3.1. Chemistry
IR spectra were obtained with a Shimadzu IRAffinity-1 FT-IR spectrometer (Shimadzu Corporation, Kyoto, Japan). Optical rotations were measured with Horiba SEPA-500 polarimeters (Horiba Ltd., Kyoto, Japan). 1H- and 13C-NMR spectra were recorded on a JEOL JNM-AL 400 NMR spectrometer (JEOL Ltd., Tokyo, Japan) at 400 MHz for 1H and 100 MHz for 13C; and a JEOL JNM-AL 300 NMR spectrometer at 300 MHz for 1H and 75 MHz for 13C (ppm, J in Hz with tetramethylsilane (TMS) as internal standard). All proton and carbon signals were assigned by extensive NMR measurements using correlation spectroscopy (COSY), Heteronuclear Multiple-Bond Correlation (HMBC), and Heteronuclear Multiple Quantum Correlation (HMQC) techniques. Mass spectra were recorded on a JEOL JMS 700 instrument (JEOL Ltd., Tokyo, Japan) with a direct inlet system operating at 70 eV.
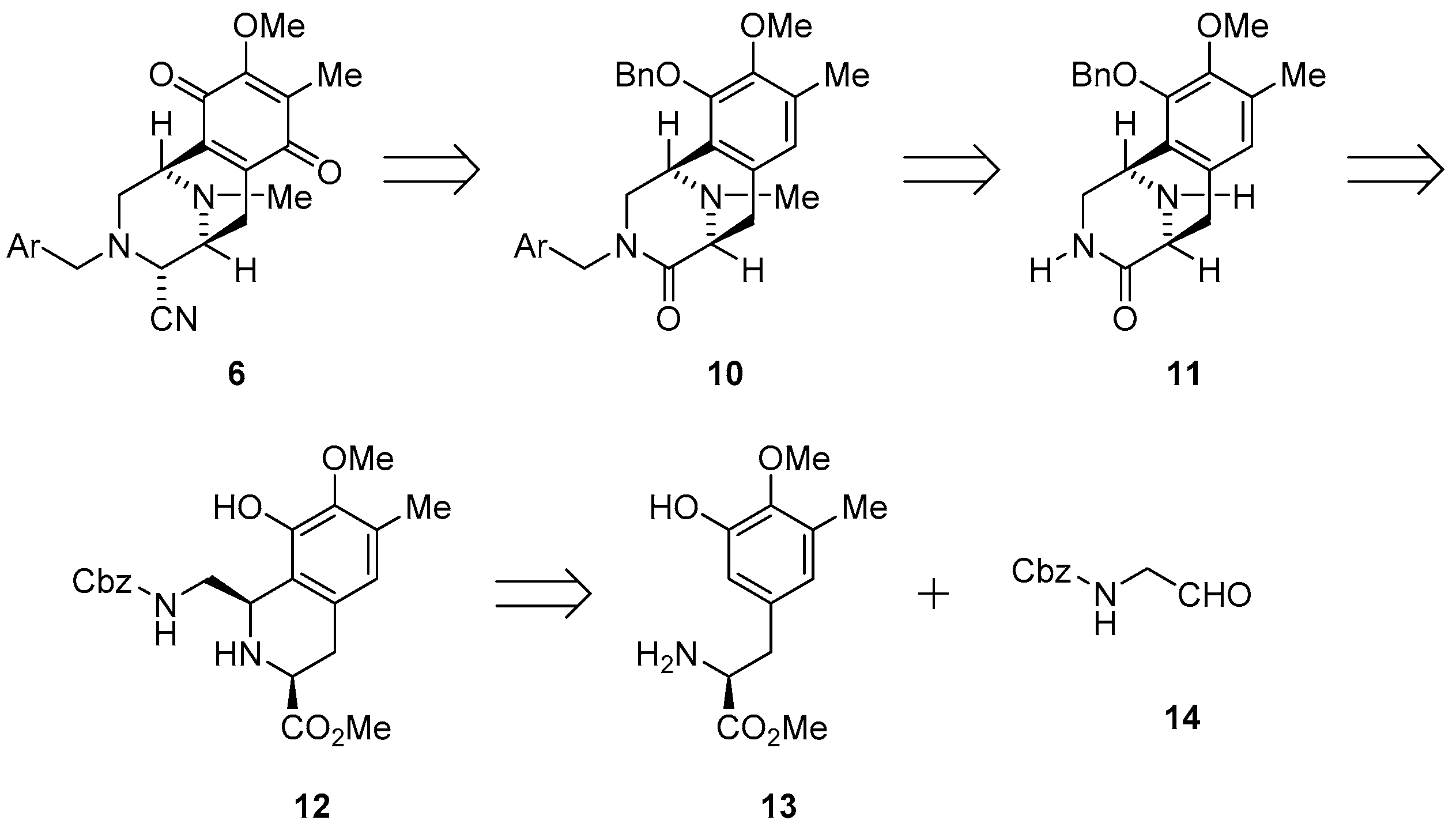
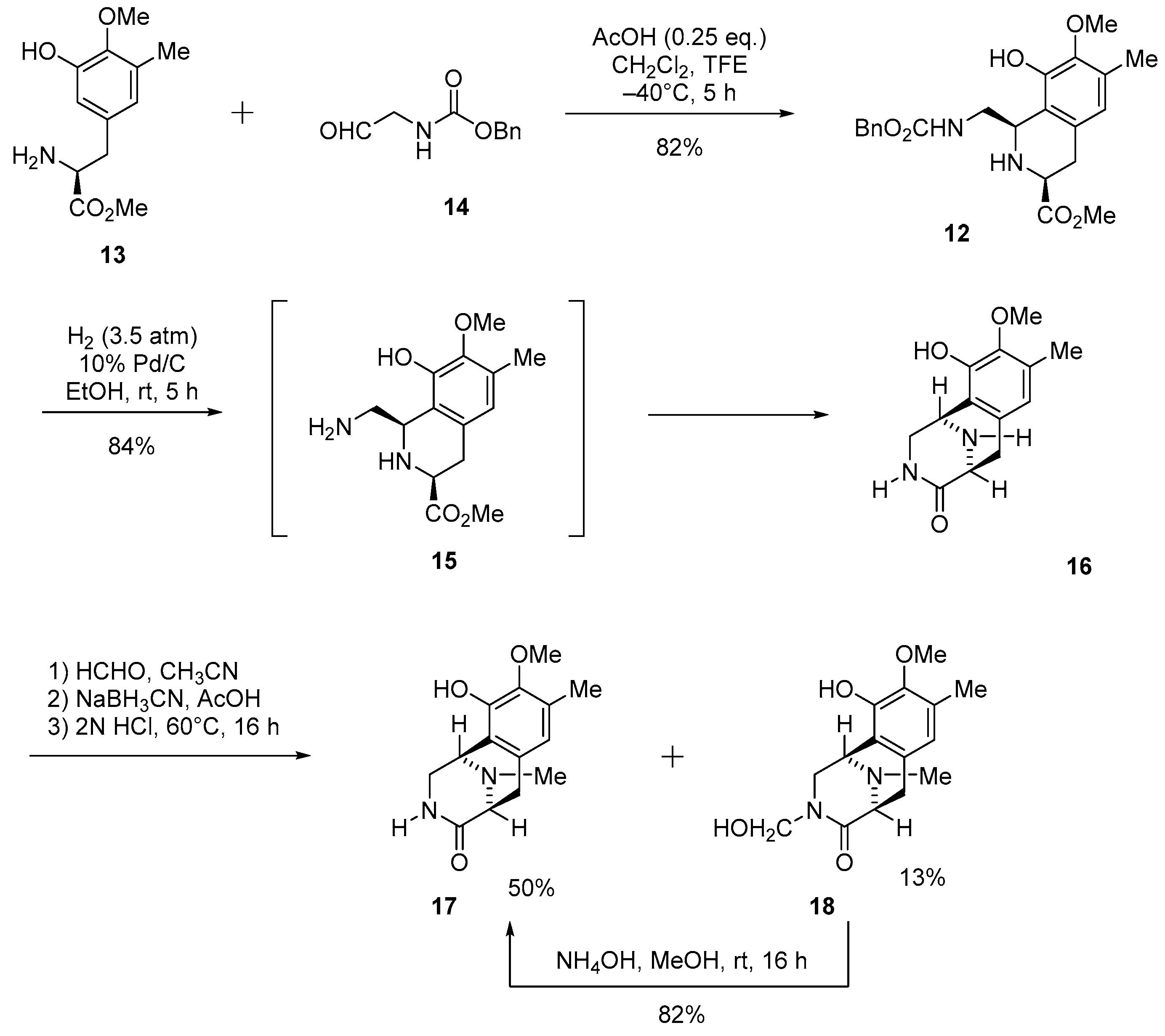
3.1.1. Synthesis of 1,2,3,4-Tetrahydroisoquinoline-3-carboxylate (12)
To a stirred solution of aldehyde 14 (2.73 g, 14.1 mmol, 1.3 eq.) and 4 Å molecular sieves (2.60 g) in CH2Cl2 (70 mL), a solution of amine 13 (2.60 g, 10.9 mmol), acetic acid (160 µL) and 2,2,2-trifluoroethanol (10 mL) was added slowly over 6 min at −40 °C. After being stirred at −40 °C for 5 h, the reaction mixture was neutralized with NaHCO3, and then filtered through a Celite pad. