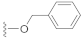
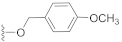
3.3. Synthesis Experiments
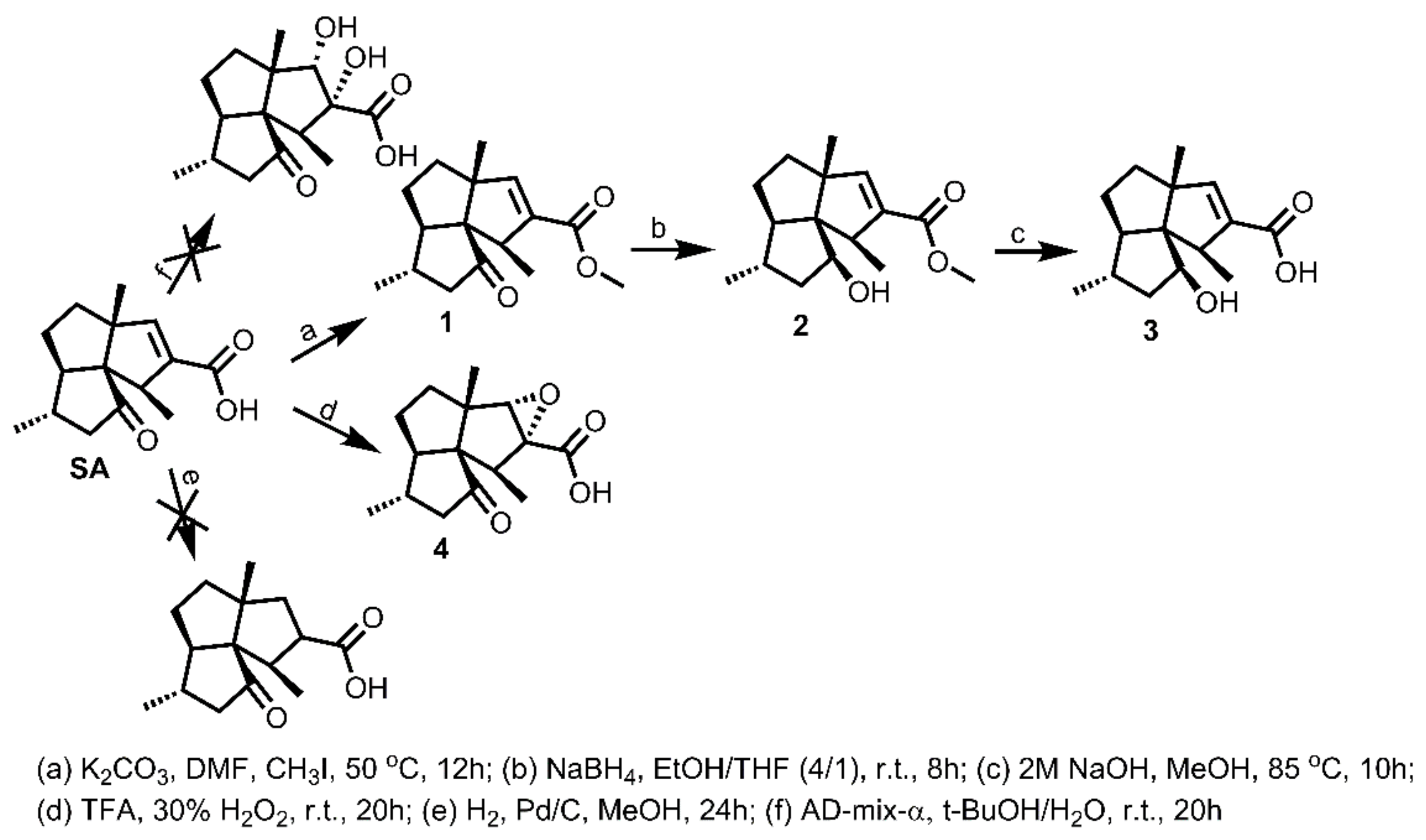
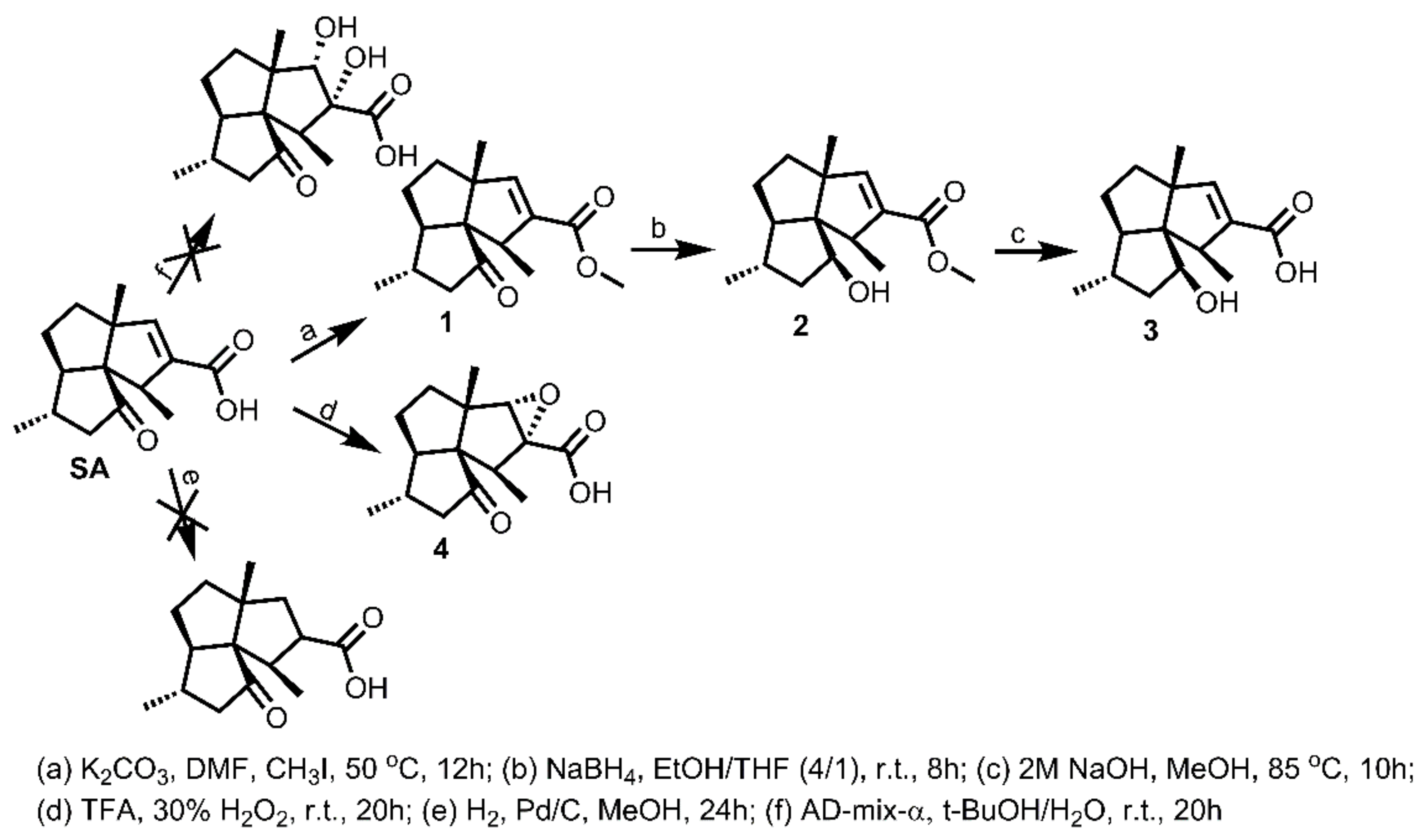
3.3.1. Synthesis of 1–4
To a solution of SA (50 mg, 0.20 mmol) in DMF (2 mL, freshly distilled from CaH2), was added K2CO3 (80 mg, 0.58 mmol), followed by the addition of iodomethane (26 μL, 0.41 mmol). The obtained mixture was then stirred under N2 atmosphere at 50 °C for 12 h. Afterwards, the reaction mixture was cooled to room temperature and quenched by brine (10 mL). The mixture was then extracted with DCM (8 mL × 3), washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (EtOAc/PE = 20:80) to yield 1 (49.8 mg, 95% yield) as an off-yellow oil. − 30.5 (c 0.09, DCM); 1H NMR (300 MHz, CDCl3) δH: 6.27 (s, 1H), 3.72 (s, 3H), 3.00 (q, J = 6.9 Hz, 1H), 2.34 (dd, J = 16.8, 6.9 Hz, 1H), 2.08 (m, 1H), 1.98 (dd, J = 16.8, 12.6 Hz, 1H), 1.75–1.79 (m, 1H), 1.54–1.67 (m, 4H), 1.19 (s, 3H), 1.10 (d, J = 6.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δC: 217.9, 165.0, 149.7, 137.1, 68.6, 62.8, 61.8, 52.1, 51.6, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 263.1650 [M+H]+ (calcd. for C16H23O3 at m/z 263.1647).
To a solution of 1 (30 mg, 0.11 mmol) in EtOH/TFH (v/v = 4/1, 5 mL), was added NaBH4 (10 mg, 0.26 mmol). The obtained mixture was then stirred under N2 atmosphere at room temperature for 8 h. Afterwards, the reaction mixture was quenched by addition of acetone (2 mL), followed by stirring for half an hour. The mixture was then evaporated to remove all solvent. Water (5 mL) was added into the residue to form a suspension, which was then extracted with DCM (5 mL × 3). The combined DCM extract was washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (EtOAc/PE = 10:90) to yield 2 (24.6 mg, 86% yield) as an off-white powder. + 93.1 (c 0.41, DCM); 1H NMR (300 MHz, CDCl3) δH: 6.29 (s, 1H), 4.15 (m, 1H), 3.73 (s, 3H), 3.09 (q, J = 6.9 Hz, 1H), 1.92 (m, 1H), 1.84–1.87 (m, 1H), 1.61–1.65 (m, 2H), 1.47–1.54 (m, 4H), 1.54–1.67 (m, 4H), 1.35 (d, J = 6.9 Hz, 3H), 1.20 (s, 3H), 1.13 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 165.4, 150.1, 139.2, 77.8, 66.4, 62.6, 57.8, 51.5, 49.7, 43.4, 40.0, 37.8, 31.6, 23.0, 20.1, 19.9; HRMS m/z 265.1799 [M + H]+ (calcd. for C16H25O3 at m/z 265.1804), 247.1694 [M − H2O + H]+ (calcd. for C16H23O2 at m/z 247.1698).
Compound 2 (20 mg, 0.076 mmol) was added into the flask containing NaOH aqueous solution (2 mol/L, 2 mL), followed by addition of methanol (0.2 mL) to form a clear solution. A condenser was connected to the flask. The solution was then stirred under N2 atmosphere at 85 °C for 10 h. Afterwards, the reaction mixture was cooled to room temperature and quenched by dropwise addition of HCl aqueous solution (1 mol/L, 5 mL). The mixture was then extracted with DCM (6 mL × 3), washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (MeOH/DCM = 4:96) to yield 3 (17.9 mg, 94% yield) as a white amorphous powder. + 110.9 (c 0.39, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.45 (s, 1H), 4.17 (m, 1H), 3.09 (q, J = 7.2 Hz, 1H), 1.92 (m, 1H), 1.85–1.87 (m, 1H), 1.65 (m, 1H), 1.48–1.52 (m, 4H), 1.37 (d, J = 6.8 Hz, 3H), 1.28 (m, 1H), 1.22 (s, 3H), 1.13 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 169.7, 152.8, 138.8, 77.8, 66.5, 62.6, 58.0, 49.4, 43.7, 40.0, 37.8, 31.6, 22.9, 20.9, 19.8; HRMS m/z 249.1499 [M − H]+ (calcd. for C15H21O3 at m/z 249.1491).
