Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
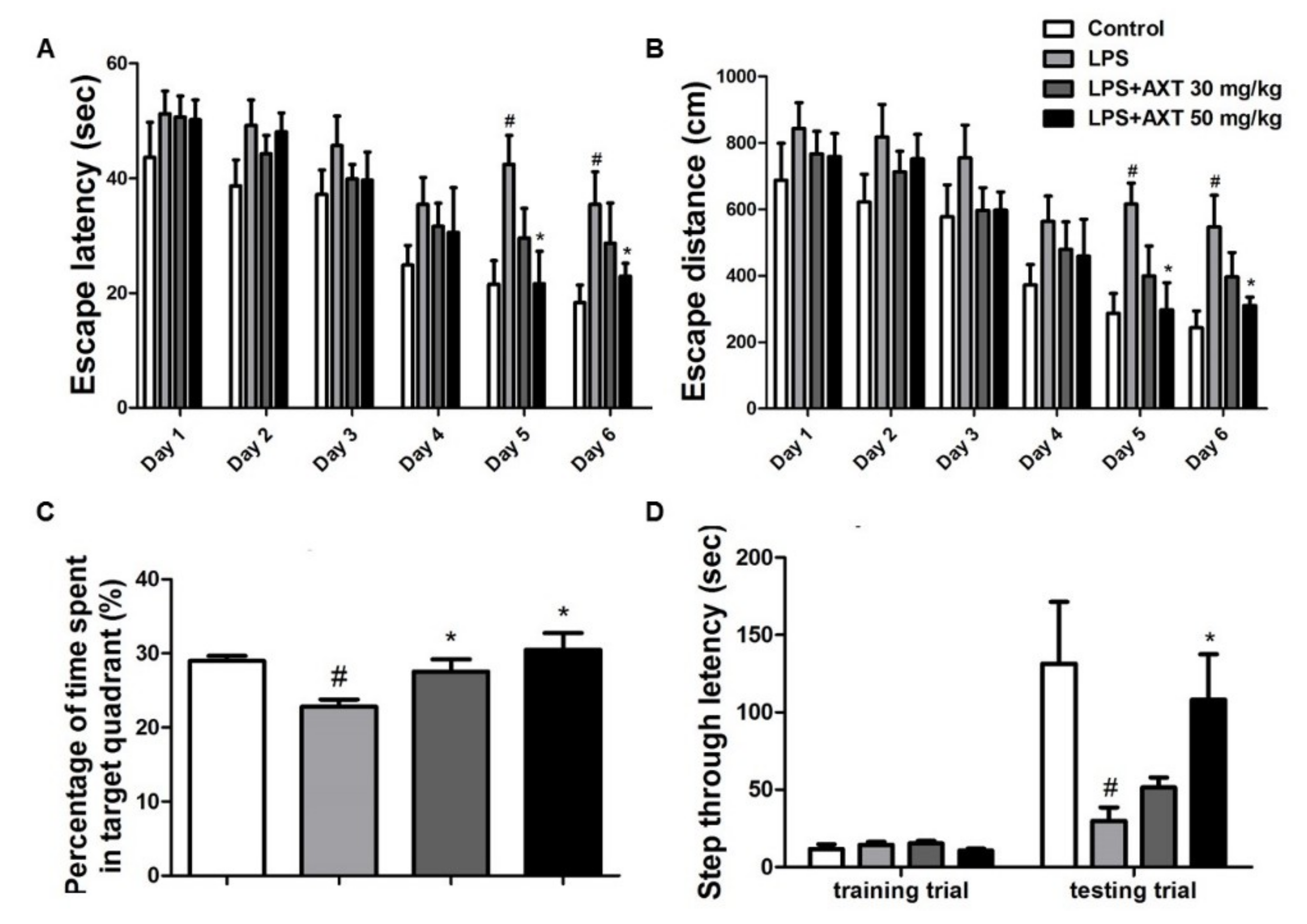
2.1. Astaxanthin Alleviates LPS-Induced Memory Impairment in Mice
2.2. Astaxanthin Downregulates LPS-Induced Aβ Burden in the Brain of Mice
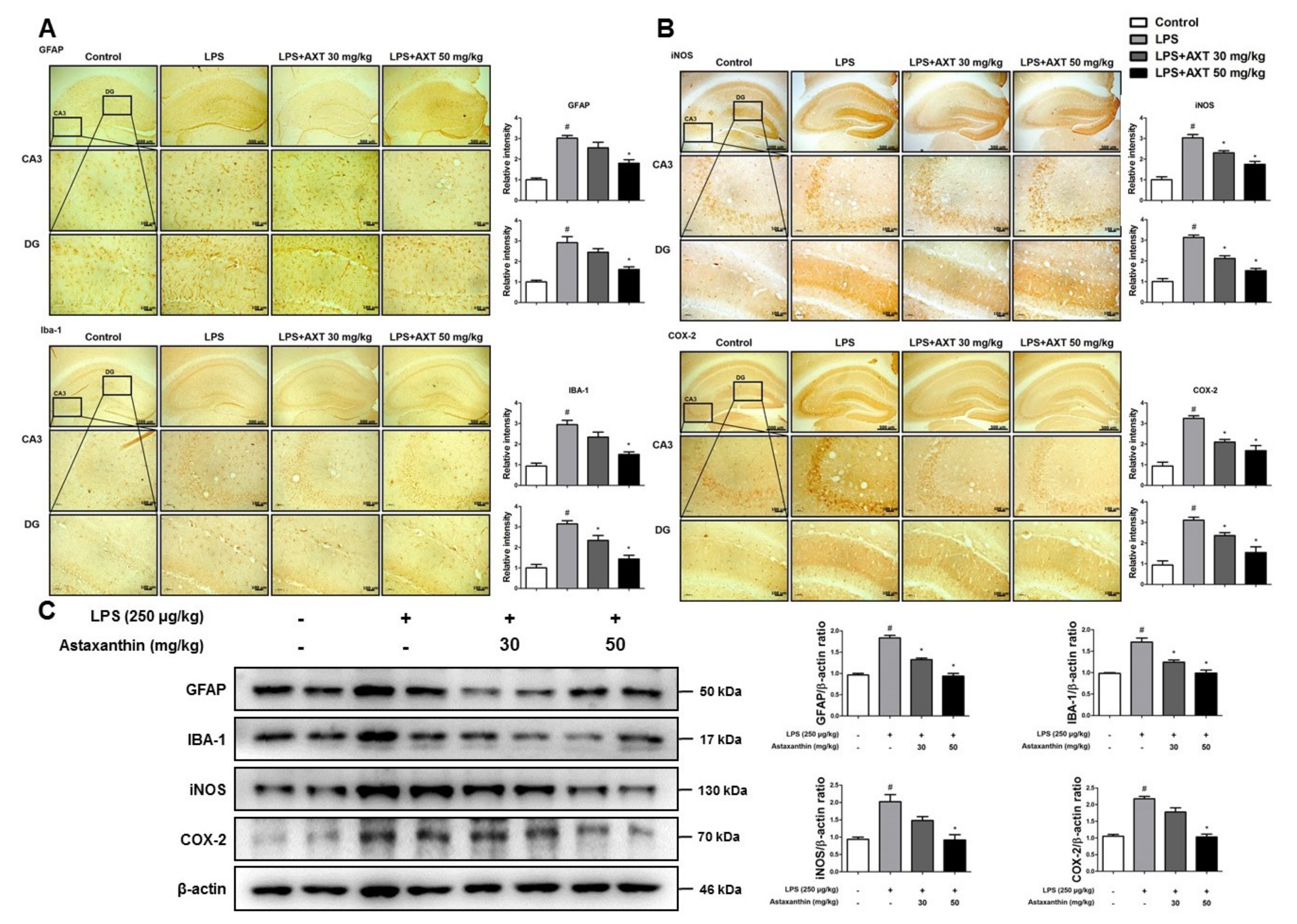
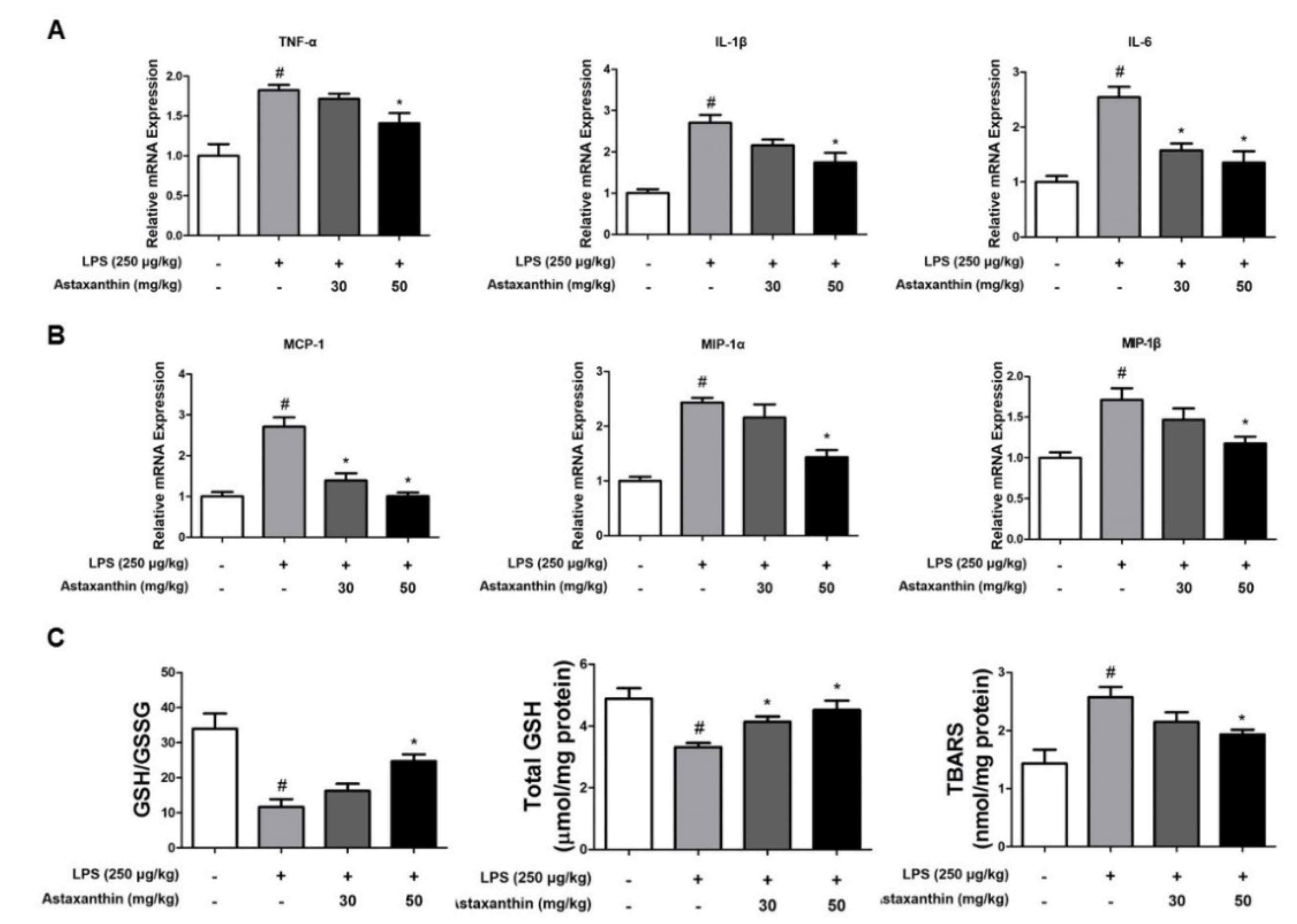
2.3. Astaxanthin Prevents LPS-Induced Neuroinflammation in the Brain of Mice
2.4. Astaxanthin Reduces LPS-Induced Oxidative Stress in the Brain of Mice
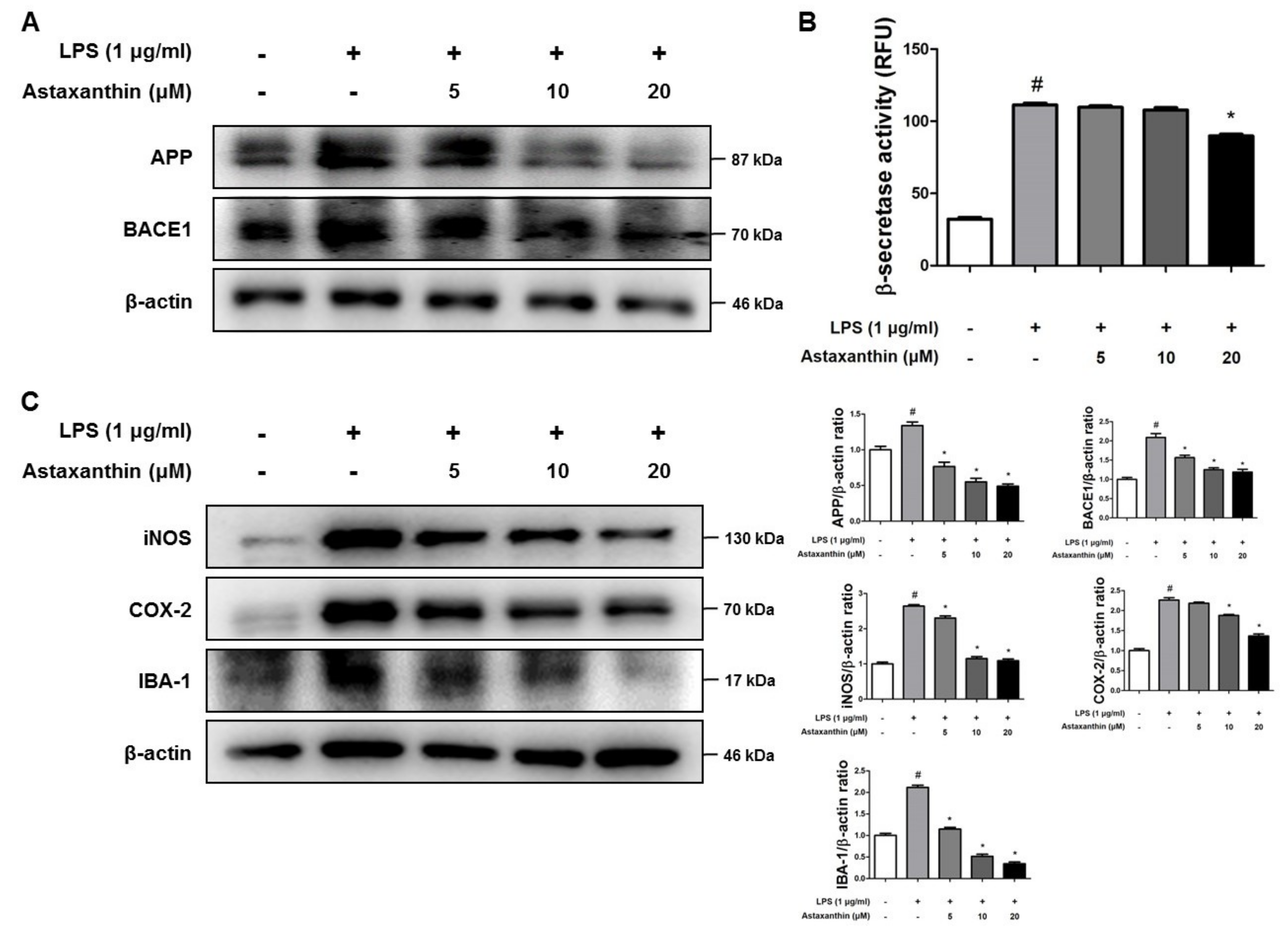
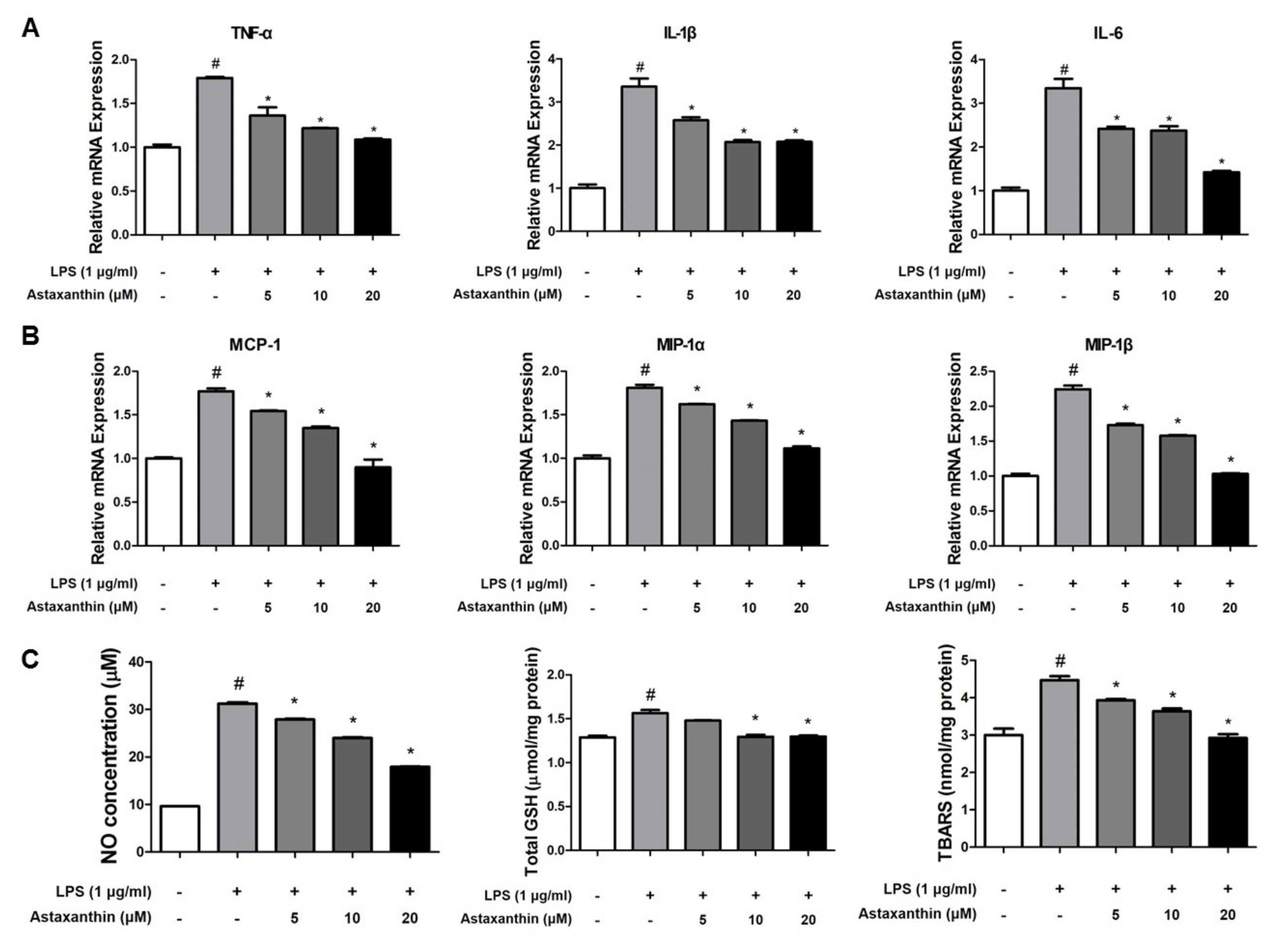
2.5. Astaxanthin Inhibits Amyloidogenesis and Neuroinflammation in Microglia BV-2 Cells
2.6. Astaxanthin Inhibits LPS-Induced Oxidative Stress in the Microglia BV-2 Cells
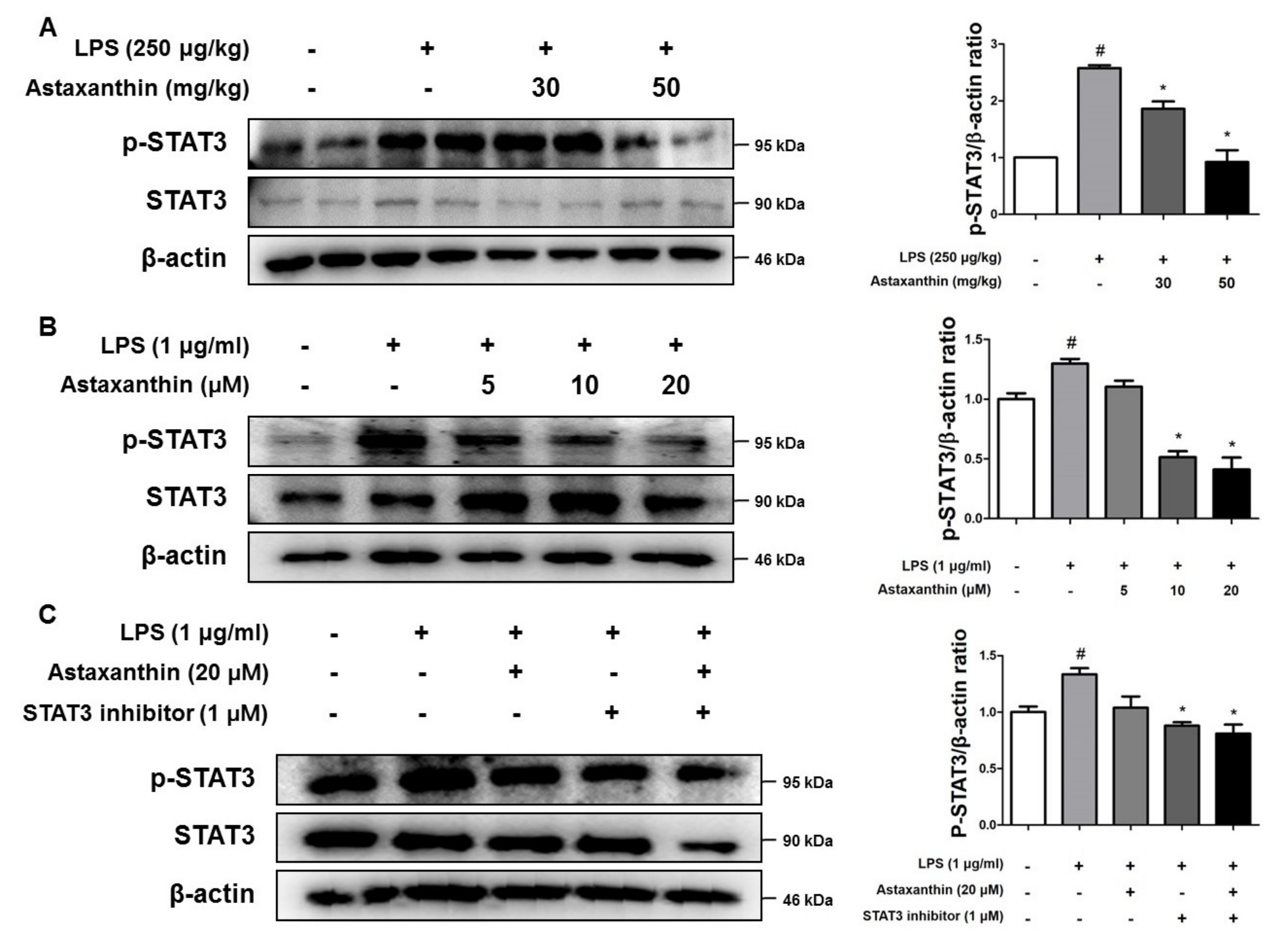
2.7. Astaxanthin Inhibits the Phosphorylation of STAT3 in the Brain of Mice and the Microglia BV-2 Cells
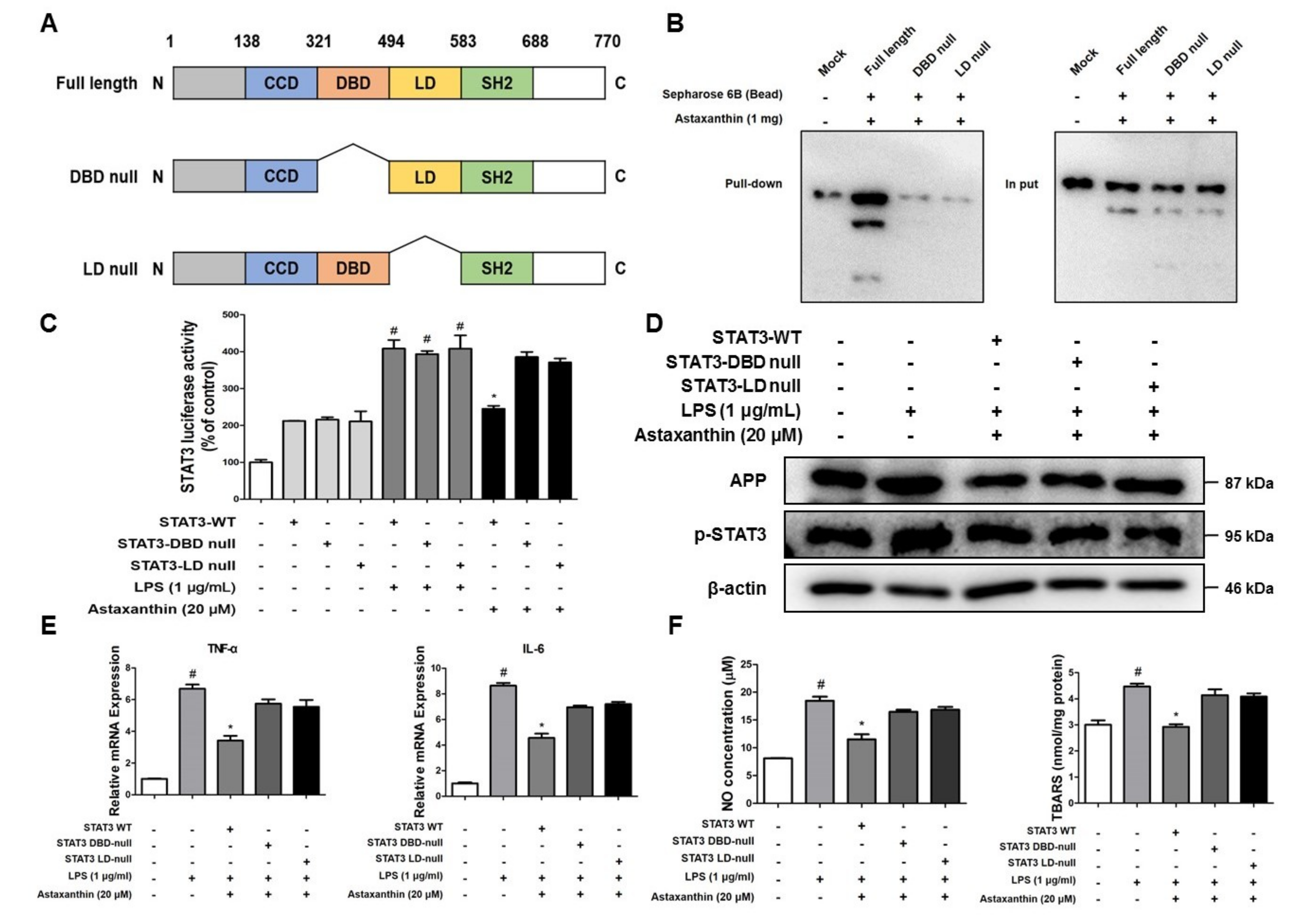
2.8. Astaxanthin Directly Binds to the DBD and LD of STAT3
3. Discussion
4. Materials and Methods
4.1. Animals Experiment and Housing Condition
4.2. Morris Water Maze
4.3. Probe Test
4.4. Passive Avoidance Test
4.5. Collection and Preservation of Brain Tissue
4.6. Microglial BV-2 Cells Cultures
4.7. Western Blot Analysis
4.8. Immunohistochemistry
4.9. Measurement of Aβ1–42
4.10. Assay of β-Secretase Activities
4.11. RNA Isolation and Quantitative Real-Time RT-PCR