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (CHCl3–EtOAc = 2:1) to afford compound 12 (3.71 g, 82%) as a pale yellow amorphous. [α] −83.9 (c 1.1, CHCl3); 1H NMR (400 MHz, C5D5N, 80 °C) δ 10.30 (1H, brs, NH or OH), 7.46–7.25 (5H, m, Bn-H), 6.54 (1H, s, 5-H), 5.35–5.25 (2H, m, 5’-H), 4.90 (1H, br t, J = 3.2 Hz, 1-H), 4.31–4.27 (1H, m, Bn-H), 4.00–3.94 (1H, m, Bn-H), 3.79–3.75 (1H, m 3-H), 3.73 (3H, s, 7-OCH3), 3.70 (3H, s, 3-COOCH3), 3.02 (2H, brd, J = 6.4 Hz, 4-H), 2.30 (3H, s, 6-CH3); 13C-NMR (400 MHz, C5D5N, 80 °C) δ174.0 (s, COOCH3), 157.6 (s, C-3’), 148.2 (s, C-8), 145.9 (s, C-7), 138.3 (s, Bn), 132.2 (s, C-4a), 129.5 (s, C-6), 128.8 (d, Bn), 128.2 (d, Bn), 128.0 (d, Bn), 122.7 (s, C-8a), 121.9 (d, C-5), 66.4 (t, C-5’), 60.2 (q, 7-OCH3), 56.0 (d, C-3), 54.2 (d, C-1), 51.8 (q, 3-COOCH3), 46.8 (t, C-1’), 33.8 (t, C-4), 15.8 (q, 6-CH3); IR (CHCl3) 3520, 3437, 3024, 3015, 2955, 2359, 2342, 1717, 1506, 1456, 1233, 1059 cm−1; FABMS m/z 415 [M + H]+; HRFABMS m/z 415.1867 ([M + H]+, calcd for C22H27N2O6 415.1869).
