Testing the Neuroprotective Properties of PCSO-524® Using a Neuronal Cell Cycle Suppression Assay



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
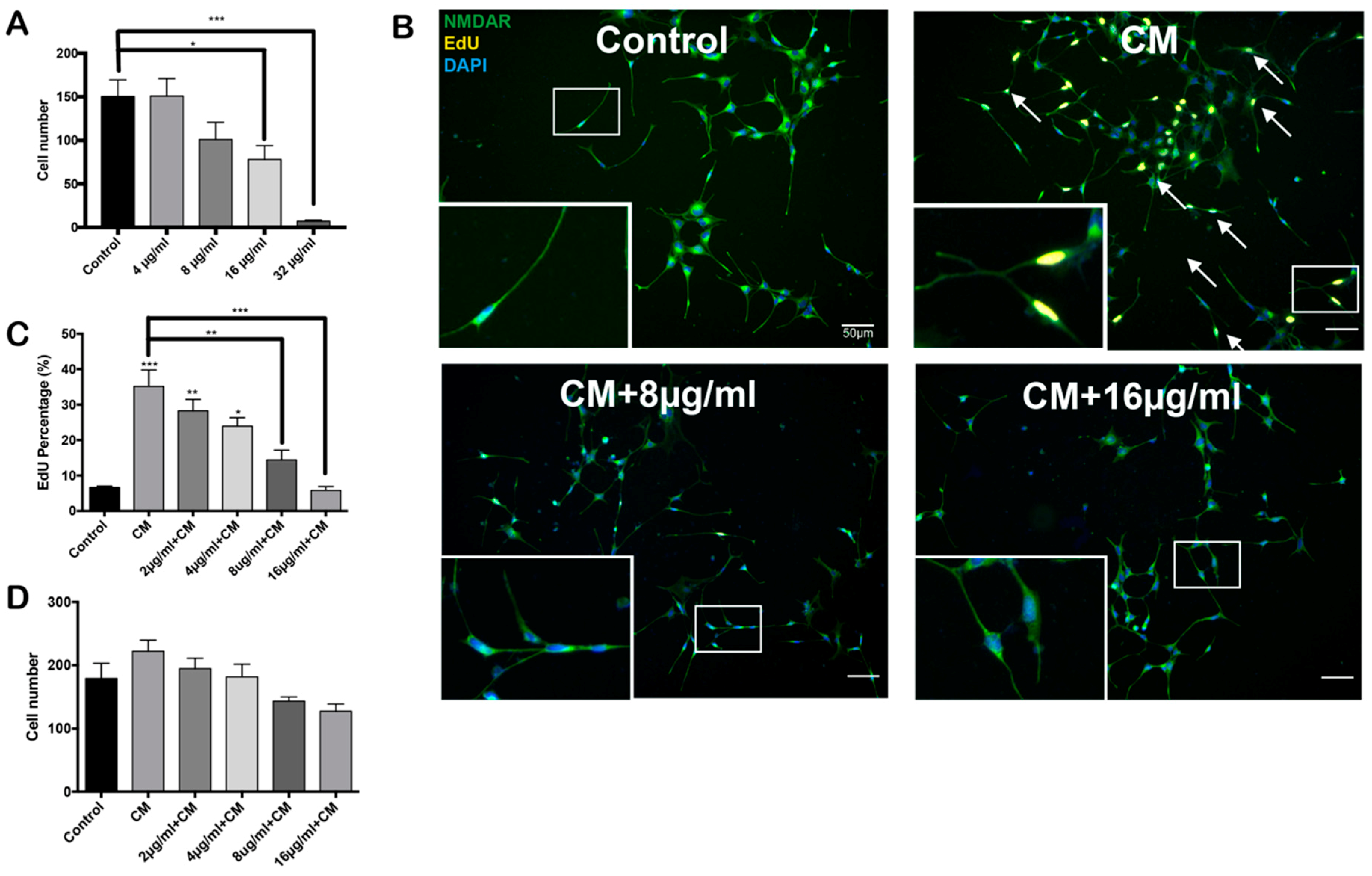
