Fucoxanthin Alleviates Oxidative Stress through Akt/Sirt1/FoxO3α Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
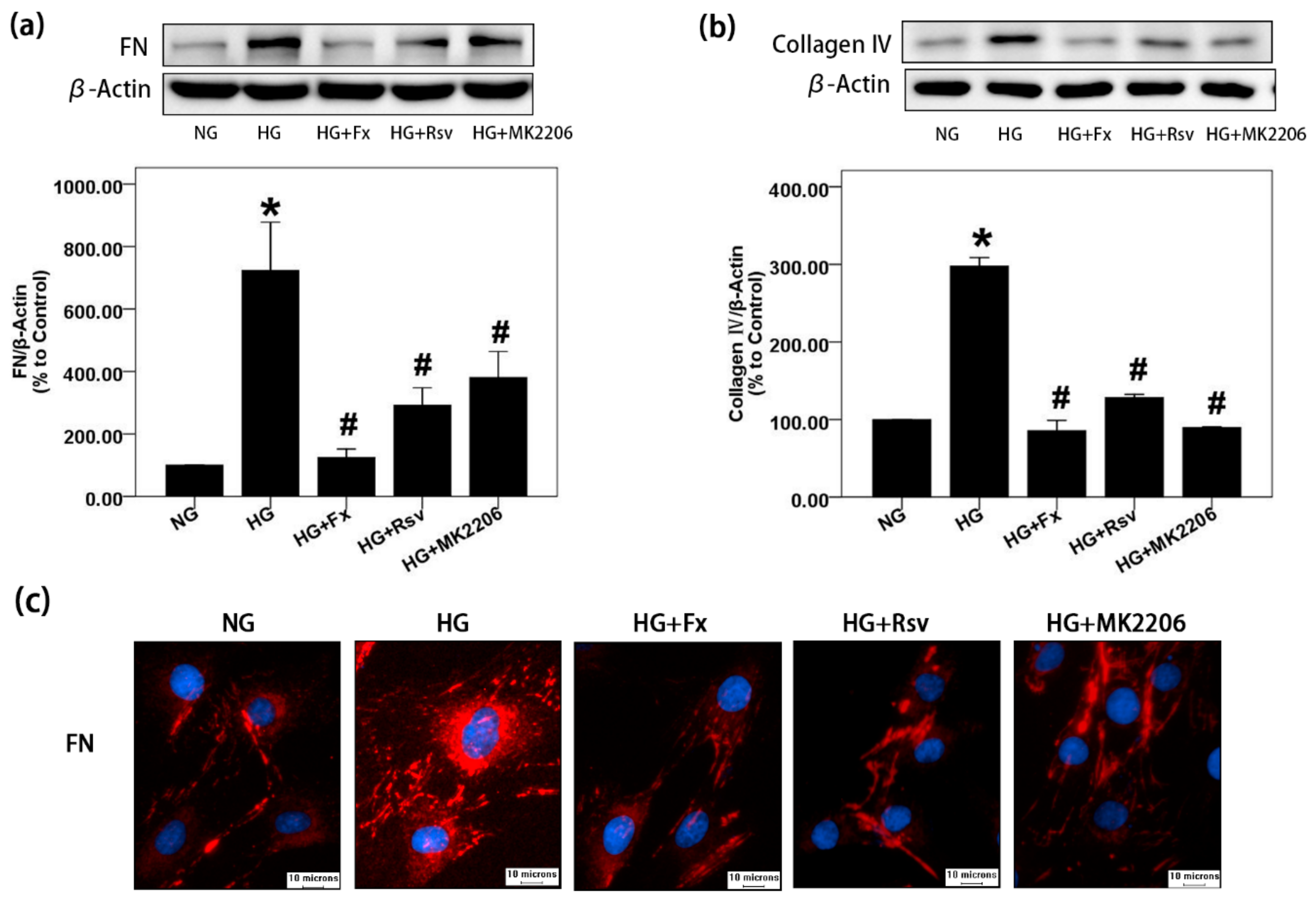
2.1. Fx Could Effectively Reverse the Increase of Extracellular Matrix Induced by HG in Gmcs
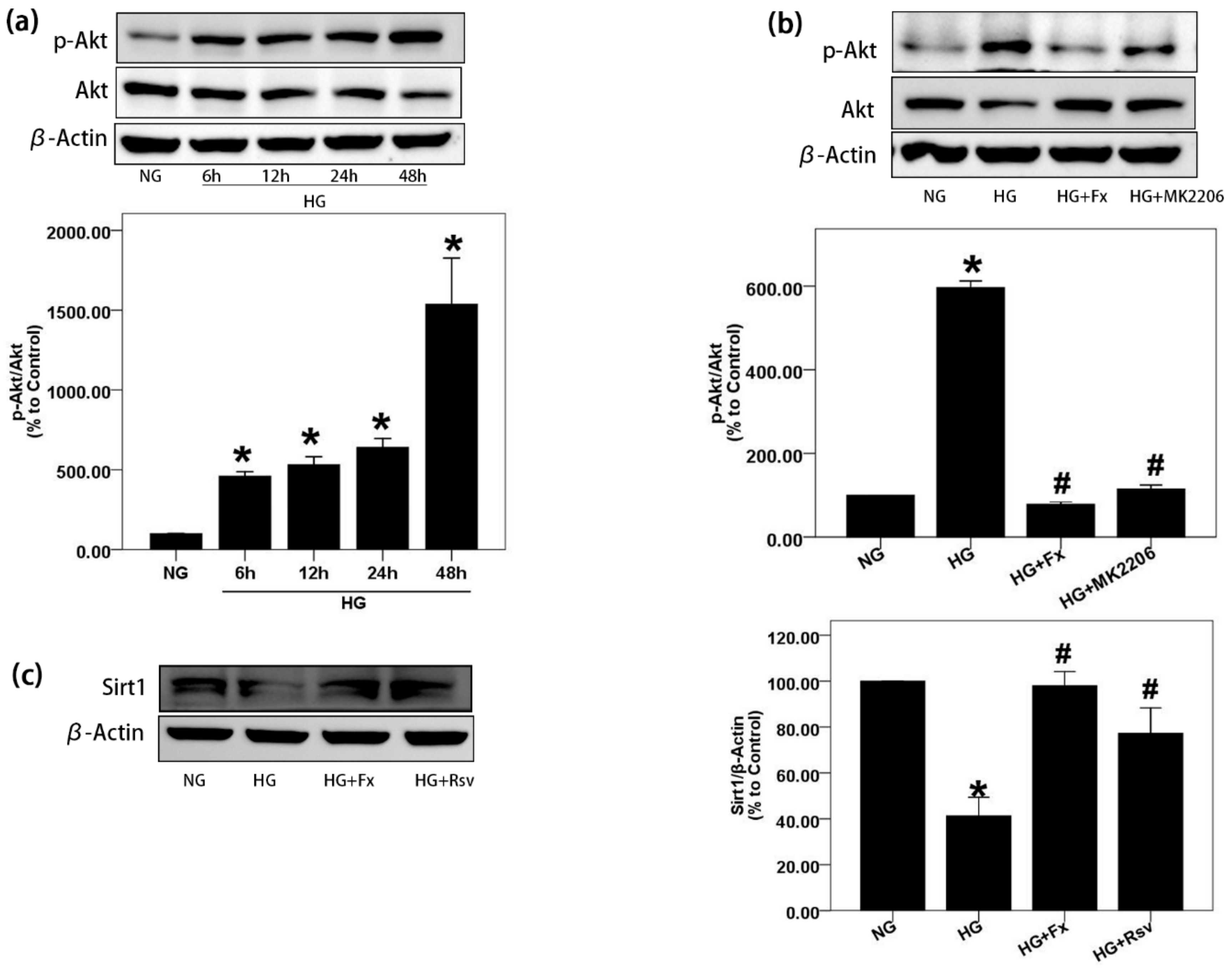
2.2. Fx Could Effectively Reverse the Activation of Akt and the Inhibition of Sirt1 Induced by HG in Gmcs
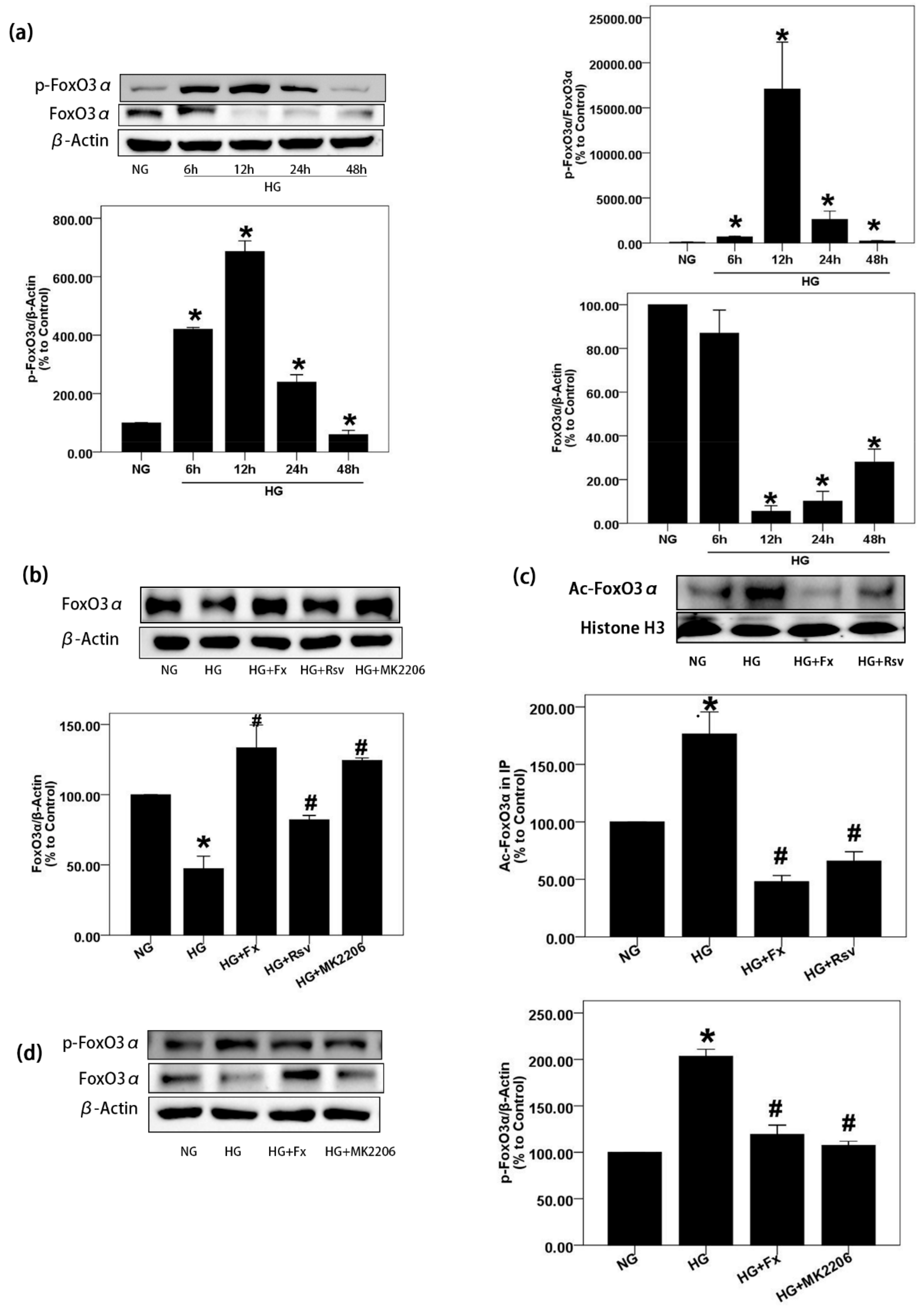
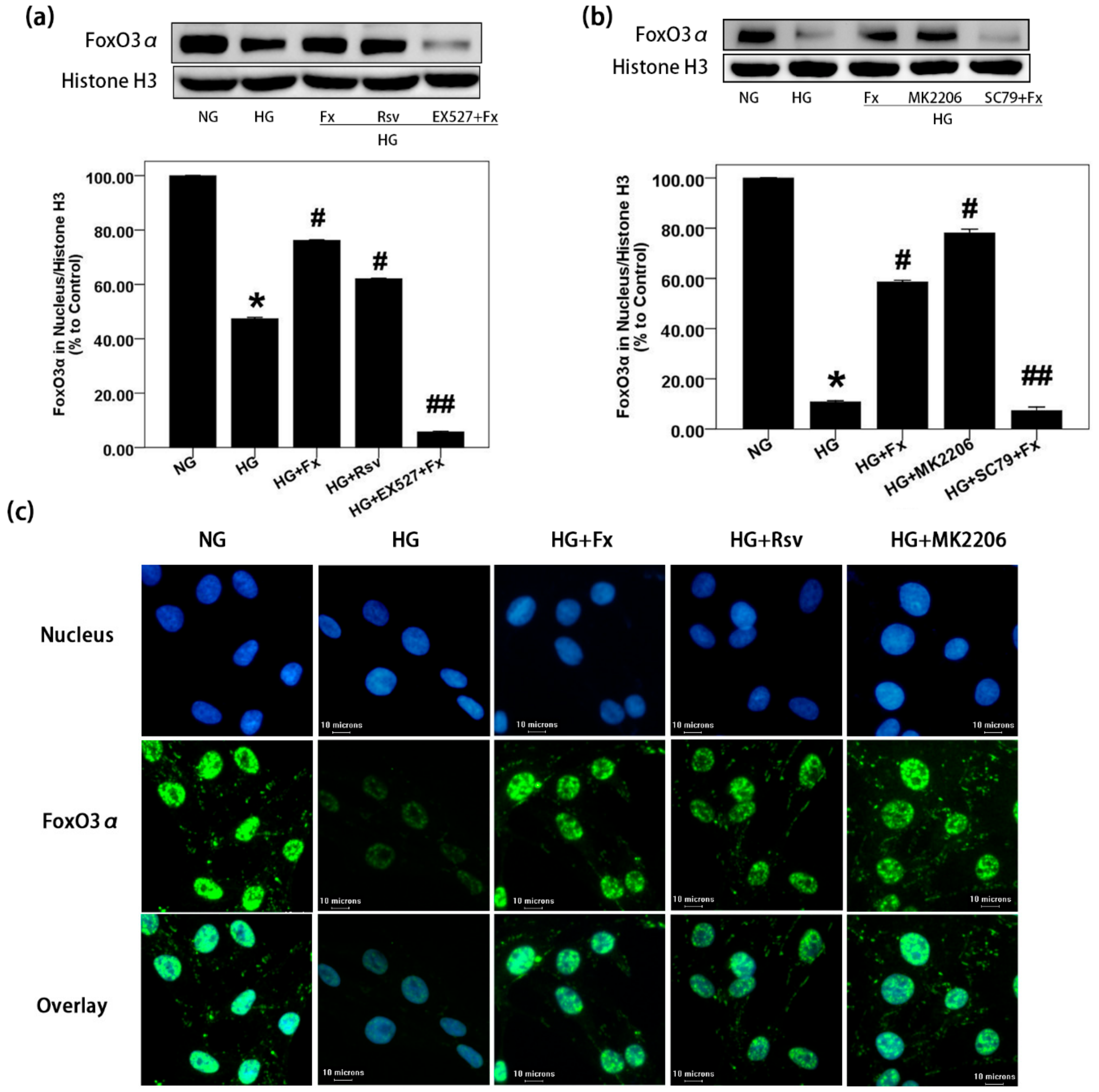
2.3. Fx Reverses the Expression of Foxo3α Inhibited by HG in Gmcs
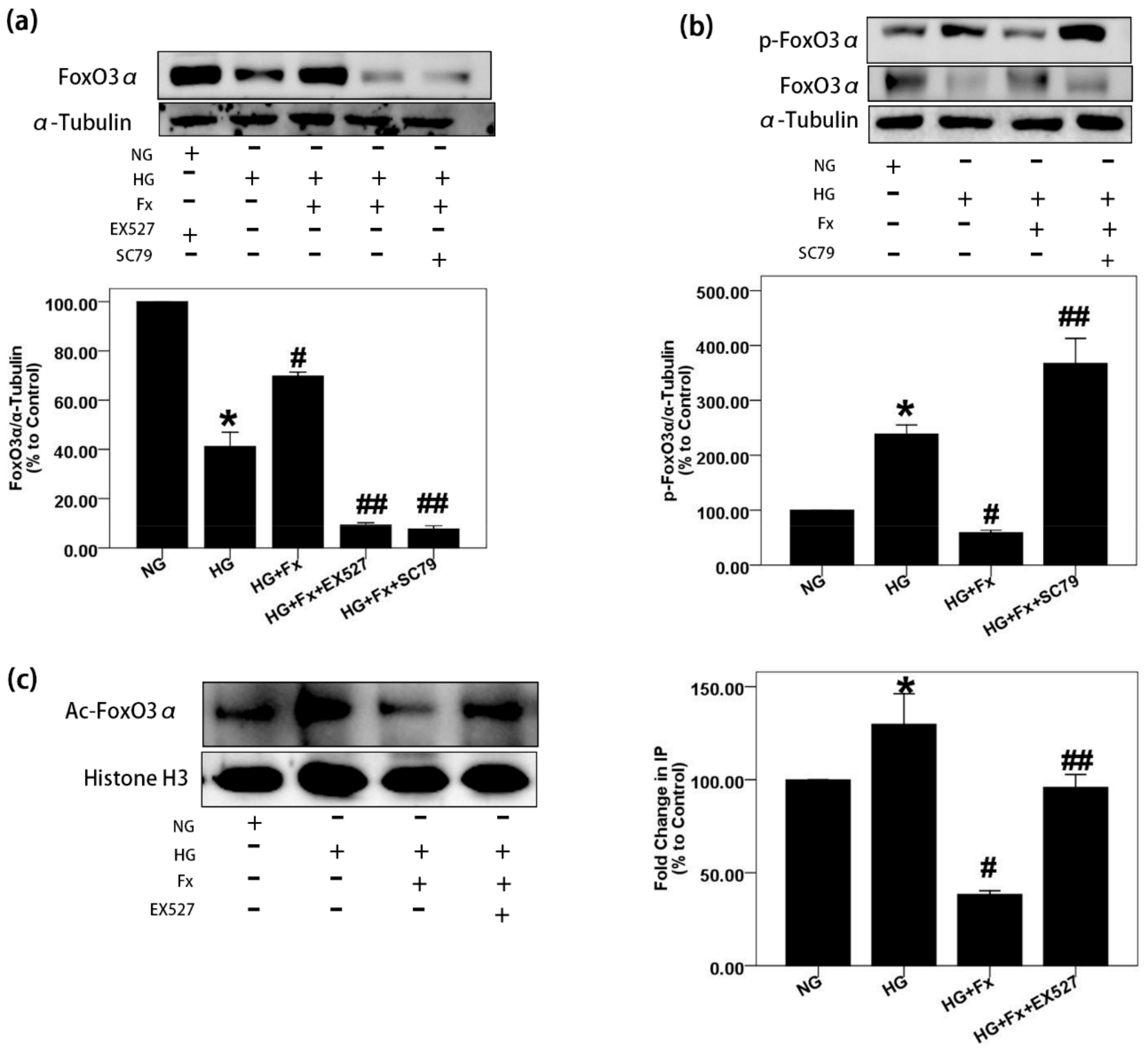
2.4. Fx Regulates the Expression of Foxo3α through the Akt and Sirt1 Signaling in Gmcs
2.5. Fx Enhances the Nuclear Transport of Foxo3α through Akt and Sirt1 Signaling in Gmcs
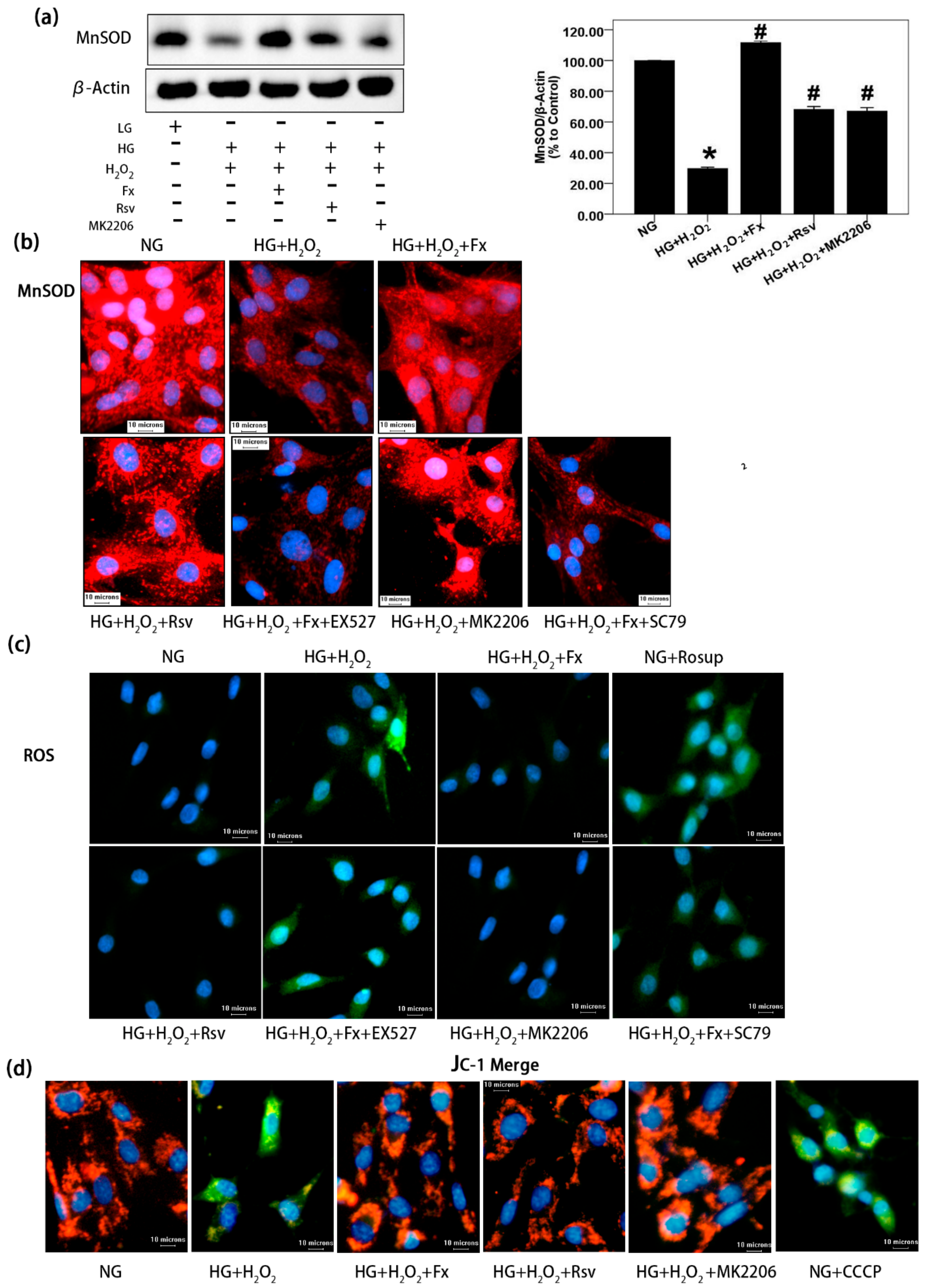
2.6. Fx Effectively Increases the Expression of Mnsod, Reduces the Oxidative Stress Level, and Improves Mitochondrial Membrane Potential in HG Induced Gmcs
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Cell Culture
4.3. Western Blot
4.4. Immunofluorescence
4.5. Determination of ROS
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Fx | fucoxanthin |
| Akt | serine-threonine kinase |
| Sirt1 | silent information regulator T1 |
| FoxO3α | forkhead box O3α |