3.1.2. Synthesis of (1R,5S)-10-Hydroxy-9-methoxy-8-methyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (16)
A solution of 12 (2.96 g, 7.14 mmol) in EtOH (370 mL) was hydrogenated over 10% Pd/C (55%wet, 1.52 g, 1.43 mmol) at 25 °C for 5 h under 3.5 atm hydrogen. The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure and the residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 9:1) to afford compound 16 (1.49 g, 84%) as a pale brown solid. [α] −177.0 (c 1.0, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 8.80 (1H, brs, 10-OH), 7.39 (1H, d, J = 4.0 Hz, 3-N-H), 6.37 (1H, s, 7-H), 4.19 (1H, d, J = 4.4 Hz, 1-H), 3.60 (3H, s, 9-OCH3), 3.53 (1H, dd, J = 11.2, 4.4 Hz, 2-H), 3.49 (1H, d, J = 6.2 Hz, 5-H), 3.07 (1H, dd, J = 11.2, 4.0 Hz, 2-H), 2.86 (1H, dd, J = 16.5, 6.2 Hz, 6-H), 2.59 (1H, d, J = 16.5 Hz, 6-H), 2.13 (3H, s, 8-CH3); 13C-NMR (100 MHz, DMSO-d6) δ 171.4 (s, C-4), 145.7 (s, C-10), 143.7 (s, C-9), 129.7 (s, C-6a), 128.7 (s, C-8), 122.9 (s, C-10a), 120.6 (d, C-7), 59.9 (q, 9-OCH3), 52.4 (d, C-5), 47.5 (t, C-2), 43.7 (d, C-1), 32.0 (t, C-6), 15.5 (q, 8-CH3); IR (KBr) 3497, 3428, 3345, 3246, 1643, 1335, 1273, 1069, 1001 cm−1; EIMS m/z (%) 248 (M+, 24), 191 (17), 190 (100), 175 (16); HREIMS m/z 248.1162 (M+, calcd for C13H16N2O3 248.1161).
3.1.3. Synthesis of (1R,5S)-10-Hydroxy-9-methoxy-8,11-dimethyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (17) and (1R,5S)-10-hydroxy-3-(hydroxymethyl)-9-methoxy-8,11-dimethyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (18)
To a stirred solution of amine 16 (248 mg, 1.00 mmol) in CH3CN (34 mL) was added 37% HCHO (1.60 mL, 20.0 mmol, 20 eq.). The reaction mixture was stirred for 15 min, after which NaCNBH3 (700 mg, 10.0 mmol, 10 eq.) was added. The reaction mixture was stirred for 15 min, after which AcOH (570 µL, 10.0 mmol, 10 eq.) was added dropwise over 3 min. The reaction mixture was stirred for 5 min, after which 2 N HCl (34 mL) was added 1 portion. The reaction was heated to 60 °C and was stirred for 16 h. The reaction was quenched with saturated NaHCO3 (200 mL) and extracted with CHCl3−MeOH = 9:1 (3 × 150 mL). The combined extracts were washed with H2O (100 mL), brine (100 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (benzene−acetone = 1:2) to afford compound 18 (39.0 mg, 13%) as a colorless solid, and with CHCl3−MeOH (6:1) to afford 17 (130 mg, 50%) as a colorless solid.