2.1. Conditioned Medium Induces Differentiated HT22 Cell Cycle Reentry
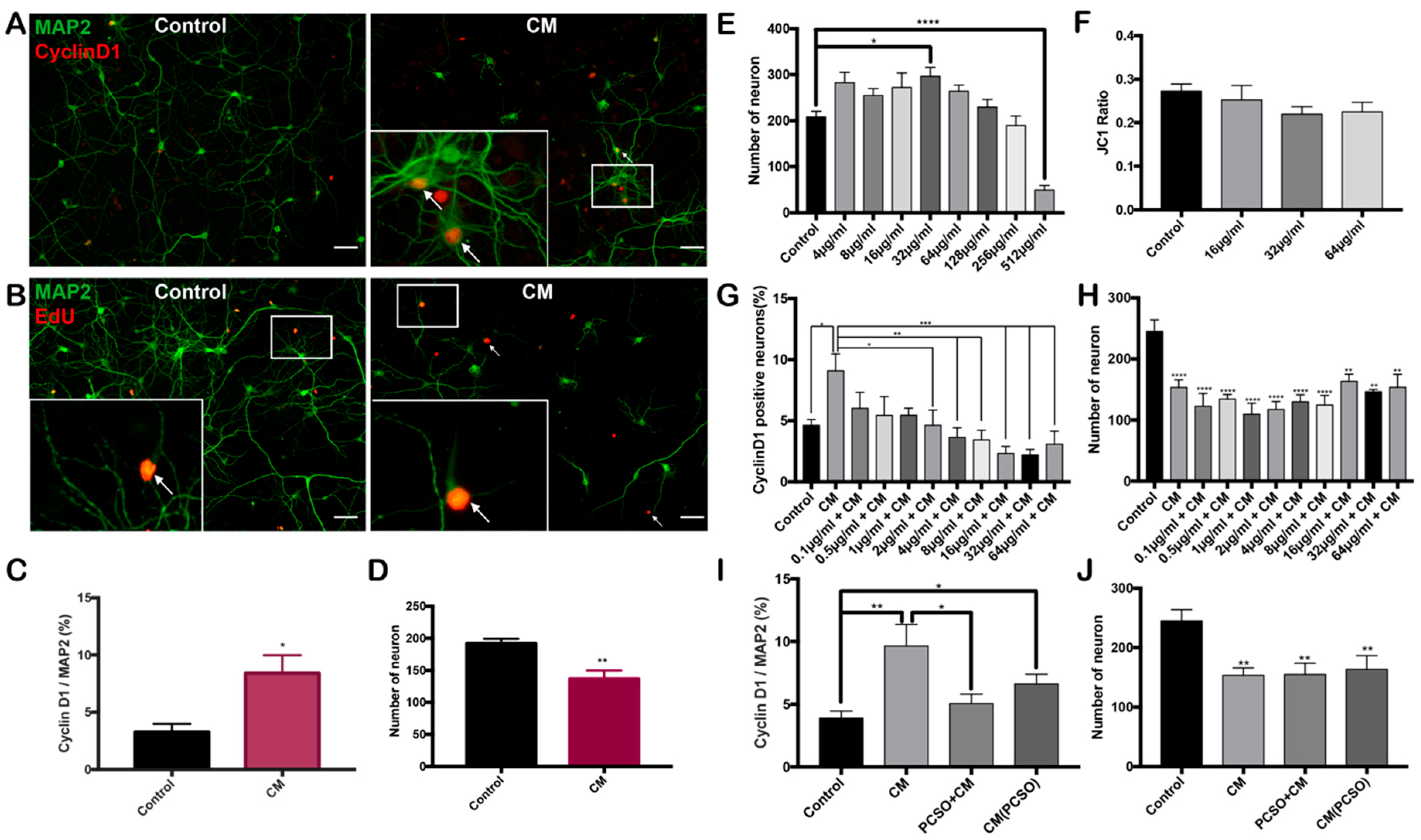
2.2. PCSO-524® Protects Against CM-Induced Cell Cycle Reentry