| HG | high glucose |
| GMCs | glomerular mesangial cells |
| DN | diabetic nephropathy |
| FN | fibronectin |
| ROS | reactive oxygen species |
| Rsv | resveratrol |
| MnSOD | manganese superoxide dismutase |
| NG | normal glucose |
| ECM | extracellular matrix |
| FBS | fetal bovine serum |
| DMEM | dulbecco’s modified eagle medium |
References
- Kato, M.; Natarajan, R. Diabetic nephropathy—Emerging epigenetic mechanisms. Nat. Rev. Nephrol. 2014, 10, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Kim-Muller, J.Y.; Zhao, S.; Srivastava, S.; Mugabo, Y.; Noh, H.L.; Kim, Y.R.; Madiraju, S.R.; Ferrante, A.W.; Skolnik, E.Y.; Prentki, M.; et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014, 20, 593–602. [Google Scholar] [CrossRef]
- Kato, M.; Yuan, H.; Xu, Z.G.; Lanting, L.; Li, S.L.; Wang, M.; Hu, M.C.; Reddy, M.A.; Natarajan, R. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3325–3335. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Yoshioka, K.; Tatematsu, S.; Hara, Y.; Minakuchi, H.; Sueyasu, K.; Washida, N.; Tokuyama, H.; Tzukerman, M.; et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J. Biol. Chem. 2010, 285, 13045–13056. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. SIRT1 in Type 2 Diabetes: Mechanisms and Therapeutic Potential. Diabetes Metab. J. 2013, 37, 315–325. [Google Scholar] [CrossRef]