17: [α] −224.0 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 6.49 (1H, s, 7-H), 5.92 (1H, brs, 3-N-H), 4.17 (1H, d, J = 4.5 Hz, 1-H), 3.91 (1H, dd, J = 11.6, 4.5 Hz, 2-H), 3.77 (3H, s, 9-OCH3), 3.57 (1H, d, J = 6.6 Hz, 5-H), 3.30 (1H, ddd, J = 11.6, 3.8, 0.9, 2-H), 3.17 (1H, dd, J = 17.0, 6.6 Hz, 6-H), 2.79 (1H, d, J = 17.0 Hz, 6-H), 2.52 (3H, s, N-CH3), 2.25 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 172.3 (s, C-4), 145.5 (s, C-10), 143.3 (s, C-9), 129.3 (s, C-6a), 129.0 (s, C-8), 121.9 (d, C-7), 119.2 (s, C-10a), 60.8 (q, 9-OCH3), 59.2 (d, C-5), 50.0 (d, C-1), 45.3 (t, C-2), 40.1 (q, N-CH3), 27.8 (t, C-6), 15.8 (q, 8-CH3); IR (KBr) 3265, 2938, 2874, 1684, 1645, 1495, 1335, 1265, 1055, 1038 cm−1; EIMS m/z (%) 262 (M+, 20), 205 (17), 204 (100), 189 (16); HREIMS m/z 262.1317 (M+, calcd for C14H18N2O3 262.1317).
18: [α] −197.7 (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 6.47 (1H, s, 7-H), 4.88 (1H, d, J = 10.4 Hz, 3-N-CH2OH), 4.56 (1H, d, J = 10.4 Hz, 3-N-CH2OH), 4.22 (1H, d, J = 4.6 Hz, 1-H), 4.08 (1H, dd, J = 11.5, 4.6 Hz, 2-H), 3.76 (3H, s, 9-OCH3), 3.61 (1H, d, J = 6.5 Hz, 5-H), 3.35 (1H, d, J = 11.5, 2-H), 3.15 (1H, dd, J = 17.0, 6.5 Hz, 6-H), 2.77 (1H, d, J = 17.0 Hz, 6-H), 2.49 (3H, s, 11-N-CH3), 2.25 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 172.1 (s, C-4), 145.6 (s, C-10), 143.4 (s, C-9), 129.4 (s, C-6a), 128.6 (s, C-8), 121.7 (d, C-7), 118.9 (s, C-10a), 71.6 (t, C-3), 60.7 (q, 9-OCH3), 59.1 (d, C-5), 50.7 (d, C-1), 50.0 (t, C-2), 39.8 (q, N-CH3), 27.1 (t, C-6), 15.7 (q, 8-CH3); IR (KBr) 3489, 3150, 2949, 2934, 1618, 1504, 1236, 1055, 1034 cm−1; EIMS m/z (%) 292 (M+, 3), 262 (17), 205 (18), 204 (100), 189 (16); HREIMS m/z 292.1424 (M+, calcd for C15H20N2O4 292.1423).
3.1.4. Synthesis of 17 from 18
To a stirred solution of lactam 18 (262 mg, 0.896 mmol) in MeOH (26 mL) was added NH4OH (10.5 mL) at room temperature (rt). The reaction mixture was stirred for 16 h. The reaction was quenched with conc. HCl at 0 °C, and then neutralized with 5% NaHCO3. The reaction mixture was diluted with H2O (200 mL) and extracted with CHCl3−MeOH = 9:1 (4×50 mL). The combined extracts were washed with brine (100 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 9:1) to afford compound 17 (19.3 mg, 82%) as a colorless solid.