4.12. Nitro Oxide and Oxidative Stress Assay
4.13. Plasmid Construction
4.14. Pull-Down Assay
4.15. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Trojanowski, J.Q. Inflammatory hypotheses: Novel mechanisms of Alzheimer’s neurodegeneration and new therapeutic targets? Neurobiol. Aging 2000, 21, 441–445; discussion 451–453. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Hauss-Wegrzyniak, B.; Wenk, G.L. Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: The role of ascorbic acid in the vehicle. Neurosci. Lett. 2002, 322, 75–78. [Google Scholar] [CrossRef]
- Herber, D.L.; Roth, L.M.; Wilson, D.; Wilson, N.; Mason, J.E.; Morgan, D.; Gordon, M.N. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp. Neurol. 2004, 190, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Zhou, W.W.; Lu, S.; Su, Y.J.; Xue, D.; Yu, X.L.; Wang, S.W.; Zhang, H.; Xu, P.X.; Xie, X.X.; Liu, R.T. Decreasing oxidative stress and neuroinflammation with a multifunctional peptide rescues memory deficits in mice with Alzheimer disease. Free Radic. Biol. Med. 2014, 74, 50–63. [Google Scholar] [CrossRef]
- Chauhan, V.; Ji, L.; Chauhan, A. Anti-amyloidogenic, anti-oxidant and anti-apoptotic role of gelsolin in Alzheimer’s disease. Biogerontology 2008, 9, 381–389. [Google Scholar] [CrossRef]