- Li, A.; Peng, R.; Sun, Y.; Liu, H.; Peng, H.; Zhang, Z. LincRNA 1700020I14Rik alleviates cell proliferation and fibrosis in diabetic nephropathy via miR-34a-5p/Sirt1/HIF-1alpha signaling. Cell Death Dis. 2018, 9, 461. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Cao, Q.; Wang, F.; Huang, L.Y.; Sang, T.T.; Liu, F.; Chen, S.Y. SIRT1 Protects Against Oxidative Stress-Induced Endothelial Progenitor Cells Apoptosis by Inhibiting FOXO3a via FOXO3a Ubiquitination and Degradation. J. Cell Physiol. 2015, 230, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Wulf, G.; Burga, L.; Cantley, L.C. Cell-to-cell variability in PI3K protein level regulates PI3K-AKT pathway activity in cell populations. Curr. Biol. 2011, 21, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Matsubara, T.; Mima, A.; Sumi, E.; Kanamori, H.; Iehara, N.; Fukatsu, A.; Yanagita, M.; Nakano, T.; Ishimoto, Y.; et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005, 68, 552–561. [Google Scholar] [CrossRef]
- Li, D.; Lu, Z.; Xu, Z.; Ji, J.; Zheng, Z.; Lin, S.; Yan, T. Spironolactone promotes autophagy via inhibiting PI3K/AKT/mTOR signalling pathway and reduce adhesive capacity damage in podocytes under mechanical stress. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, S.; Previn, R.; Chen, D.; Jin, Y.; Zhou, G. Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis? Int. J. Mol. Sci. 2018, 19, 3794. [Google Scholar] [CrossRef]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Akasaki, Y.; Alvarez-Garcia, O.; Saito, M.; Carames, B.; Iwamoto, Y.; Lotz, M.K. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014, 66, 3349–3358. [Google Scholar] [CrossRef]
- Xia, S.; Wang, K.; Wan, L.; Li, A.; Hu, Q.; Zhang, C. Production, characterization, and antioxidant activity of fucoxanthin from the marine diatom Odontella aurita. Mar. Drugs 2013, 11, 2667–2681. [Google Scholar] [CrossRef]
- Asai, A.; Sugawara, T.; Ono, H.; Nagao, A. Biotransformation of fucoxanthinol into amarouciaxanthin A in mice and HepG2 cells: Formation and cytotoxicity of fucoxanthin metabolites. Drug Metab. Dispos. 2004, 32, 205–211. [Google Scholar] [CrossRef]
- Mikami, K.; Hosokawa, M. Biosynthetic pathway and health benefits of fucoxanthin, an algae-specific xanthophyll in brown seaweeds. Int. J. Mol. Sci. 2013, 14, 13763–13781. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Murakami-Funayama, K.; Miyashita, K. Anti-obesity and anti-diabetic effects of fucoxanthin on diet-induced obesity conditions in a murine