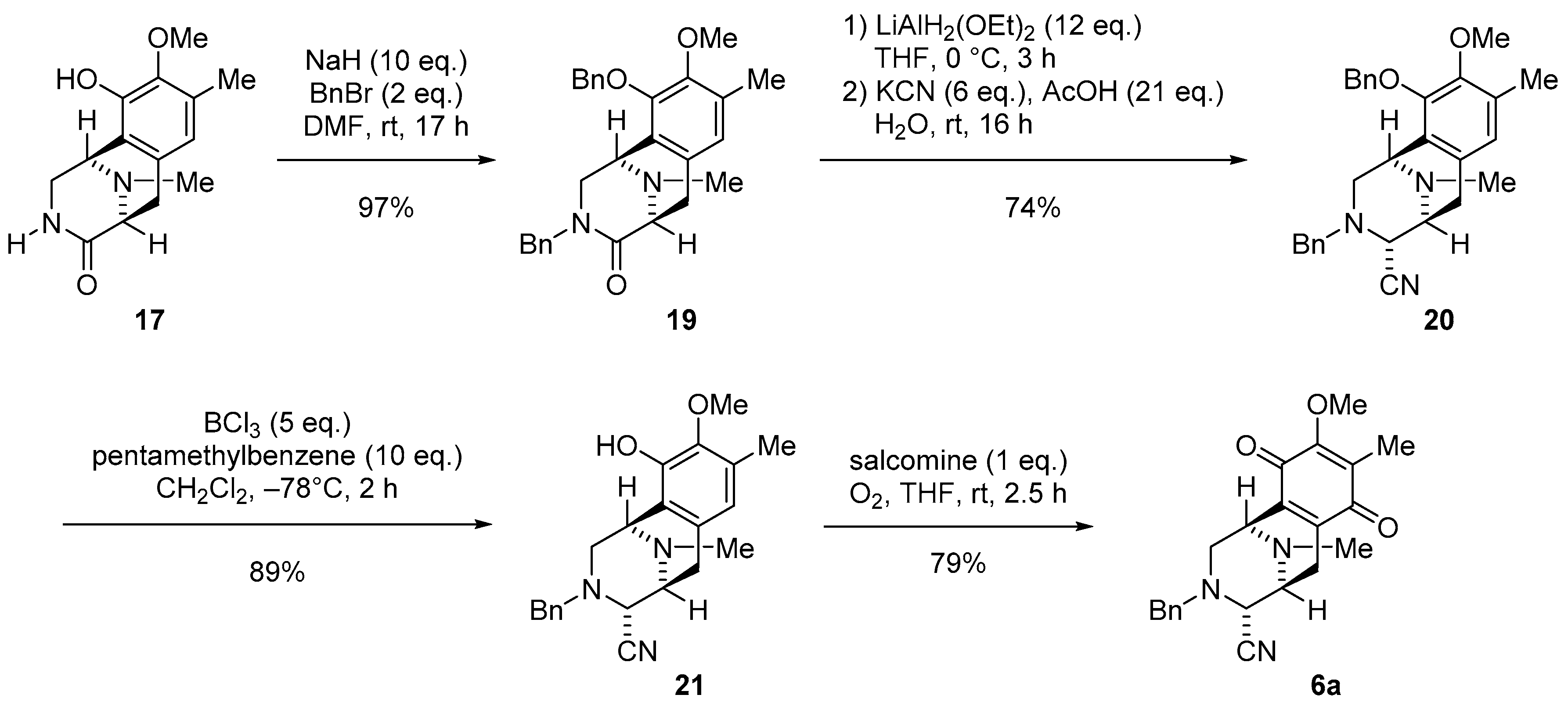
3.1.5. Synthesis of (1R,5S)-3-Benzyl-10-(benzyloxy)-9-methoxy-8,11-dimethyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (19)
To a stirred solution of lactam 17 (10.0 mg, 38.0 µmol) and benzyl bromide (10.0 µL, 76.0 µmol, 2.0 eq.) in DMF (1 mL) was added NaH (60% oil dispersion, 15.2 mg, 381 µmol, 10.0 eq.) at 0 °C. The reaction mixture was stirred at 25 °C for 17 h. The reaction mixture was diluted with H2O (5 mL) and extracted with Et2O (3 × 10 mL). The combined extracts were dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 99:1) to afford compound 19 (16.4 mg, 97%) as a yellow oil. [α] −77.5 (c 1.2, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.34–7.22 (5H, m, 10-O-Bn-H), 7.13–7.04 (3H, m, 3-N-Bn-H), 6.75–6.73 (2H, m, 3-N-Bn-H), 6.75 (1H, s, 7-H), 4.98 (1H, d, J = 11.5 Hz, 10-OCH2Ph), 4.84 (1H, d, J = 15.1 Hz, 3-N-CH2Ph), 4.74 (1H, d, J = 11.5 Hz, 10-OCH2Ph), 4.10 (1H, d, J = 15.1 Hz, 3-N-CH2Ph), 3.89 (1H, brd, J = 4.4 Hz, 1-H), 3.68–3.65 (1H, m, 2-H), 3.68 (1H, d, J = 6.3 Hz, 5-H), 3.67 (3H, s, 9-OCH3), 3.16 (1H, dd, J = 17.1, 6.3 Hz, 6-H), 2.92 (1H, d, J = 11.7 Hz, 2-H), 2.86 (1H, d, J = 17.1 Hz, 6-H), 2.29 (3H, s, 11-N-CH3), 2.29 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 170.2 (s, C-4), 149.3 (s, C-9), 148.3 (s, C-10), 137.3 (s, Bn), 136.3 (s, Bn), 131.2 (d, C-8), 128.4 (d×2, Bn), 128.0 (d×2, Bn), 128.2 (s, C-6a), 127.1 (d, Bn), 126.8 (d, Bn), 126.0 (d, C-7), 125.6 (s, C-10a), 74.1 (t, 10-OCH2Ph), 59.9 (q, 9-OCH3), 59.3 (d, C-5), 51.5 (d, C-1), 50.5 (t, C-2), 48.5 (t, 3-N-CH2Ar), 39.7 (q, 11-N-CH3), 27.4 (t, C-6), 15.6 (q, 8-CH3); IR (CHCl3) 3009, 2940, 1636, 1493, 1454, 1337, 1059, 698 cm−1; EIMS m/z (%) 442 (M+, 30), 351 (10), 295 (23), 294 (100), 204 (30), 203 (37), 91 (11); HREIMS m/z 442.2254 (M+, calcd for C28H30N2O3 442.2256).