- Monje, M.L.; Mizumatsu, S.; Fike, J.R.; Palmer, T.D. Irradiation induces neural precursor-cell dysfunction. Nat. Med. 2002, 8, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Rosi, S.; Vazdarjanova, A.; Ramirez-Amaya, V.; Worley, P.F.; Barnes, C.A.; Wenk, G.L. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience 2006, 142, 1303–1315. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation 2009, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Cho, I.H.; Kim, H.S.; Jung, J.E.; Kim, J.E.; Lee, K.H.; Park, T.; Yang, Y.M.; Seong, S.Y.; Ye, S.K.; et al. Anti-inflammatory effects of 8-hydroxydeoxyguanosine in LPS-induced microglia activation: Suppression of STAT3-mediated intercellular adhesion molecule-1 expression. Exp. Mol. Med. 2006, 38, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef]
- Aid, S.; Langenbach, R.; Bosetti, F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J. Neuroinflamm. 2008, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Zhang, H.; Li, X.; Zhan, H.; Chen, F.; Han, L.; Xu, Y.; Cao, X. Ampelopsin attenuates lipopolysaccharide-induced inflammatory response through the inhibition of the NF-kappaB and JAK2/STAT3 signaling pathways in microglia. Int. Immunopharmacol. 2017, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Hwang, C.J.; Lee, D.Y.; Gu, S.M.; Lee, H.P.; Choi, D.Y.; Oh, K.W.; Han, S.B.; Hong, J.T. (E)-2-Methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates LPS-Mediated Memory Impairment by