model. Mol. Med. Rep. 2009, 2, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, Y.; Yuan, Y.; Xu, B.; Gao, Q.; Chen, P.; Zhang, T.; Guan, T. PC-1 NF suppresses high glucose-stimulated inflammation and extracellular matrix accumulation in glomerular mesangial cells via the Wnt/beta-catenin signaling. Exp. Ther. Med. 2019, 18, 2029–2036. [Google Scholar] [CrossRef]
- Ruankham, W.; Suwanjang, W.; Wongchitrat, P.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Sesamin and sesamol attenuate H2O2 -induced oxidative stress on human neuronal cells via the SIRT1-SIRT3-FOXO3a signaling pathway. Nutr. Neurosci. 2019. [Google Scholar] [CrossRef]
- Shemesh, I.I.; Rozen-Zvi, B.; Kalechman, Y.; Gafter, U.; Sredni, B. AS101 prevents diabetic nephropathy progression and mesangial cell dysfunction: Regulation of the AKT downstream pathway. PLoS ONE 2014, 9, e114287. [Google Scholar] [CrossRef]
- Yerra, V.G.; Kalvala, A.K.; Kumar, A. Isoliquiritigenin reduces oxidative damage and alleviates mitochondrial impairment by SIRT1 activation in experimental diabetic neuropathy. J. Nutr. Biochem. 2017, 47, 41–52. [Google Scholar] [CrossRef]
