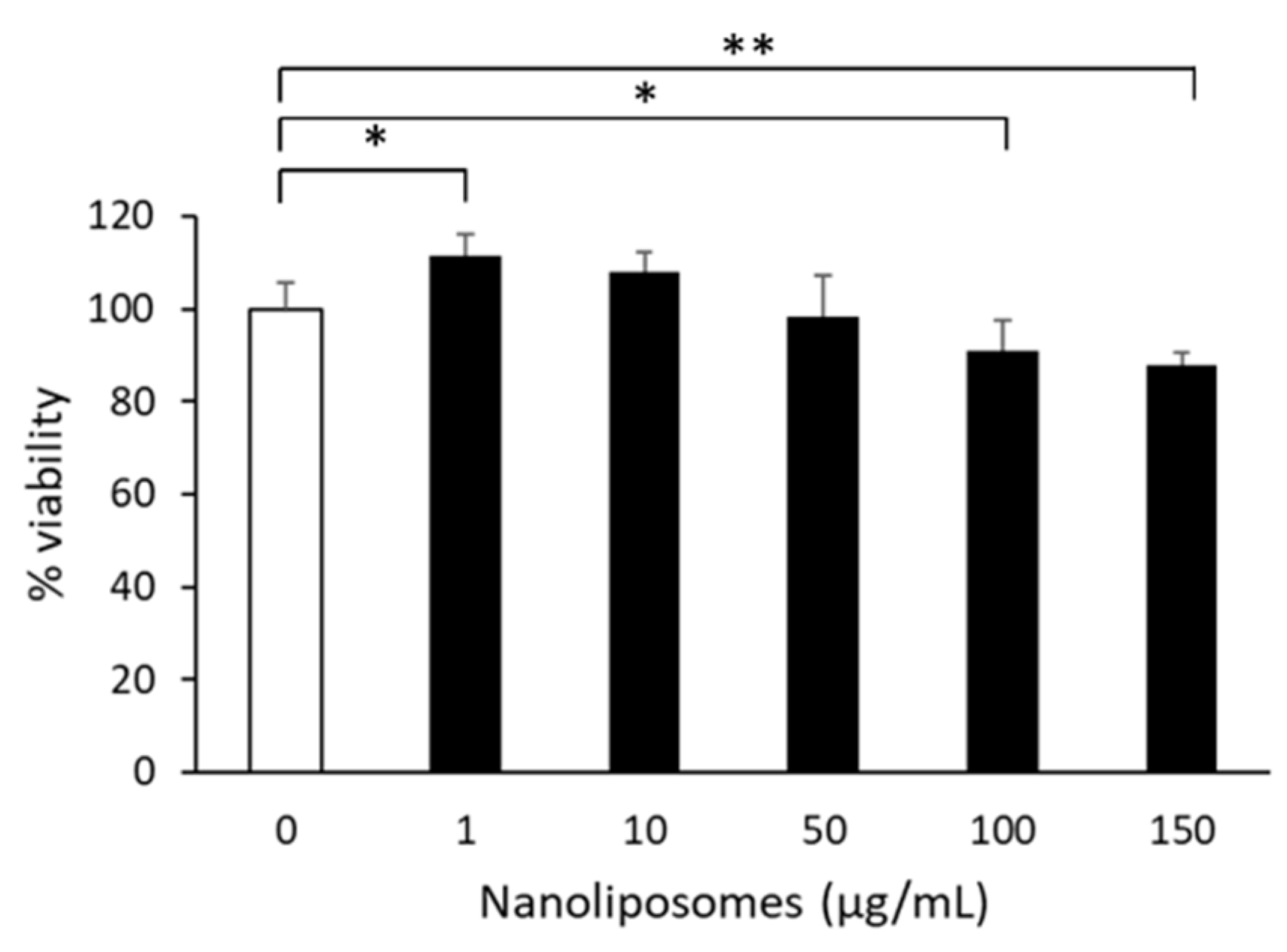
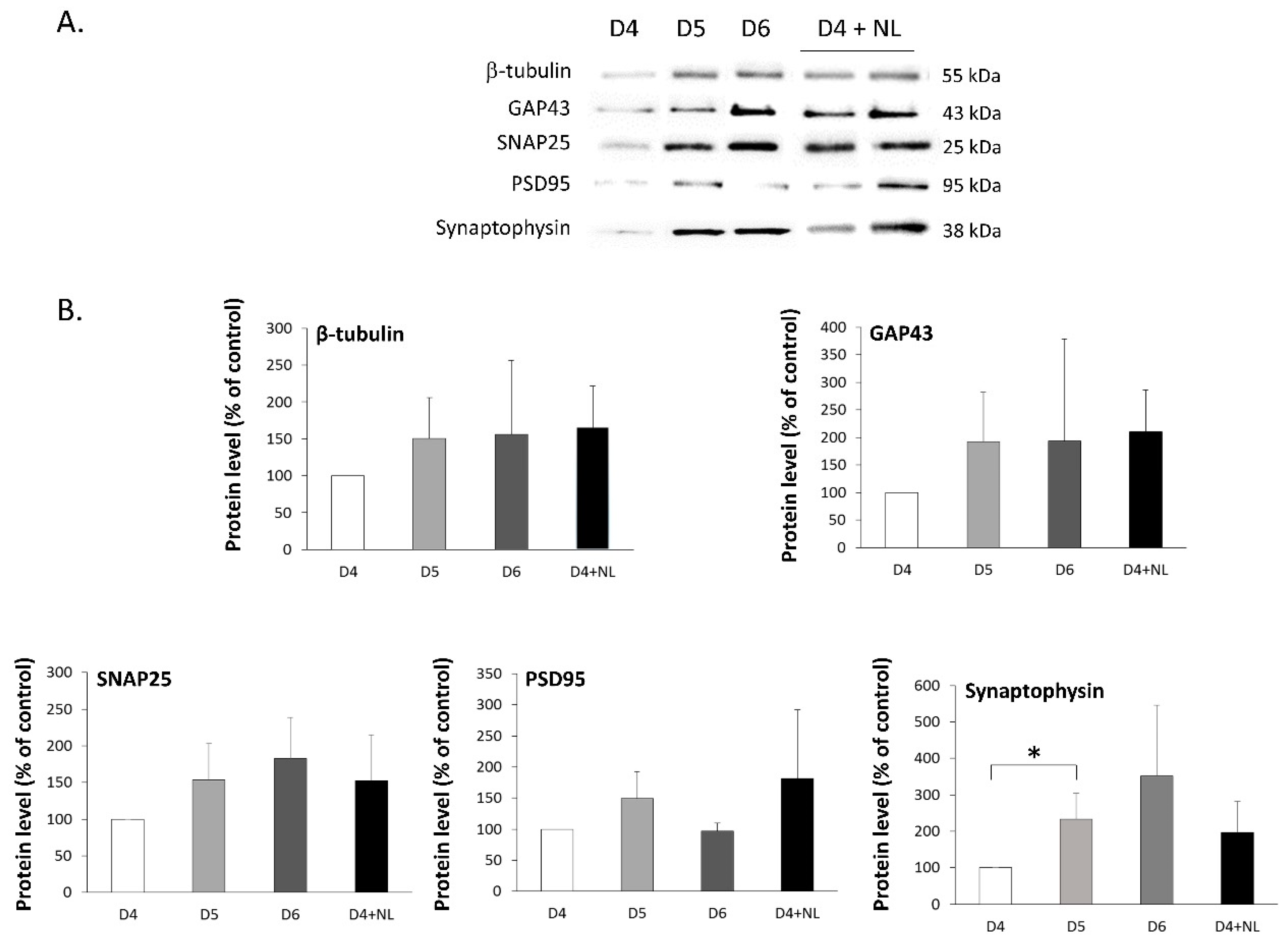
2.5. Treatment of Primary Cultured Neurons with Nanoliposomes
3. Materials and Methods
3.1. Fatty Acids Composition
3.2. Lipid Classes
3.3. Preparation of Nanoliposomes
3.4. Liposome Size and Electrophoretic Mobility Measurements
3.5. Stability of Nanoliposomes
3.6. Transmission Electron Microscopy (TEM)
3.7. Atomic Force Microscopy Imaging
3.8. Membrane Fluidity of Nanoliposomes
3.9. Cell Culture
3.10. Immunoblotting
3.11. Immunofluorescence Analyses
3.12. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calder, P.C. Omega-3 Fatty Acids and Inflammatory Processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Calviello, G. Long-chain omega-3 fatty acids and cancer: Any cause for concern? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kitajka, K.; Sinclair, A.J.; Weisinger, R.S.; Weisinger, H.S.; Mathai, M.; Jayasooriya, A.P.; Halver, J.E.; Puskas, L.G. Effects of dietary omega-3 polyunsaturated fatty acids on brain gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10931–10936. [Google Scholar] [CrossRef] [PubMed]
- Guirland, C.; Zheng, J.Q. Membrane Lipid Rafts and Their Role in Axon Guidance. In Axon Growth and Guidance; Bagnard, D., Ed.; Springer: New York, NY, USA, 2007; Volume 621, pp. 144–154. ISBN 978-0-387-76714-7. [Google Scholar]
- Cao, D.; Xue, R.; Xu, J.; Liu, Z. Effects of docosahexaenoic acid on the survival and neurite outgrowth of rat cortical neurons in primary cultures. J. Nutr. Biochem. 2005, 16, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, A.; Lavado, E.M.; Granda, B.; Velasco, A.; Medina, J.M. Neuronal differentiation is triggered by oleic acid synthesized and released by astrocytes: Oleic acid induces neuronal differentiation. J. Neurochem. 2008, 79, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, E.; Hashimoto, M.; Shido, O. Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 2006, 139, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Cansev, M.; Wurtman, R.J. Oral supplementation with docosahexaenoic acid and uridine-5′-monophosphate increases dendritic spine density in adult gerbil hippocampus. Brain Res. 2007, 1182, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Dietary (n-3) Fatty Acids and Brain Development. J. Nutr. 2007, 137, 855–859. [Google Scholar] [CrossRef]
- Shindou, H.; Koso, H.; Sasaki, J.; Nakanishi, H.; Sagara, H.; Nakagawa, K.M.; Takahashi, Y.; Hishikawa, D.; Iizuka-Hishikawa, Y.; Tokumasu, F.; et al. Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem. 2017, 292, 12054–12064. [Google Scholar] [CrossRef]
- Li, D.; Wahlqvist, M.L.; Sinclair, A.J. Advances in n-3 polyunsaturated fatty acid nutrition. Asia Pac. J. Clin. Nutr. 2019, 28, 1–5. [Google Scholar]
- Hishikawa, D.; Valentine, W.J.; Iizuka-Hishikawa, Y.; Shindou, H.; Shimizu, T. Metabolism and functions of docosahexaenoic acid-containing membrane glycerophospholipids. FEBS Lett. 2017, 591, 2730–2744. [Google Scholar] [CrossRef] [PubMed]
- Colin, J.; Thomas, M.H.; Gregory-Pauron, L.; Pinçon, A.; Lanhers, M.C.; Corbier, C.; Claudepierre, T.; Yen, F.T.; Oster, T.; Malaplate-Armand, C. Maintenance of membrane organization in the aging mouse brain as the determining factor for preventing receptor dysfunction and for improving response to anti-Alzheimer treatments. Neurobiol. Aging 2017, 54, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Fanni, J.; Parmentier, M.; Regnault, P. Procédé d’extraction d’huile par voie enzymatique et obtention d’hydrolysats protéiques à fonctionnalités dirigées. Brev. FR 2002, 2, 703. [Google Scholar]
- Maherani, B.; Arab-Tehrany, E.; Mozafari, M.R.; Gaiani, C.; Linder, M. Liposomes: A Review of Manufacturing Techniques and Targeting Strategies. Curr. Nanosci. 2011, 7, 436–452. [Google Scholar] [CrossRef]