To a solution of SA (15 mg, 0.06 mmol) in TFA (trifluoroacetic acid, 1 mL), was added dropwise the 30% H2O2 (1.5 mL) at 0 °C. The obtained mixture was then stirred under N2 atmosphere from 0 °C to room temperature for 20 h. Afterwards, the reaction mixture was quenched by Na2SO3 aqueous solution (4 mL) at 0 °C and diluted with H2O (3 mL) to a pH around 1.0. The mixture was then extracted with DCM (8 mL × 3), washed successively with brine (15 mL × 3) and water (15 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (MeOH/DCM = 10:90) to yield 4 (13.5 mg, 85% yield) as a white amorphous powder. + 46.7 (c 0.28, DCM); 1H NMR (400 MHz, CDCl3) δH: 3.56 (s, 1H), 2.74 (q, J = 6.9 Hz, 1H), 2.39 (dd, J = 16.8, 6.4 Hz, 1H), 2.00 (m, 3H), 1.78–1.86 (m, 4H), 1.11 (s, 3H), 1.06 (d, J = 6.8 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 216.5, 170.9, 70.8, 70.8, 67.9, 61.1, 55.9, 49.8, 44.2, 35.7, 32.2, 27.6, 22.6, 19.3, 10.9; HRMS m/z 246.1260 [M − H2O]− (calcd. for C15H18O3 at m/z 246.1256), 263.1246 [M − H]− (calcd. for C15H19O4 at m/z 263.1283).
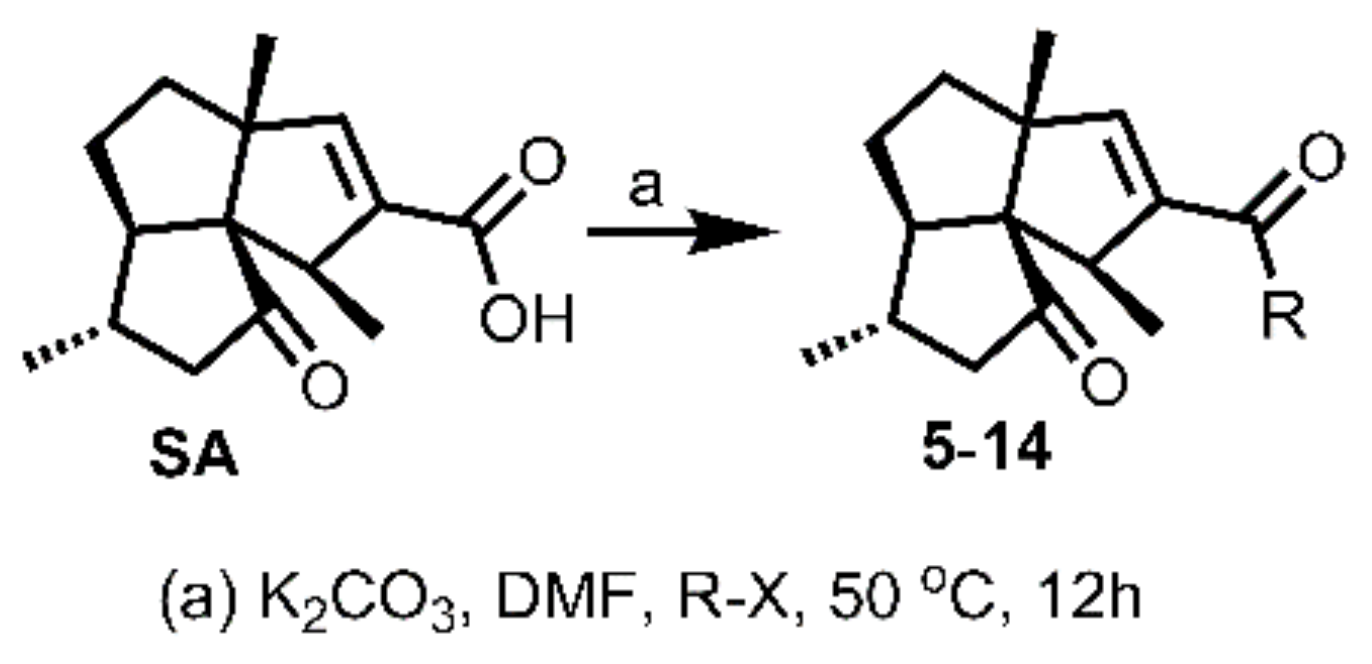
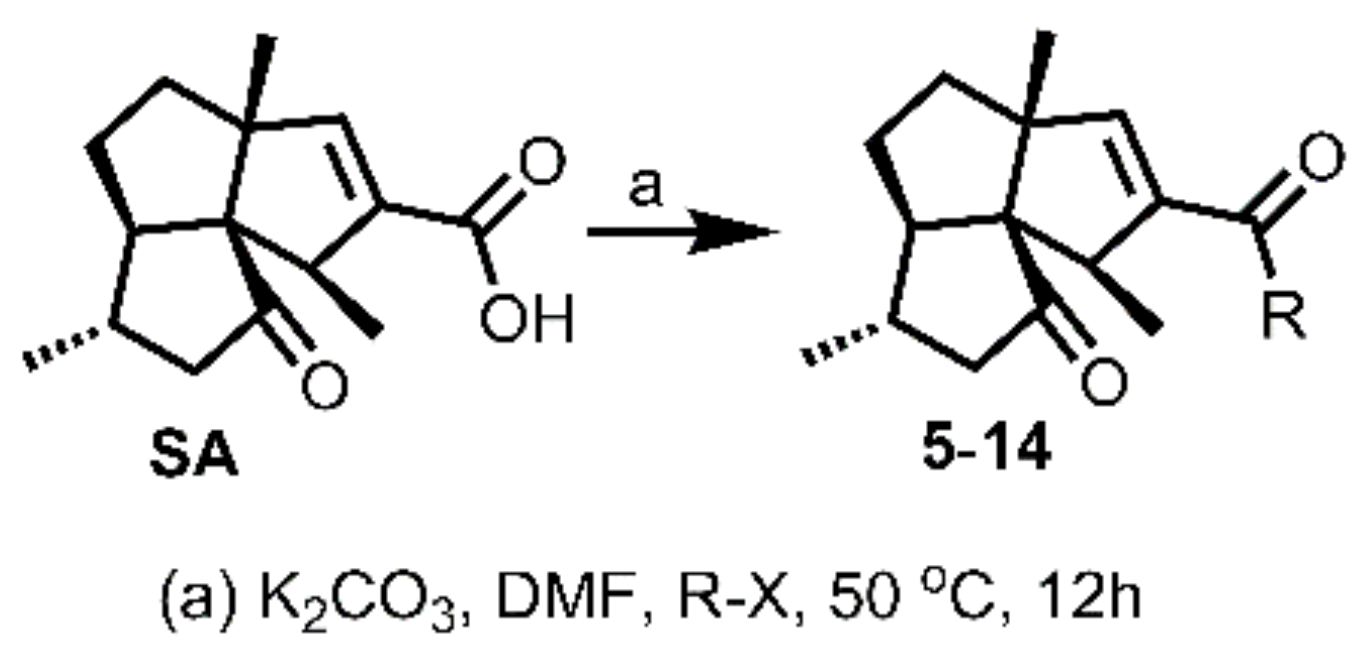
3.3.2. General Procedure for Synthesis of 5–14
To a solution of SA (15 mg, 0.06 mmol) in DMF (1 mL, freshly distilled from CaH2), was added K2CO3 (25 mg, 0.18 mmol), followed by the addition of benzyl halide derivative (0.12 mmol) for 5–12, or 2-(bromomethyl)pyridine, 4-(bromomethyl)pyridine, for 13–14, respectively. The obtained mixture was then stirred under N2 atmosphere at 50 °C for 12 h. Afterwards, the reaction mixture was cooled to room temperature and quenched by brine (5 mL). The mixture was then extracted with DCM (5 mL × 3), washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (EtOAc/PE ranged from 10:90 to 30:70 depending on the TLC analysis) to yield 5–14.