- Yang, K.S.; Lim, J.H.; Kim, T.W.; Kim, M.Y.; Kim, Y.; Chung, S.; Shin, S.J.; Choi, B.S.; Kim, H.W.; Kim, Y.S.; et al. Vascular endothelial growth factor-receptor 1 inhibition aggravates diabetic nephropathy through eNOS signaling pathway in db/db mice. PLoS ONE 2014, 9, e94540. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-beta1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2017, 102, 77–86. [Google Scholar] [CrossRef]
- Xie, X.; Peng, J.; Huang, K.; Huang, J.; Shen, X.; Liu, P.; Huang, H. Polydatin ameliorates experimental diabetes-induced fibronectin through inhibiting the activation of NF-kappaB signaling pathway in rat glomerular mesangial cells. Mol. Cell Endocrinol. 2012, 362, 183–193. [Google Scholar] [CrossRef]
- Chen, S.J.; Lee, C.J.; Lin, T.B.; Liu, H.J.; Huang, S.Y.; Chen, J.Z.; Tseng, K.W. Inhibition of Ultraviolet B-Induced Expression of the Proinflammatory Cytokines TNF-α and VEGF in the Cornea by Fucoxanthin Treatment in a Rat Model. Mar. Drugs 2016, 14, 13. [Google Scholar]
- Rodriguez-Luna, A.; Avila-Roman, J.; Oliveira, H.; Motilva, V.; Talero, E. Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes. Mar. Drugs 2019, 17, 451. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.-J.; Jeon, Y.