- Arab Tehrany, E.; Kahn, C.J.F.; Baravian, C.; Maherani, B.; Belhaj, N.; Wang, X.; Linder, M. Elaboration and characterization of nanoliposome made of soya; rapeseed and salmon lecithins: Application to cell culture. Colloids Surf. B Biointerfaces 2012, 95, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Latifi, S.; Tamayol, A.; Habibey, R.; Sabzevari, R.; Kahn, C.; Geny, D.; Eftekharpour, E.; Annabi, N.; Blau, A.; Linder, M.; et al. Natural lecithin promotes neural network complexity and activity. Sci. Rep. 2016, 6, 25777. [Google Scholar] [CrossRef] [PubMed]
- Benedet, J.A.; Umeda, H.; Shibamoto, T. Antioxidant Activity of Flavonoids Isolated from Young Green Barley Leaves toward Biological Lipid Samples. J. Agric. Food Chem. 2007, 55, 5499–5504. [Google Scholar] [CrossRef]
- Chansiri, G.; Lyons, R.T.; Patel, M.V.; Hem, S.L. Effect of surface charge on the stability of oil/water emulsions during steam sterilization. J. Pharm. Sci. 1999, 88, 454–458. [Google Scholar] [CrossRef]
- Belhaj, N.; Arab-Tehrany, E.; Linder, M. Oxidative kinetics of salmon oil in bulk and in nanoemulsion stabilized by marine lecithin. Process Biochem. 2010, 45, 187–195. [Google Scholar] [CrossRef]
- Leekumjorn, S.; Cho, H.J.; Wu, Y.; Wright, N.T.; Sum, A.K.; Chan, C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 1508–1516. [Google Scholar] [CrossRef]
- Arimura, N.; Kaibuchi, K. Neuronal polarity: From extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.; Matouba, E.; Fanni, J.; Parmentier, M. Enrichment of salmon oil with n-3 PUFA by lipolysis, filtration and enzymatic re-esterification. Eur. J. Lipid Sci. Technol. 2002, 104, 455–462. [Google Scholar] [CrossRef]
- Ackman, R.G. Remarks on official methods employing boron trifluoride in the preparation of methyl esters of the fatty acids of fish oils. J. Am. Oil Chem. Soc. 1998, 75, 541–545. [Google Scholar] [CrossRef]