Compound 5 (19.5 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 96% yield. − 93.1 (c 0.18, DCM); 1H NMR (400 MHz, CDCl3) δH: 7.33–7.39 (m, 5H), 6.33 (s, 1H), 5.19 (d, J = 12.4 Hz, 1H ), 5.14 (d, J = 12.4 Hz, 1H ), 3.03 (q, J = 6.8 Hz, 1H), 2.34 (dd, J = 16.8, 6.8 Hz, 1H), 2.07 (m, 1H), 1.98 (dd, J = 16.8, 12.8 Hz, 1H), 1.78 (m, 1H), 1.62–1.67 (m, 2H), 1.54–1.57 (m, 2H), 1.20 (s, 3H), 1.12 (d, J = 7.2 Hz, 3H), 1.10 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 217.9, 164.4, 150.1, 137.1, 136.1, 128.7, 128.7, 128.4, 128.3, 128.3, 68.6, 66.3, 62.8, 61.8, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 337.1811 [M − H]− (calcd. for C22H25O3 at m/z 337.1804).
Compound 6 (23.5 mg), an off-white powder, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 98% yield. − 95.0 (c 0.89, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.40 (d, J = 8.1 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 6.32 (s, 1H), 5.16 (d, J = 12.3 Hz, 1H), 5.12 (d, J = 12.3 Hz, 1H), 3.04 (q, J = 7.2 Hz, 1H), 2.34 (dd, J = 16.8, 6.6 Hz, 1H), 1.94–2.07 (m, 2H), 1.75–1.78 (m, 1H), 1.53–1.66 (m, 4H), 1.32 (s, 9H), 1.19 (s, 3H), 1.12 (d, J = 6.6 Hz, 3H), 1.10 (d, J = 5.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.5, 151.3, 149.9, 137.1, 133.1, 128.2, 128.2, 125.6, 125.6, 68.6, 66.1, 62.8, 61.8, 52.0, 50.1, 38.4, 34.7, 33.5, 31.5, 31.5, 31.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 395.2586 [M + H]+ (calcd. for C26H35O3 at m/z 395.2586).
Compound 7 (24.0 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 96% yield. − 76.1 (c 0.91, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.50 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 6.32 (s, 1H), 5.14 (d, J = 12.6 Hz, 1H), 5.08 (d, J = 12.6 Hz, 1H), 3.03 (q, J = 7.2 Hz, 1H), 2.35 (dd, J = 16.8, 6.6 Hz, 1H), 2.07 (m, 1H), 1.98 (dd, J = 16.5, 12.6 Hz, 1H), 1.77 (m, 1H), 1.54–1.68 (m, 4H), 1.20 (s, 3H), 1.11 (d, J = 6.3 Hz, 6H ); 13C NMR (75 MHz, CDCl3) δC: 217.8, 164.2, 150.3, 136.9, 135.1, 131.8, 131.8, 130.0, 130.0, 122.4, 68.6, 65.4, 62.8, 61.8, 52.0, 50.0, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 417.1069 [M + H]+ (calcd. for C22H26BrO3 at m/z 417.1065).
Compound 8 (23.5 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 97% yield. − 68.6 (c 0.08, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.34 (d, J = 7.5 Hz, 2H), 7.23 (m, 1H), 6.29 (s, 1H), 5.43 (s, 2H), 2.99 (q, J = 6.9 Hz, 1H), 2.33 (dd, J = 16.5, 6.6 Hz, 1H), 2.06 (m, 1H), 1.97 (dd, J = 16.5, 12.6 Hz, 1H), 1.72–1.78 (m, 1H), 1.51–1.66 (m, 4H), 1.17 (s, 3H), 1.09 (d, J = 7.2 Hz, 3H), 1.09 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.3, 150.3, 137.1, 136.7, 131.6, 130.6, 130.6, 128.6, 128.6, 68.6, 65.4, 62.7, 61.7, 61.2, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 17.9; HRMS m/z 370.1357 [M − HCl]−, 405.1028 [M − H]− (calcd. for C22H23ClO3 and C22H23Cl2O3 at m/z 370.1336 and 405.1025, respectively).
Compound 9 (22.4 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 90% yield. − 91.4 (c 0.17, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.49–7.52 (m, 1H), 7.30–7.43 (m, 8H), 6.22 (s, 1H), 5.12 (s, 2H), 2.98 (q, J = 7.2 Hz, 1H), 2.34 (dd, J = 16.5, 6.6 Hz, 1H), 2.03–2.08 (m, 1H), 1.98 (dd, J = 16.5, 12.6 Hz, 1H), 1.72–1.76 (m, 1H), 1.54–1.66 (m, 4H), 1.19 (s, 3H), 1.11 (d, J = 6.6 Hz, 3H ), 1.08 (d, J = 7.2 Hz, 3H ); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.2, 149.8, 142.4, 140.6, 137.0, 133.4, 130.4, 129.6, 129.2, 129.2, 128.4, 128.3, 128.3, 127.7, 127.5, 68.5, 64.5, 62.8, 61.8, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 415.2269 [M + H]+ (calcd. for C28H31O3 at m/z 415.2273).