-J. Protective effect of fucoxanthin isolated from Sargassum siliquastrum on UV-B induced cell damage. J. Photochem. Photobiol. B Biol. 2009, 95, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Beppu, F.; Niwano, Y.; Tsukui, T.; Hosokawa, M.; Miyashita, K. Single and repeated oral dose toxicity study of fucoxanthin (FX), a marine carotenoid, in mice. J. Toxicol. Sci. 2009, 34, 501–510. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Xie, X.; Peng, J.; Chang, X.; Huang, K.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Activation of RhoA/ROCK regulates NF-kappaB signaling pathway in experimental diabetic nephropathy. Mol. Cell Endocrinol. 2013, 369, 86–97. [Google Scholar] [CrossRef]
- Xie, X.; Chen, Q.; Tao, J. Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Mar. Drugs 2018, 16, 117. [Google Scholar] [CrossRef]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 719–731. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Tsai, K.L.; Sun, Y.J.; Huang, C.Y.; Yang, J.Y.; Hung, M.C.; Hsiao, C.D. Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007, 35, 6984–6994. [Google Scholar] [CrossRef]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Woo, S.R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Chen, C.; Hao, J.; Huang, J.; Wang, S.; Liu, P.; Huang, H. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-beta1 in rat glomerular messangial cells. Mol. Cell Endocrinol. 2015, 399, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.