2.3. PCSO-524® Shows Neuroprotective Effect on Post-mitotic Neurons
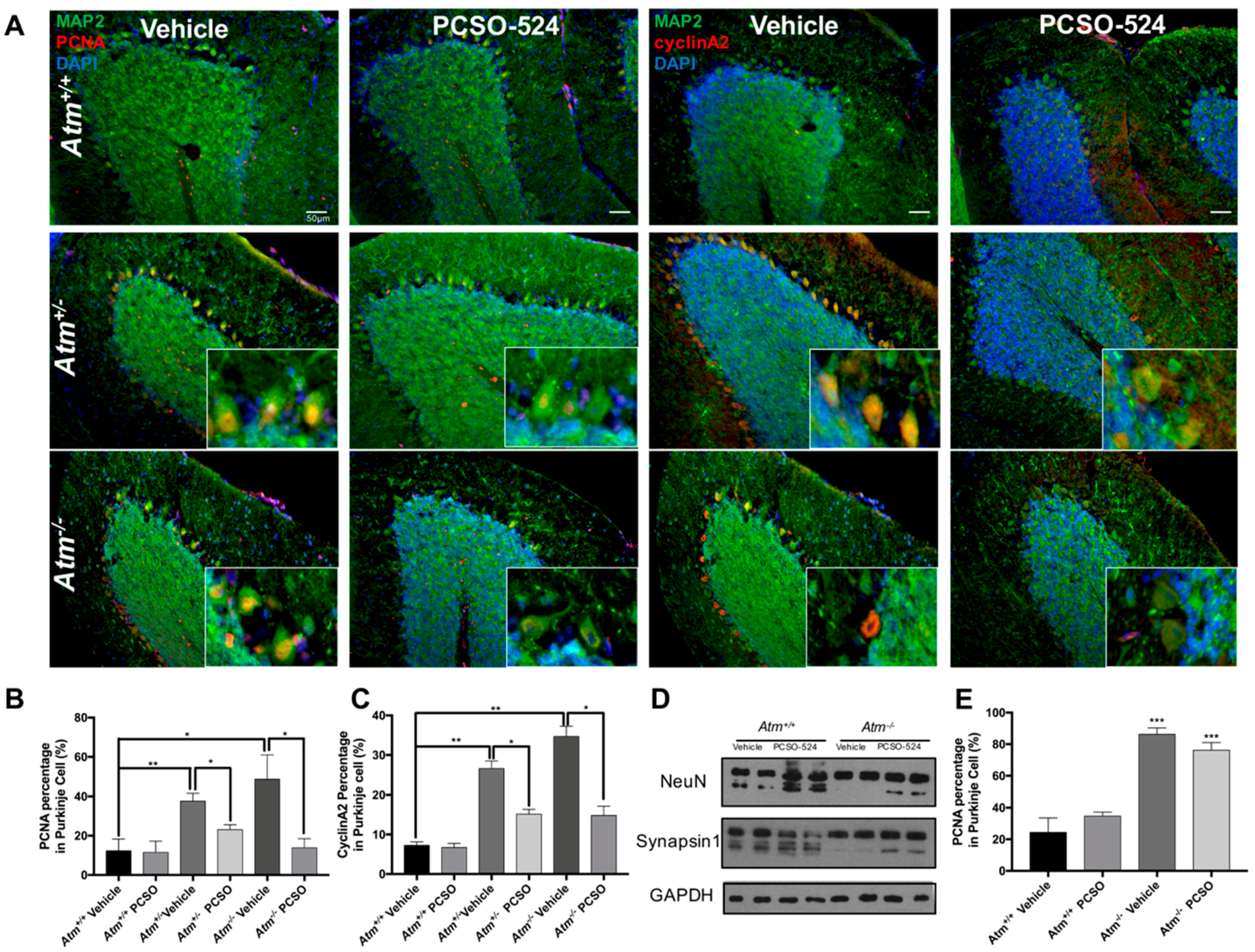
2.4. PCSO-524® Showed Protective Effect but Not Reversible Effect In Vivo
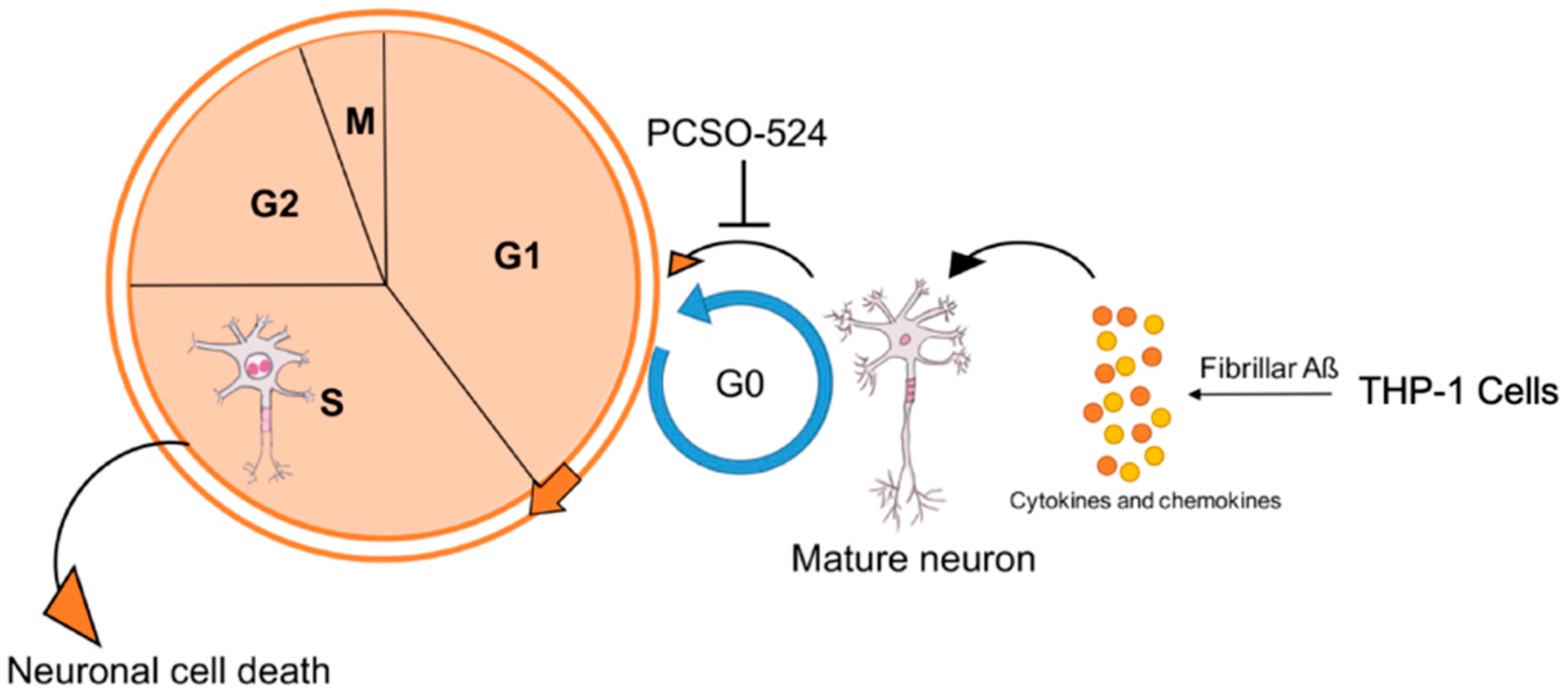
3. Discussion
4. Materials and Methods
4.1. Animals
- PGK35:
- 5′-GGA AAA GCG CCT CCC CTA CCC-3′
- Bal AT9:
- 5′-CCT CCT CAT ATT TGT AAC ACG CTG-3′
- Bal AT12:
- 5′-TGT AAT GTG CCT TAA AGA ACC TGG-3′.