Compound 10 (21.7 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 15:85) from the reaction residue with 97% yield. − 126.2 (c 0.11, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.36 (s, 1H), 7.30–7.32 (m, 2H), 7.23–7.26 (m, 1H), 6.34 (s, 1H), 5.16 (d, J = 12.6 Hz, 1H), 5.10 (d, J = 12.6 Hz, 1H), 3.04 (q, J = 7.2 Hz, 1H), 2.36 (dd, J = 16.5, 6.6 Hz, 1H), 2.06–2.11 (m, 1H), 2.00 (dd, J = 16.5, 12.6 Hz, 1H), 1.78–1.81 (m, 1H), 1.55–1.65 (m, 4H), 1.21 (s, 3H), 1.12 (d, J = 6.6 Hz, 3H ), 1.10 (d, J = 6.3 Hz, 3H ); 13C NMR (75 MHz, CDCl3) δC: 217.8, 164.2, 150.4, 138.1, 136.8, 134.5, 130.0, 128.5, 128.3, 126.3, 68.6, 65.4, 62.8, 61.8, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 371.1425 [M − H]− (calcd. for C22H24ClO3 at m/z 371.1414).
Compound 11 (20.9 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 15:85) from the reaction residue with 95% yield. − 83.0 (c 0.09, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.29 (m, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.85–6.90 (m, 2H), 6.33 (s, 1H), 5.17 (d, J = 12.6 Hz, 1H), 5.11 (d, J = 12.6 Hz, 1H), 3.81 (s, 3H), 3.04 (q, J = 7.2 Hz, 1H), 2.34 (dd, J = 16.5, 6.9 Hz, 1H), 2.05–2.10 (m, 1H), 2.00 (dd, J = 16.5, 12.6 Hz, 1H), 1.76–1.78 (m, 1H), 1.53–1.69 (m, 4H), 1.20 (s, 3H), 1.12 (d, J = 6.9 Hz, 3H ), 1.10 (d, J = 6.3 Hz, 3H ); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.3, 159.8, 150.1, 137.6, 137.1, 129.8, 120.4, 113.8, 113.7, 68.6, 66.1, 62.8, 61.8, 55.4, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 367.1913 [M − H]− (calcd. for C23H27O4 at m/z 367.1910).
Compound 12 (21.1 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 20:80) from the reaction residue with 96% yield. − 68.3 (c 0.16, DCM); 1H NMR (300 MHz, CDCl3) δH: 7.31 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.29 (s, 1H), 5.13 (d, J = 12.0 Hz, 1H), 5.07 (d, J = 12.0 Hz, 1H), 3.81 (s, 3H), 3.01 (q, J = 6.9 Hz, 1H), 2.34 (dd, J = 16.5, 6.9 Hz, 1H), 2.04–2.09 (m, 1H), 1.98 (dd, J = 16.5, 12.6 Hz, 1H), 1.74–1.78 (m, 1H), 1.56–1.66 (m, 4H), 1.18 (s, 3H), 1.10 (d, J = 7.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.5, 159.7, 149.9, 137.1, 130.2, 130.2, 128.2, 114.0, 114.0, 68.6, 66.1, 62.8, 61.8, 55.4, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 386.2336 [M + NH4]+ (calcd. for C23H32NO4 at m/z 386.2331).
Compound 13 (23.8 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 25:75) from the reaction residue with 95% yield. − 96.0 (c 0.17, DCM); 1H NMR (300 MHz, CDCl3) δH: 8.59–8.61 (m, 1H), 7.69–7.75 (m, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.22–7.26 (m, 1H), 6.39 (s, 1H), 5.29 (s, 2H), 3.07 (q, J = 6.8 Hz, 1H), 2.36 (dd, J = 16.8, 6.8 Hz, 1H), 2.07–2.11 (m, 1H), 2.00 (dd, J = 16.8, 12.8 Hz, 1H), 1.78–1.83 (m, 1H), 1.55–1.70 (m, 4H), 1.20 (s, 3H), 1.14 (d, J = 7.5 Hz, 3H), 1.12 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 217.8, 164.1, 156.0, 150.4, 149.6, 136.9, 136.8, 123.0, 121.9, 68.6, 66.9, 62.8, 61.9, 52.0, 50.1, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 338.1754 [M − H]− (calcd. for C21H24NO3 at m/z 338.1756).
Compound 14 (24.0 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 30:70) from the reaction residue with 96% yield. − 106.8 (c 0.09, DCM); 1H NMR (300 MHz, CDCl3) δH: 8.52 (d, J = 5.7 Hz, 2H), 7.26 (d, J = 5.4 Hz, 2H), 6.40 (s, 1H), 5.22 (d, J = 13.8 Hz, 1H), 5.16 (d, J = 13.8 Hz, 1H), 3.06 (q, J = 6.8 Hz, 1H), 2.37 (dd, J = 16.8, 6.8 Hz, 1H), 2.07–2.12 (m, 1H), 2.00 (dd, J = 16.8, 12.8 Hz, 1H), 1.78–1.82 (m, 1H), 1.57–1.70 (m, 4H), 1.23 (s, 3H), 1.14 (d, J = 6.9 Hz, 3H), 1.12 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 217.7, 163.9, 150.8, 150.2, 150.2, 145.1, 136.6, 122.0, 122.0, 68.6, 64.3, 62.8, 61.9, 52.0, 50.0, 38.4, 33.5, 28.4, 23.7, 20.0, 18.0; HRMS m/z 338.1759 [M − H]− (calcd. for C21H24NO3 at m/z 338.1756).