-F.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Mene, P.; Stoppacciaro, A. Isolation and propagation of glomerular mesangial cells. Methods Mol. Biol. 2009, 466, 3–17. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Jin, L.; Zheng, D.; Tang, X.; Yang, J.; Fan, L.; Xie, X. Fucoxanthin Alleviates Oxidative Stress through Akt/Sirt1/FoxO3α Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Mar. Drugs 2019, 17, 702. https://doi.org/10.3390/md17120702
Yang G, Jin L, Zheng D, Tang X, Yang J, Fan L, Xie X. Fucoxanthin Alleviates Oxidative Stress through Akt/Sirt1/FoxO3α Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Marine Drugs. 2019; 17(12):702. https://doi.org/10.3390/md17120702
Chicago/Turabian StyleYang, Guanyu, Lin Jin, Dongxiao Zheng, Xiaoliang Tang, Junwei Yang, Lingxuan Fan, and Xi Xie. 2019. "Fucoxanthin Alleviates Oxidative Stress through Akt/Sirt1/FoxO3α Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs" Marine Drugs 17, no. 12: 702. https://doi.org/10.3390/md17120702
APA StyleYang, G., Jin, L., Zheng, D., Tang, X., Yang, J., Fan, L., & Xie, X. (2019). Fucoxanthin Alleviates Oxidative Stress through Akt/Sirt1/FoxO3α Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Marine Drugs, 17(12), 702. https://doi.org/10.3390/md17120702