Inhibition of STAT3 Pathway. Neuromolecular Med. 2017, 19, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.M.; Kim, J.A.; Hwang, C.J.; Jin, P.; Baek, M.K.; Lee, J.M.; Hong, J.E.; Lee, S.M.; Han, S.B.; Oh, K.W.; et al. Neuroinflammatory and Amyloidogenic Activities of IL-32beta in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.H.; Yeap, Y.Y.; Ong, L.S.; Jans, D.A.; Bogoyevitch, M.A. Oxidative stress impairs multiple regulatory events to drive persistent cytokine-stimulated STAT3 phosphorylation. Biochim. Biophys. Acta 2014, 1843, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Jung, Y.Y.; Hwang, C.J.; Lee, H.P.; Sok, C.H.; Kim, J.H.; Lee, S.M.; Seo, H.O.; Hyun, B.K.; Choi, D.Y.; et al. Inhibitory effect of ent-Sauchinone on amyloidogenesis via inhibition of STAT3-mediated NF-kappaB activation in cultured astrocytes and microglial BV-2 cells. J. Neuroinflamm. 2014, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Sivakumaran, V.; Duhe, R.J.; Aon, M.A.; Paolocci, N.; Booz, G.W. Depletion of cellular glutathione modulates LIF-induced JAK1-STAT3 signaling in cardiac myocytes. Int. J. Biochem. Cell Biol. 2012, 44, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Themanns, M.; Mueller, K.M.; Kessler, S.M.; Golob-Schwarzl, N.; Mohr, T.; Kaltenecker, D.; Bourgeais, J.; Paier-Pourani, J.; Friedbichler, K.; Schneller, D.; et al. Hepatic Deletion of Janus Kinase 2 Counteracts Oxidative Stress in Mice. Sci. Rep. 2016, 6, 34719. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.X.; Edwards, B.; Lee, S.; Finelli, M.J.; Davies, B.; Davies, K.E.; Oliver, P.L. Neuron-specific antioxidant OXR1 extends survival of a mouse model of amyotrophic lateral sclerosis. Brain J. Neurol. 2015, 138, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 2009, 14, 206–222. [Google Scholar] [CrossRef]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Hwang, C.J.; Lee, H.P.; Kim, H.S.; Han, S.B.; Hong, J.T. Inhibitory effect of ethanol extract of Nannochloropsis oceanica on lipopolysaccharide-induced neuroinflammation, oxidative stress, amyloidogenesis and memory impairment. Oncotarget 2017, 8, 45517–45530. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hwang, C.J.; Lee, H.P.; Kim, C.S.; Son, D.J.; Ham, Y.W.; Hellstrom, M.; Han, S.B.; Kim, H.S.; Park, E.K.; et al. Inhibitory effect of punicalagin on lipopolysaccharide-induced neuroinflammation, oxidative stress and memory impairment via inhibition of nuclear factor-kappaB. Neuropharmacology 2017, 117, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Martin, R.; Kohler, G.; Chan, C. Palmitate induces transcriptional regulation of BACE1 and presenilin by STAT3 in neurons mediated by astrocytes. Exp. Neurol. 2013, 248, 482–490. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Badshah, H.; Ali, T.; Kim, M.O. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFkappaB signaling pathway. Sci. Rep. 2016, 6, 24493. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Zhang, X.S.; Zhang, X.; Wu, Q.; Li, W.; Wang, C.X.; Xie, G.B.; Zhou, X.M.; Shi, J.X.; Zhou, M.L. Astaxanthin offers neuroprotection and reduces neuroinflammation in experimental subarachnoid hemorrhage. J. Surg. Res. 2014, 192, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Pepeu, G.; Casamenti, F. Experimental brain inflammation and neurodegeneration as model of Alzheimer’s disease: Protective effects of selective COX-2 inhibitors. Int. J. Immunopathol. Pharmacol. 2003, 16, 31–40. [Google Scholar] [PubMed]
- Nathan, C.; Calingasan, N.; Nezezon, J.; Ding, A.; Lucia, M.S.; La Perle, K.; Fuortes, M.; Lin, M.; Ehrt, S.; Kwon, N.S.; et al. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J. Exp. Med. 2005, 202, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Ho, L.; Valdellon, J.; Borchelt, D.; Kelley, K.; Spielman, L.; Aisen, P.S.; Pasinetti, G.M. Cyclooxygenase (COX)-2 and cell cycle activity in a transgenic mouse model of Alzheimer’s disease neuropathology. Neurobiol. Aging 2002, 23, 327–334. [Google Scholar] [CrossRef]