4.2. Drugs and Antibodies
4.3. Maintenance and Differentiation of HT22 Cells
4.4. Embryonic Cortical Neuron Culture
4.5. THP-1 Cell Culture and Conditioned Medium
4.6. Histochemistry and Immunocytochemistry
4.7. Western Blot of Cultured Cell Lysates
4.8. ELISA
4.9. JC1 Dye Incorporation
4.10. Drug Treatment In Vivo
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, H.G.; Casadesus, G.; Zhu, X.; Castellani, R.J.; McShea, A.; Perry, G.; Petersen, R.B.; Bajic, V.; Smith, M.A. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem. Int. 2009, 54, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Herrup, K. Loss of neuronal cell cycle control in ataxia-telangiectasia: A unified disease mechanism. J. Neurosci. 2005, 25, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, G.U.; Breunig, J.J.; Depboylu, C.; Rouaux, C.; Michel, P.P.; Alvarez-Fischer, D.; Boutillier, A.L.; Degregori, J.; Oertel, W.H.; Rakic, P.; et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 3585–3590. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pizarro, J.G.; Beas-Zarate, C.; Pallas, M.; Camins, A. Role of cell cycle re-entry in neurons: A common apoptotic mechanism of neuronal cell death. Neurotox. Res. 2012, 22, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Norambuena, A.; Wallrabe, H.; McMahon, L.; Silva, A.; Swanson, E.; Khan, S.S.; Baerthlein, D.; Kodis, E.; Oddo, S.; Mandell, J.W.; et al. mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement. 2017, 13, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Seward, M.E.; Swanson, E.; Norambuena, A.; Reimann, A.; Cochran, J.N.; Li, R.; Roberson, E.D.; Bloom, G.S. Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 2013, 126 Pt 5, 1278–1286. [Google Scholar] [CrossRef]
- Herrup, K.; Busser, J.C. The induction of multiple cell cycle events precedes target-related neuronal death. Development 1995, 121, 2385–2395. [Google Scholar] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Farinelli, S.E.; Greene, L.A. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J. Biol. Chem. 1996, 271, 8161–8169. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Levine, B.; Ferrari, G.; Greene, L.A. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 1997, 17, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Morris, E.J.; Greene, L.A.; Geller, H.M. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J. Neurosci. 1997, 17, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Landreth, G.E. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist 2002, 8, 276–283. [Google Scholar] [PubMed]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Maphis, N.; Xu, G.; Varvel, N.H.; Kokiko-Cochran, O.N.; Weick, J.P.; Staugaitis, S.M.; Cardona, A.; Ransohoff, R.M.; Herrup, K.; et al. Microglial derived tumor necrosis factor-alpha drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol. Dis. 2014, 62, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Bhaskar, K.; Patil, A.R.; Pimplikar, S.W.; Herrup, K.; Lamb, B.T. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 2008, 28, 10786–10793. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Conde, J.R.; Fendrick, S.E.; Flanary, B.E.; Mariani, C.L. Role of microglia in the central nervous system’s immune response. Neurol. Res. 2005, 27, 685–691. [Google Scholar] [PubMed]
- Combs, C.K.; Johnson, D.E.; Cannady, S.B.; Lehman, T.M.; Landreth, G.E. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J. Neurosci. 1999, 19, 928–939. [Google Scholar] [CrossRef]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Combs, C.; Cannady, S.B.; Geldmacher, D.S.; Herrup, K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol. Aging 2000, 21, 797–806. [Google Scholar] [CrossRef]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, A.; Giuffrida Stella, A.M.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999, 13, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Floden, A.M.; Li, S.; Combs, C.K. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J. Neurosci. 2005, 25, 2566–2575. [Google Scholar] [CrossRef] [PubMed]