- Hasan, M.; Belhaj, N.; Benachour, H.; Barberi-Heyob, M.; Kahn, C.J.F.; Jabbari, E.; Linder, M.; Arab-Tehrany, E. Liposome encapsulation of curcumin: Physico-chemical characterizations and effects on MCF7 cancer cell proliferation. Int. J. Pharm. 2014, 461, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Gbogouri, G.A.; Linder, M.; Fanni, J.; Parmentier, M. Analysis of lipids extracted from salmon (Salmo salar) heads by commercial proteolytic enzymes. Eur. J. Lipid Sci. Technol. 2006, 108, 766–775. [Google Scholar] [CrossRef]
- Hasan, M.; Ben Messaoud, G.; Michaux, F.; Tamayol, A.; Kahn, C.J.F.; Belhaj, N.; Linder, M.; Arab-Tehrany, E. Chitosan-coated liposomes encapsulating curcumin: Study of lipid–polysaccharide interactions and nanovesicle behavior. RSC Adv. 2016, 6, 45290–45304. [Google Scholar] [CrossRef]
- Colas, J.C.; Shi, W.; Rao, V.M.; Omri, A.; Mozafari, M.R.; Singh, H. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron 2007, 38, 841–847. [Google Scholar] [CrossRef]
- Maherani, B.; Arab-tehrany, E.; Kheirolomoom, A.; Reshetov, V.; Stebe, M.J.; Linder, M. Optimization and characterization of liposome formulation by mixture design. Analyst 2012, 137, 773–786. [Google Scholar] [CrossRef]
- Colin, J.; Allouche, A.; Chauveau, F.; Corbier, C.; Pauron-Gregory, L.; Lanhers, M.C.; Claudepierre, T.; Yen, F.T.; Oster, T.; Malaplate-Armand, C. Improved Neuroprotection Provided by Drug Combination in Neurons Exposed to Cell-Derived Soluble Amyloid-β Peptide. J. Alzheimer’s Dis. 2016, 52, 975–987. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Salmon Lecithin |
|---|---|
| Total phospholipids (%) | 67.7 ± 1.1 |
| Phosphatidylcholine, PC (%) | 42.4 ± 0.5 |
| Phosphatidylethanolamine, PE (%) | 7.7 ± 0.1 |
| Phosphatidylserine, PS (%) | 9.1 ± 0.1 |
| Phosphatidylinositol, PI (%) | 13.0 ± 0.3 |
| Sphingomyelin, SPM (%) | 1.5 ± 0.1 |
| Lysophosphosphatidylcholine, LPC (%) | 2.8 ± 0.1 |
| Other phospholipids (%) | 23.5 ± 0.2 |