- Hwang, D.Y.; Chae, K.R.; Kang, T.S.; Hwang, J.H.; Lim, C.H.; Kang, H.K.; Goo, J.S.; Lee, M.R.; Lim, H.J.; Min, S.H.; et al. Alterations in behavior, amyloid beta-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer’s disease. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 805–813. [Google Scholar]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Christen, Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621S–629S. [Google Scholar] [CrossRef]
- Thomas, T.; Thomas, G.; McLendon, C.; Sutton, T.; Mullan, M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996, 380, 168–171. [Google Scholar] [CrossRef]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L. Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K.; St Clair, D.K. Beta-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Yu, L.J.; Hui, X.C.; Wu, Z.Z.; Yin, K.L.; Yang, H.; Xu, Y. Hydroxy-safflor yellow A attenuates Abeta(1)(-)(4)(2)-induced inflammation by modulating the JAK2/STAT3/NF-kappaB pathway. Brain Res. 2014, 1563, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Eufemi, M.; Cocchiola, R.; Romaniello, D.; Correani, V.; Di Francesco, L.; Fabrizi, C.; Maras, B.; Schinina, M.E. Acetylation and phosphorylation of STAT3 are involved in the responsiveness of microglia to beta amyloid. Neurochem. Int. 2015, 81, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Ohishi, M.; Mori, H.; Murakami, M.; Chinen, T.; Aki, D.; Hanada, T.; Takeda, K.; Akira, S.; Hoshijima, M.; et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 