- Gil, A. Polyunsaturated fatty acids and inflammatory diseases. Biomed. Pharmacother. 2002, 56, 388–396. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505s–1519s. [Google Scholar] [CrossRef] [PubMed]
- Calon, F. Omega-3 Polyunsaturated Fatty Acids in Alzheimer’s Disease: Key Questions and Partial Answers. Curr. Alzheimer Res. 2011, 8, 470–478. [Google Scholar] [CrossRef]
- Hamazaki, T.; Hashimoto, M. Neuroprotective and Ameliorative Actions of Polyunsaturated Fatty Acids Against Neuronal Diseases—Evidence From Basic to Clinical Studies: Preface. J. Pharmacol. Sci. 2011, 116, 149. [Google Scholar] [CrossRef]
- Mickleborough, T.D.; Sinex, J.A.; Platt, D.; Chapman, R.F.; Hirt, M. The effects PCSO-524 (R), a patented marine oil lipid and omega-3 PUFA blend derived from the New Zealand green lipped mussel (Perna canaliculus), on indirect markers of muscle damage and inflammation after muscle damaging exercise in untrained men: A randomized, placebo controlled trial. J. Int. Soc. Sport Nutr. 2015, 12, 10. [Google Scholar]
- Miller, T.E.; Ormrod, D. The Anti-Inflammatory Activity of Perna-Canaliculus (Nz Green Lipped Mussel). N. Z. Med. J. 1980, 92, 187–193. [Google Scholar]
- Brien, S.; Prescott, P.; Coghlan, B.; Bashir, N.; Lewith, G. Systematic review of the nutritional supplement Perna Canaliculus (green-lipped mussel) in the treatment of osteoarthritis. Qjm-Int. J. Med. 2008, 101, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Coulson, S.; Vecchio, P.; Gramotnev, H.; Vitetta, L. Green-lipped mussel (Perna canaliculus) extract efficacy in knee osteoarthritis and improvement in gastrointestinal dysfunction: A pilot study. Inflammopharmacology 2012, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L.; Suo, W.Z. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009, 84, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Boghaert, E.R.; Simpson, J.; Jacob, R.J.; Lacey, T.; Walsh, J.W.; Zimmer, S.G. The effect of dibutyryl camp (dBcAMP) on morphological differentiation, growth and invasion in vitro of a hamster brain-tumor cell line: A comparative study of dBcAMP effects in 2- and 3-dimensional cultures. Int. J. Cancer 1991, 47, 610–618. [Google Scholar] [CrossRef]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 Is a Nuclear-Protein Required for Cell-Cycle Progression in G(1). Gene Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Lijam, N.; Paylor, R.; McDonald, M.P.; Crawley, J.N.; Deng, C.X.; Herrup, K.; Stevens, K.E.; Maccaferri, G.; McBain, C.J.; Sussman, D.J.; et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 1997, 90, 895–905. [Google Scholar] [CrossRef]
- McDonald, D.R.; Bamberger, M.E.; Combs, C.K.; Landreth, G.E. beta-amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP-1 monocytes. J. Neurosci. 1998, 18, 4451–4460. [Google Scholar] [CrossRef]
- Yates, S.L.; Burgess, L.H.; Kocsis-Angle, J.; Antal, J.M.; Dority, M.D.; Embury, P.B.; Piotrkowski, A.M.; Brunden, K.R. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J. Neurochem. 2000, 74, 1017–1025. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (Delta psi m) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Reers, M.; Smiley, S.T.; MottolaHartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. Mitochondrial membrane potential monitored by JC-1 dye. Method Enzymol. 1995, 260, 406–417. [Google Scholar]