3.1.6. Synthesis of (1R,4R,5S)-3-Benzyl-10-(benzyloxy)-9-methoxy-8,11-dimethyl-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (20)
To a solution of lactam 19 (45.7 mg, 103 µmol) in THF (2.5 mL) at 0 °C was slowly added LiAlH2(OEt)2 (1.0 mol/L in CH2Cl2, 1.20 mL, 1.20 mmol, 12 eq.) over 10 min. The reaction mixture was stirred at 0 °C for 3 h. The reaction mixture was quenched with AcOH (120 µL, 2.15 mmol, 20.8 eq.), followed by the addition of KCN (40.4 mg, 620 µmol, 6.0 eq.) in H2O (1.0 mL), and stirring was continued for 16 h at 25 °C. The reaction mixture was neutralized with 5% NaHCO3 solution and diluted with saturated Rochell’s salt aq., and the mixture was stirred for 1.5 h. The reaction mixture was extracted with CHCl3 (3 × 30 mL). The combined extracts were washed with brine (30 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 4:1) to afford compound 20 (33.2 mg, 74%) as a colorless amorphous. [α] −48.4 (c 1.5, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.36–7.25 (5H, m, 10-O-Bn-H), 7.17–7.14 (3H, m, 3-N-Bn-H), 6.90–6.88 (2H, m, 3-N-Bn-H), 6.69 (1H, s, 7-H), 5.04 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 4.85 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 3.91 (1H, brs, 1-H), 3.83 (3H, s, 9-OCH3), 3.65 (1H, s, 4-H), 3.52 (2H, s, 3-N-CH2Ph), 3.21 (1H, d, J = 7.6 Hz, 5-H), 3.02 (1H, dd, J = 17.6, 7.6 Hz, 6-H), 2.81 (1H, dd, J = 11.2, 3.0 Hz, 2-H), 2.51 (1H, brd, J = 11.2, Hz, 2-H), 2.35 (1H, d, J = 17.6 Hz, 6-H), 2.32 (3H, s, 8-CH3), 2.15 (3H, s, 11-N-CH3); 13C-NMR (100 MHz, CDCl3) δ 148.8 (s, C-9), 148.3 (s, C-10), 137.4 (s, Bn), 137.0 (s, Bn), 130.2 (s, C-6a), 130.0 (s, C-8), 128.4 (d×2, Bn), 128.3 (d, Bn), 128.1 (d×2, Bn), 127.2 (d, Bn), 126.5 (s, C-10a), 124.2 (d, C-7), 116.5 (s, 4-CN), 74.4 (t, 10-OCH2Ph), 60.0 (q, 9-OCH3), 59.1 (d, C-4), 58.9 (d, 3-N-CH2Ph), 55.4 (d, C-5), 53.4 (t, C-2), 52.8 (d, C-1), 41.2 (q, 11N-CH3), 25.0 (t, C-6), 15.8 (q, 8-CH3); IR (CHCl3) 3015, 2936, 2826, 2359, 2342, 2226, 1321, 1227, 1061, 1028, 700 cm−1; EI-MS m/z (%) 453 (M+, 2), 295 (27), 294 (100), 204 (21), 203 (20); HREIMS m/z 453.2416 (M+, calcd for C29H31N3O2 453.2416).
3.1.7. Synthesis of (1R,4R,5S)-3-Benzyl-10-hydroxy-9-methoxy-8,11-dimethyl-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (21)
To a solution of 20 (20.0 mg, 44.1 μmol) and pentamethylbenzene (65.4 mg, 441 μmol, 10.0 eq.) in CH2Cl2 (6.0 mL) was added BCl3 (1.0 mol/L in CH2Cl2, 220 µL, 220 µmol, 5 eq.) at −78 °C and the mixture was stirred for 2 h. The reaction mixture was diluted with CH2Cl2 (5.0 mL) and quenched with saturated NaHCO3 solution at 0 °C. The mixture was extracted with CH2Cl2 (3 × 25 mL). The combined extracts were dried over Na2SO4 and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 21 (14.3 mg, 89%) as a colorless amorphous. [α] −127.4 (c 1.5, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.19–7.15 (3H, m, 3-N-Bn-H), 6.96–6.92 (2H, m, 3-N-Bn-H) 6.48 (1H, s, 7-H), 5.67 (1H, brs, 10-OH), 4.09 (1H, brs, 1-H), 3.78 (3H, s, 9-OCH3), 3.65 (1H, s, 4-H), 3.62 (1H, d, J = 7.8 Hz, 3-N-CH2Ph), 3.54 (1H, d, J = 7.8 Hz, 3-N-CH2Ph), 3.27 (1H, brd, J = 7.5 Hz, 5-H), 3.06 (1H, dd, J = 17.6, 7.5 Hz, 6-H), 2.96 (1H, dd, J = 11.2, 2.9 Hz, 2-H), 2.72 (1H, d, J = 11.2 Hz, 2-H), 2.38 (3H, s, 11-N-CH3), 2.31 (3H, s, 8-CH3), 2.38-2.17 (1H, m, overlapped, 6-H); 13C-NMR (100 MHz, CDCl3) δ 145.4 (s, C-10), 142.8 (s, C-9), 137.1 (s, Bn), 130.8 (s, C-6a), 128.4 (d, Bn), 128.3 (d, Bn), 128.3 (s, C-8), 127.3 (s, Bn), 120.4 (d, C-7), 119.4 (s, C-10a), 116.6 (4-CN), 60.8 (q, 9-OCH3), 59.0 (t, 3-N-CH2Ph), 58.6 (d, C-4), 55.4 (d, C-5), 53.0 (t, C-2), 52.5 (d, C-1), 41.5 (q, 11-N-CH3), 25.1 (t, C-6), 15.8 (q, 8-CH3); IR (CHCl3) 3534, 3019, 2928, 2359, 1454, 1418, 1227, 1059, 1026 cm−1; EIMS m/z (%) 363 (M+, 2), 205 (23), 204 (100), 189 (10); HREIMS m/z 363.1943 (M+, calcd for C22H25N3O2 363.1947).