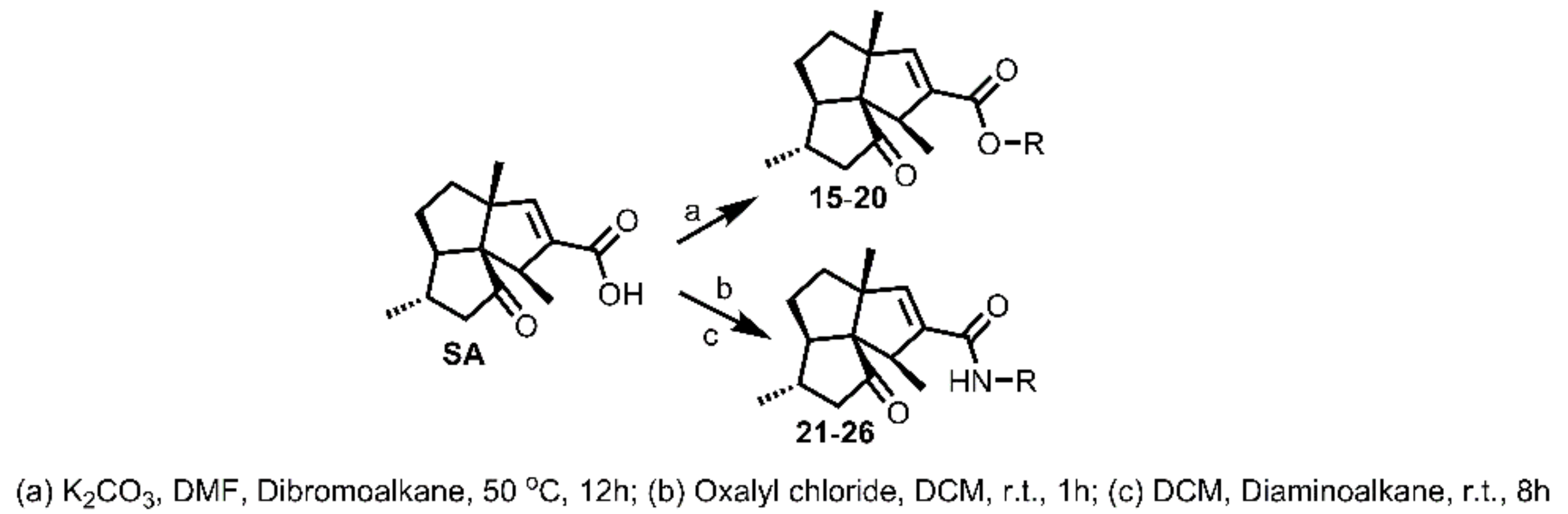
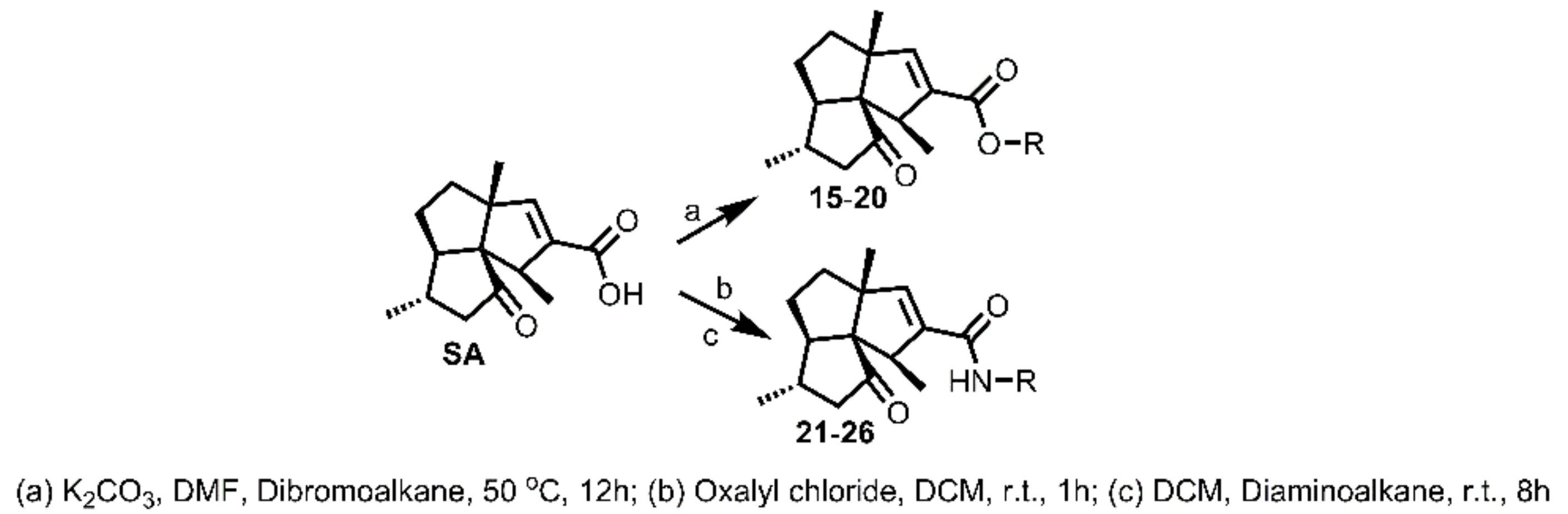
3.3.3. General Procedure for Synthesis of 15–20
To a solution of SA (15 mg, 0.06 mmol) in DMF (1 mL, freshly distilled from CaH2), was added K2CO3 (25 mg, 0.18 mmol), followed by the addition of dibromoalkane (0.24 mmol). The obtained mixture was then stirred under N2 atmosphere at 50 °C for 12 h. Afterwards, the reaction mixture was cooled to room temperature and quenched by brine (5 mL). The mixture was then extracted with DCM (5 mL × 3), washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (EtOAc/PE ranged from 5:95 to 10:90 depending on the TLC analysis) to yield 15–20.
Compound 15 (21.1 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 92% yield. − 68.4 (c 0.07, DCM); 1H NMR (300 MHz, CDCl3) δH: 6.27 (s, 1H), 4.07–4.25 (m, 2H), 3.45 (t, J = 6.3 Hz, 2H), 3.00 (q, J = 6.9 Hz, 1H), 2.35 (dd, J = 16.5, 6.6 Hz, 1H), 2.02–2.20 (m, 1H), 1.94–2.00 (m, 2H), 1.76–1.88 (m, 3H), 1.57–1.67 (m, 5H), 1.20 (s, 3H), 1.05–1.15 (m, 6H); 13C NMR (75 MHz, CDCl3) δC: 217.9, 164.6, 149.8, 137.1, 68.6, 63.5, 62.8, 61.8, 52.0, 50.1, 38.4, 33.5, 33.3, 29.5, 28.4, 27.4, 23.7, 20.0, 18.0; HRMS m/z 400.1477 [M + NH4]+ (calcd. for C19H31BrNO3 at m/z 400.1487).