- Li, L.; Cheung, T.; Chen, J.; Herrup, K. A comparative study of five mouse models of Alzheimer’s disease: Cell cycle events reveal new insights into neurons at risk for death. Int. J. Alzheimers Dis. 2011, 2011, 171464. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Bhaskar, K.; Kounnas, M.Z.; Wagner, S.L.; Yang, Y.; Lamb, B.T.; Herrup, K. NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J. Clin. Investig. 2009, 119, 3692–3702. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Cramer, P.E.; Varvel, N.H.; Reed-Geaghan, E.; Jiang, Q.; Szabo, A.; Herrup, K.; Lamb, B.T.; Landreth, G.E. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 2012, 33, e21–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hui, C.W.; Li, J.; Herrup, K. The interaction of the atm genotype with inflammation and oxidative stress. PLoS ONE 2014, 9, e85863. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.W.; Herrup, K. Individual Cytokines Modulate the Neurological Symptoms of ATM Deficiency in a Region Specific Manner. eNeuro 2015, 2, ENEURO.0032-15.2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lum, J.H.; Ng, C.K.; McKay, J.; Butt, Y.K.; Wong, M.S.; Lo, S.C. Pain Controlling and Cytokine-regulating Effects of Lyprinol, a Lipid Extract of Perna Canaliculus, in a Rat Adjuvant-induced Arthritis Model. Evid. Based Complement. Alternat. Med. 2009, 6, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, M.W.; Macrides, T.A.; Kalafatis, N.; Betts, W.H.; Haynes, D.R.; Broadbent, J. Anti-inflammatory activity of a lipid fraction (lyprinol) from the NZ green-lipped mussel. Inflammopharmacology 1997, 5, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Busser, J.; Geldmacher, D.S.; Herrup, K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci. 1998, 18, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Arendt, T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer’s disease. J. Alzheimers Dis. 2002, 4, 243–247. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Herrup, K. Cdk5 nuclear localization is p27-dependent in nerve cells: Implications for cell cycle suppression and caspase-3 activation. J. Biol. Chem. 2010, 285, 14052–14061. [Google Scholar] [CrossRef]
- Hui, C.W.; Zhang, Y.; Herrup, K. Non-Neuronal Cells Are Required to Mediate the Effects of Neuroinflammation: Results from a Neuron-Enriched Culture System. PLoS ONE 2016, 11, e0147134. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Obrietan, K.; van den Pol, A.N. GABAB receptor-mediated regulation of glutamate-activated calcium transients in hypothalamic and cortical neuron development. J. Neurophysiol. 1999, 82, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kauwe, J.S.; Bailey, M.H.; Ridge, P.G.; Perry, R.; Wadsworth, M.E.; Hoyt, K.L.; Staley, L.A.; Karch, C.M.; Harari, O.; Cruchaga, C.; et al. Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: Significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014, 10, e1004758. [Google Scholar] [CrossRef] [PubMed]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Buck, S.B.; Bradford, J.; Gee, K.R.; Agnew, B.J.; Clarke, S.T.; Salic, A. Detection of S-phase cell cycle progression using 5-ethynyl-2’-deoxyuridine incorporation with click chemistry an alternative to using 5-bromo-2’-deoxyuridine antibodies. Biotechniques 2008, 44, 927–929. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, B.; Zhang, Y.; Herrup, K. Testing the Neuroprotective Properties of PCSO-524® Using a Neuronal Cell Cycle Suppression Assay. Mar. Drugs 2019, 17, 79. https://doi.org/10.3390/md17020079
Zhu B, Zhang Y, Herrup K. Testing the Neuroprotective Properties of PCSO-524® Using a Neuronal Cell Cycle Suppression Assay. Marine Drugs. 2019; 17(2):79. https://doi.org/10.3390/md17020079
Chicago/Turabian StyleZhu, Beika, Yang Zhang, and Karl Herrup. 2019. "Testing the Neuroprotective Properties of PCSO-524® Using a Neuronal Cell Cycle Suppression Assay" Marine Drugs 17, no. 2: 79. https://doi.org/10.3390/md17020079
APA StyleZhu, B., Zhang, Y., & Herrup, K. (2019). Testing the Neuroprotective Properties of PCSO-524® Using a Neuronal Cell Cycle Suppression Assay. Marine Drugs, 17(2), 79. https://doi.org/10.3390/md17020079