2003, 4, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Beaulieu-Abdelahad, D.; Abdullah, L.; Bachmeier, C.; Ait-Ghezala, G.; Reed, J.; Verma, M.; Crawford, F.; Mullan, M. Anti-inflammatory activity of anatabine via inhibition of STAT3 phosphorylation. Eur. J. Pharmacol. 2013, 698, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Fernandez, J.A.; Maccioni, A.A.; Jimenez, J.M.; Maccioni, R.B. Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch. Med. Res. 2008, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.H.; Lee, Y.S.; Im, J.H.; Ham, Y.W.; Lee, H.P.; Han, S.B.; Hong, J.T. Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway. Mar. Drugs 2019, 17, 123. https://doi.org/10.3390/md17020123
Han JH, Lee YS, Im JH, Ham YW, Lee HP, Han SB, Hong JT. Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway. Marine Drugs. 2019; 17(2):123. https://doi.org/10.3390/md17020123
Chicago/Turabian StyleHan, Ji Hye, Yong Sun Lee, Jun Hyung Im, Young Wan Ham, Hee Pom Lee, Sang Bae Han, and Jin Tae Hong. 2019. "Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway" Marine Drugs 17, no. 2: 123. https://doi.org/10.3390/md17020123
APA StyleHan, J. H., Lee, Y. S., Im, J. H., Ham, Y. W., Lee, H. P., Han, S. B., & Hong, J. T. (2019). Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway. Marine Drugs, 17(2), 123. https://doi.org/10.3390/md17020123