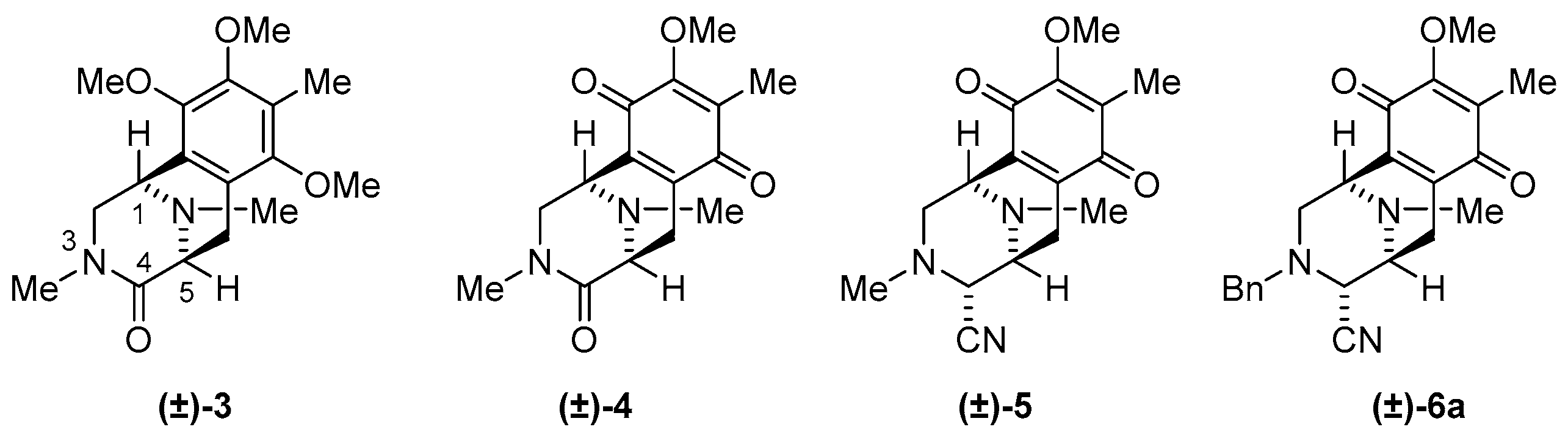
3.1.8. Synthesis of (1R,4R,5S)-3-Benzyl-9-methoxy-8,11-dimethyl-7,10-dioxo-1,2,3,4,5,6,7,10-octahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (6a)
To a solution of phenol 21 (10.0 mg, 27.5 μmol) in THF (1 mL) was added salcomine (8.90 mg, 27.5 μmol, 1.0 eq.) at 25 °C, and the reaction mixture was stirred for 2.5 h under O2 atmosphere. The reaction mixture was filtered through a cellulose pad and washed with EtOAc. The filtrate was concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CH2Cl2−MeOH = 99:1) to afford compound 6a (8.20 mg, 79%) as a dark red amorphous. 99%ee. The ee value was determined by HPLC analysis using CHIRALPAK IC [hexane/EtOH = 80/20, flow 1.0 mL/min, tr (minor) = 7.08 min, tr (major) = 7.77 min]; [α] −38.0 (c 0.3, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.25–7.13 (5H, m, 3-N-Bn-H), 4.01 (3H, s, 9-OCH3), 3.87 (1H, brs, 1-H), 3.66 (1H, d, J = 13.2 Hz, 3-N-CH2Ph), 3.54 (1H, d, J = 13.2 Hz, 3-N-CH2Ph), 3.54 (1H, d, J = 2.0 Hz, 4-H), 3.27 (1H, brd, J = 7.4 Hz, 5-H), 2.95 (1H, dd, J = 11.6, 3.2 Hz, 2-H), 2.70 (1H, dd, J = 20.5, 7.4 Hz, 6-H), 2.58 (1H, d, J = 11.6 Hz, 2-H), 2.32 (3H, s, 11-N-CH3), 2.10 (1H, d, J = 20.5 Hz, 6-H), 2.01 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 186.9 (s, C-7), 182.3 (s, C-10), 155.4 (s, C-9), 141.0 (s, C-6a), 137.4 (C-10a), 136.2 (s, Bn), 128.7 (d, Bn), 128.6 (d, Bn), 128.6 (s, C-8), 127.9 (d, Bn), 115.8 (s, 4-CN), 61.0 (q, 9-OCH3), 58.9 (t, C-12), 57.7 (d, C-4), 54.5 (d, C-5), 51.7 (t, C-2), 51.3 (d, C-1), 41.5 (q, 11N-CH3), 20.8 (t, C-6), 8.7 (q, 8-CH3); IR (CHCl3) 3024, 2928, 2855, 2384, 2228, 1653, 1308, 1234, 1155, 1024 cm−1; EIMS m/z (%) 377 (M+, 12), 220 (18), 219 (100), 218 (99), 204 (29), 176 (13), 91 (21); HREIMS m/z 377.1737 (M+, calcd for C22H23N3O3 377.1739).