| Triglycerides, TAGs (%) | 31.2 ± 0.8 |
| Cholesterol, CHOL (%) | 1.2 ± 0.1 |
| Free fatty acids, FFA (%) | ND |
| Saturated Fatty Acids (SFA) | Monounsaturated Fatty Acids (MUFA) | Polyunsaturated Fatty Acids (PUFA) | |||
|---|---|---|---|---|---|
| Fatty acid | % (SD) * | Fatty acid | % (SD) | Fatty acid | % (SD) |
| C14:0 | 2.72 (0.04) | C14:1n9 | 0.33 (0.02) | C18:2n6 | 1.20 (0.04) |
| C15:0 | 1.13 (0.01) | C16:1n7 | 3.23 (0.07) | C18:3n3 | 0.35 (0.01) |
| C16:0 | 20.67 (0.15) | C18:1n9 | 13.87 (0.13) | C20:4n6 | 3.52 (0.02) |
| C17:0 | 0.88 (0.05) | C20:1n9 | 2.11 (0.06) | C20:5n3 1 | 11.03 (0.13) |
| C18:0 | 6.10 (0.05) | C22:1n9 | 0.79 (0.08) | C22:5n3 | 3.93 (0.33) |
| C20:0 | 0.35 (0.03) | C22:6n3 2 | 26.26 (0.09) | ||
| C22:0 | 1.55 (0.06) | ||||
| SFA | 33.40 | MUFA | 20.32 | PUFA | 46.28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malaplate, C.; Poerio, A.; Huguet, M.; Soligot, C.; Passeri, E.; Kahn, C.J.F.; Linder, M.; Arab-Tehrany, E.; Yen, F.T. Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons. Mar. Drugs 2019, 17, 406. https://doi.org/10.3390/md17070406
Malaplate C, Poerio A, Huguet M, Soligot C, Passeri E, Kahn CJF, Linder M, Arab-Tehrany E, Yen FT. Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons. Marine Drugs. 2019; 17(7):406. https://doi.org/10.3390/md17070406
Chicago/Turabian StyleMalaplate, Catherine, Aurelia Poerio, Marion Huguet, Claire Soligot, Elodie Passeri, Cyril J. F. Kahn, Michel Linder, Elmira Arab-Tehrany, and Frances T. Yen. 2019. "Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons" Marine Drugs 17, no. 7: 406. https://doi.org/10.3390/md17070406
APA StyleMalaplate, C., Poerio, A., Huguet, M., Soligot, C., Passeri, E., Kahn, C. J. F., Linder, M., Arab-Tehrany, E., & Yen, F. T. (2019). Neurotrophic Effect of Fish-Lecithin Based Nanoliposomes on Cortical Neurons. Marine Drugs, 17(7), 406. https://doi.org/10.3390/md17070406