Compound 16 (21.4 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 90% yield. − 48.2 (c 0.14, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.27 (s, 1H), 4.09–4.17 (m, 2H), 3.42 (t, J = 6.8 Hz, 2H), 3.00 (q, J = 7.2 Hz, 1H), 2.35 (dd, J = 16.8, 6.8 Hz, 1H), 2.05–2.07 (m, 1H), 2.00 (dd, J = 16.8, 12.8 Hz, 1H), 1.84–1.94 (m, 3H), 1.65–1.73 (m, 5H), 1.50–1.53 (m, 3H), 1.20 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 218.0, 164.6, 149.6, 137.3, 68.6, 64.1, 62.8, 61.7, 52.0, 50.1, 38.4, 33.6, 33.5, 32.4, 28.4, 27.9, 24.8, 23.7, 20.0, 18.0; HRMS m/z 395.1221 [M − H]+ (calcd. for C20H28BrO3 at m/z 395.1222).
Compound 17 (22.9 mg), a colorless oil, was purified by silica gel CC (EtOAc/PE 10:90) from the reaction residue with 93% yield. − 59.6 (c 0.09, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.27 (s, 1H), 4.06–4.18 (m, 2H), 3.41 (t, J = 6.8 Hz, 2H), 3.00 (q, J = 7.6 Hz, 1H ), 2.35 (dd, J = 16.8, 6.8 Hz, 1H ), 2.05–2.09 (m, 1H), 1.99 (dd, J = 16.8, 12.8 Hz, 1H), 1.84–1.91 (m, 2H), 1.76–1.80 (m, 1H), 1.61–1.71 (m, 6H), 1.40–1.50 (m, 4H), 1.20 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 218.0, 164.7, 149.5, 137.3, 68.6, 64.3, 62.8, 61.7, 52.0, 50.1, 38.4, 33.9, 33.5, 32.7, 28.6, 28.4, 27.9, 25.4, 23.7, 20.0, 18.0; HRMS m/z 409.1381 [M − H]+ (calcd. for C21H30BrO3 at m/z 409.1379).
Compound 18 (23.2 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 5:95) from the reaction residue with 91% yield. − 79.1 (c 0.39, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.27 (s, 1H), 4.07–4.15 (m, 2H), 3.41 (t, J = 6.8 Hz, 2H), 3.00 (q, J = 6.8 Hz, 1H ), 2.35 (dd, J = 16.8, 6.8 Hz, 1H), 2.05–2.09 (m, 1H), 1.99 (dd, J = 16.8, 12.8 Hz, 1H), 1.78–1.89 (m, 4H), 1.64–1.68 (m, 5H), 1.50–1.60 (m, 2H), 1.35–1.38 (m, 4H), 1.20 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 218.0, 164.7, 149.4, 137.4, 68.6, 64.4, 62.8, 61.7, 52.0, 50.1, 38.4, 34.0, 33.5, 32.8, 28.6, 28.5, 28.4, 28.1, 26.0, 23.7, 20.0, 18.0; HRMS m/z 423.1527 [M − H]+ (calcd. for C22H32BrO3 at m/z 423.1535).
Compound 19 (23.4 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 5:95) from the reaction residue with 89% yield. − 124.5 (c 0.96, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.27 (s, 1H), 4.05–4.17 (m, 2H), 3.40 (t, J = 6.8 Hz, 2H), 3.00 (q, J = 7.2 Hz, 1H ), 2.35 (dd, J = 16.4, 6.8 Hz, 1H ), 2.05–2.09 (m, 1H), 1.99 (dd, J = 16.4, 12.4, 1H), 180–1.89 (m, 2H), 1.57–1.79 (m, 9H), 1.32–1.41 (m, 6H), 1.20 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 218.0, 164.8, 149.4, 137.4, 68.6, 64.5, 62.8, 61.7, 52.0, 50.1, 38.4, 34.1, 33.5, 32.9, 29.2, 28.7, 28.7, 28.4, 28.2, 26.0, 23.7, 20.0, 18.0; HRMS m/z 439.1854 [M + H]+ (calcd. for C23H36BrO3 at m/z 439.1848).
Compound 20 (24.4 mg), an off-yellow oil, was purified by silica gel CC (EtOAc/PE 5:95) from the reaction residue with 90% yield. − 108.7 (c 0.08, DCM); 1H NMR (400 MHz, CDCl3) δH: 6.26 (s, 1H), 4.05–4.16 (m, 2H), 3.40 (t, J = 6.8 Hz, 2H), 3.00 (q, J = 6.8 Hz, 1H), 2.34 (dd, J = 16.8, 6.8 Hz, 1H ), 2.05–2.09 (m, 1H), 1.99 (dd, J = 16.8, 12.8 Hz, 1H), 175–1.89 (m, 3H), 1.53–1.67 (m, 7H), 1.30–1.44 (m, 9H), 1.20 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 1.10 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC: 218.0, 164.8, 149.4, 137.4, 68.6, 64.6, 62.8, 61.7, 52.0, 50.1, 38.4, 34.2, 33.5, 32.9, 29.4, 29.3, 28.8, 28.7, 28.4, 28.2, 26.1, 23.7, 20.0, 18.0; HRMS m/z 453.2013 [M + H]+ (calcd. for C24H38BrO3 at m/z 453.2004).
3.3.4. General Procedure for Syntheses of 21–26
To a solution of SA (20 mg, 0.08 mmol) in DCM (1 mL, freshly distilled from CaH2), was added oxalyl chloride (17.5 μL, 0.20 mmol). The obtained mixture was then stirred under N2 atmosphere at room temperature for 1 h. Afterwards, the reaction mixture was evaporated in vacuo to remove the DCM and left oxalyl chloride to give the SA acid chloride, which was used directly for the next step directly. To the flask containing SA acid chloride was added 1 mL freshly distilled DCM, followed by diaminoalkane (0.4 mmol). The mixture was then stirred under N2 atmosphere at room temperature for 10h and quenched with brine (5 mL). The mixture was then extracted with DCM (5 mL x 3), washed successively with brine (10 mL × 3) and water (10 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel CC (MeOH/DCM 3:97) to yield 21–26.
Compound 21 (21.6 mg, 89% isolated yield) was obtained as a colorless oil. − 55.9 (c 0.07, DCM); 1H NMR (300 MHz, CDCl3) δH: 6.94 (t, J = 5.4 Hz, 1H), 5.80 (s, 1H), 3.39 (m, 2H), 3.05 (q, J = 7.2 Hz, 1H), 2.83 (t, J = 6.3 Hz, 2H), 2.32 (dd, J = 16.8, 6.9 Hz, 1H), 2.08 (m, 1H), 1.97 (dd, J = 16.8, 12.3 Hz, 1H), 1.61–1.77 (m, 5H), 1.52–1.59 (m, 2H), 1.16 (s, 3H), 1.09 (d, J = 6.6 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.4, 165.3, 141.4, 141.3, 68.7, 62.7, 61.8, 52.0, 50.1, 40.1, 38.6, 38.1, 33.4, 31.5, 28.3, 24.3, 20.0, 17.8; HRMS m/z 305.2228 [M + H]+ (calcd. for C18H29N2O2 at m/z 305.2229).
Compound 22 (24.5 mg, 92% isolated yield) was obtained as an off-yellow oil. − 35.1 (c 0.13, DCM); 1H NMR (300 MHz, CDCl3) δH: 5.79 (t, J = 6.0, 1H), 5.74 (s, 1H), 3.29 (m, 2H), 3.07 (q, J = 6.9 Hz, 1H), 2.68 (t, J = 6.9 Hz, 2H), 2.34 (dd, J = 16.8, 6.6 Hz, 1H), 2.09 (m, 1H), 1.97 (dd, J = 16.8, 12.3 Hz, 1H), 1.49–1.76 (m, 11H), 1.17 (s, 3H), 1.08 (d, J = 6.3 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.3, 165.2, 141.5, 140.9, 68.7, 62.7, 61.9, 52.1, 50.1, 42.1, 39.4, 38.6, 33.3, 33.3, 29.6, 28.2, 24.4, 24.3, 19.9, 17.7; HRMS m/z 333.2532 [M + H]+ (calcd. for C20H33N2O2 at m/z 333.2542).
Compound 23 (25.4 mg, 88% isolated yield) was obtained as an off-yellow oil. − 97.0 (c 0.29, DCM); 1H NMR (300 MHz, CDCl3) δH: 5.78 (t, J = 5.4 Hz, 1H), 5.74 (s, 1H), 3.26 (m, 2H), 3.06 (q, J = 6.9 Hz, 1H), 2.66 (t, J = 6.6 Hz, 2H), 2.33 (dd, J = 16.8, 6.9 Hz, 1H), 2.08 (m, 1H), 1.96 (dd, J = 16.8, 12.6 Hz, 1H), 1.62–1.77 (m, 3H), 1.40–1.59 (m, 6H), 1.30 (br.s, 6H), 1.16 (s, 3H), 1.09 (d, J = 6.6 Hz, 3H), 1.05 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.3, 165.1, 141.5, 140.8, 68.7, 62.7, 61.9, 52.1, 50.1, 42.1, 39.5, 38.6, 33.5, 33.3, 29.7, 29.2, 28.2, 27.0, 26.8, 24.3, 19.9, 17.7; HRMS m/z 361.2852 [M + H]+ (calcd. for C22H37N2O2 at m/z 361.2855).
Compound 24 (27.8 mg, 93% isolated yield) was obtained as a colorless oil. − 110.8 (c 0.53, DCM); 1H NMR (300 MHz, CDCl3) δH: 5.74 (m, 2H), 3.26 (m, 2H), 3.07 (q, J = 7.2 Hz, 1H), 2.68 (t, J = 6.6 Hz, 2H), 2.34 (dd, J = 16.8, 6.9 Hz, 1H), 2.08 (m, 1H), 1.96–2.02 (m, 1H), 1.63–1.77 (m, 3H), 1.41–1.60 (m, 6H), 1.29 (br.s, 8H), 1.17 (s, 3H), 1.09 (d, J = 6.6 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.3, 165.1, 141.6, 140.8, 68.7, 62.7, 61.9, 52.1, 50.1, 42.1, 39.5, 38.6, 33.4, 33.3, 29.7, 29.4, 29.3, 28.2, 27.0, 26.9, 24.4, 19.9, 17.7; HRMS m/z 375.2997 [M + H]+ (calcd. for C23H39N2O2 at m/z 375.3012).
Compound 25 (27.4 mg, 85% isolated yield) was obtained as an off-yellow oil. − 128.5 (c 0.38, DCM); 1H NMR (300 MHz, CDCl3) δH: 5.73 (m, 2H), 3.26 (m, 2H), 3.07 (q, J = 7.2 Hz, 1H), 2.67 (t, J = 6.9 Hz, 2H), 2.34 (dd, J = 16.8, 6.6Hz, 1H), 2.08 (m, 1H), 1.97 (dd, J = 16.8, 12.6 Hz, 1H), 1.38–1.70 (m, 9H), 1.26 (br.s, 12H), 1.17 (s, 3H), 1.09 (d, J = 6.3 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.3, 165.1, 141.6, 140.8, 68.7, 62.7, 61.9, 52.1, 50.1, 42.3, 39.6, 39.6, 38.6, 33.7, 33.3, 29.7, 29.6, 29.6, 29.4, 28.2, 27.1, 27.0, 24.4, 19.9, 17.7; HRMS m/z 403.3310 [M + H]+ (calcd. for C25H43N2O2 at m/z 403.3325).
Compound 26 (32.0 mg, 93% isolated yield) was calculated as an off-yellow oil. − 152.1 (c 0.17, DCM); 1H NMR (300 MHz, CDCl3) δH: 5.69–5.74 (m, 2H), 3.27 (m, 2H), 3.07 (q, J = 7.2 Hz, 1H), 2.67 (t, J = 6.9 Hz, 2H), 2.34 (dd, J = 16.8, 6.9 Hz, 1H), 2.09 (m, 1H), 1.97 (dd, J = 16.8, 12.3 Hz, 1H), 1.41–1.73 (m, 9H), 1.25 (br.s, 16H), 1.17 (s, 3H), 1.09 (d, J = 6.6 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC: 218.3, 165.1, 141.6, 140.8, 68.7, 62.7, 61.9, 52.1, 50.1, 42.3, 39.6, 38.6, 33.7, 33.5, 33.3, 29.8, 29.7, 29.7, 29.6, 29.6, 29.4, 28.2, 27.1, 27.0, 24.4, 19.9, 17.7; HRMS m/z 431.3620 [M + H]+ (calcd. for C27H47N2O2 at m/z 431.3638).

 ,
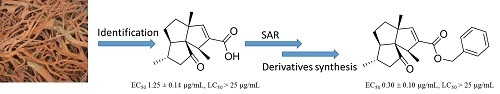
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}