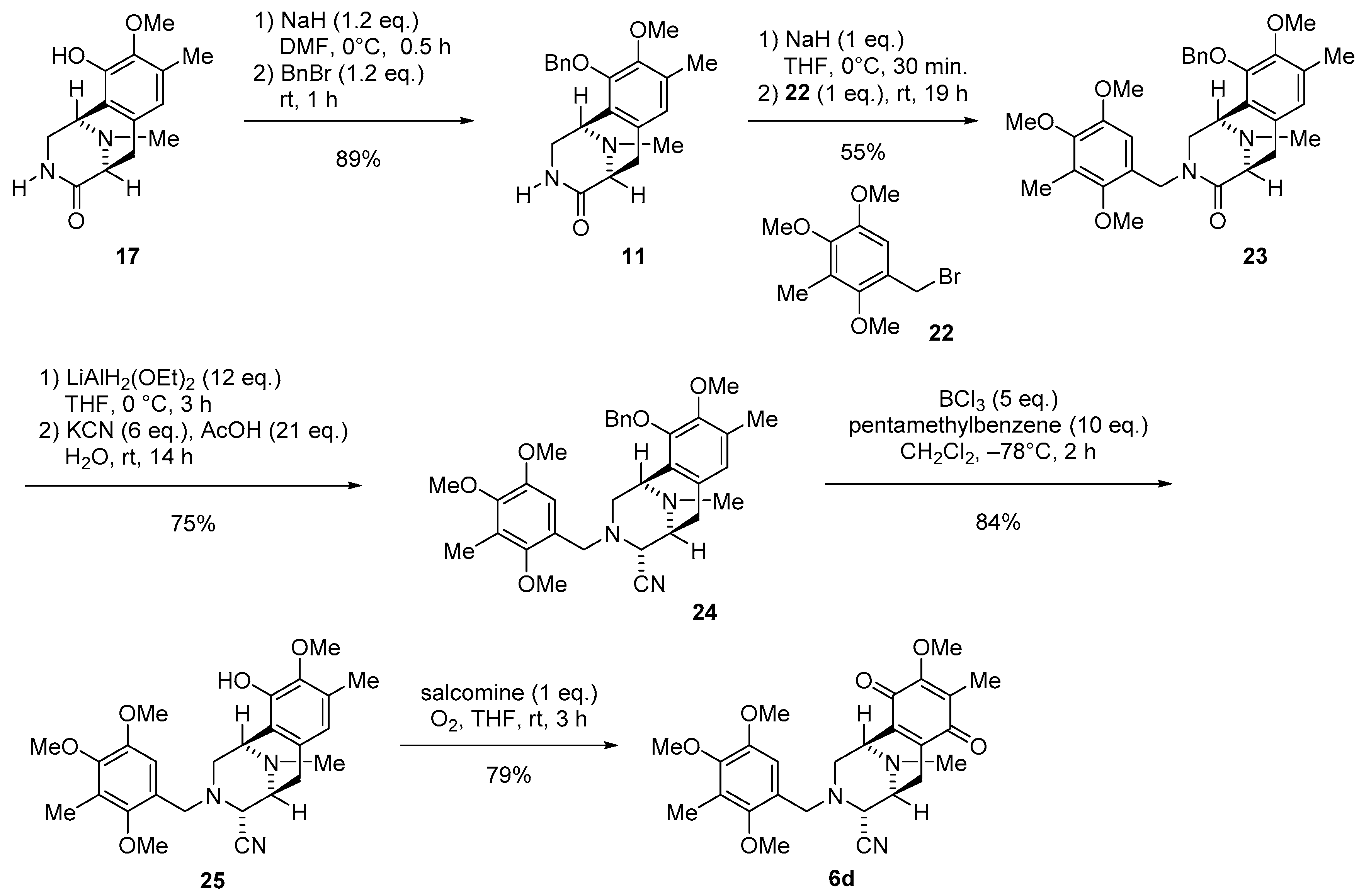
3.1.9. Synthesis of (1R,5S)-10-(Benzyloxy)-9-methoxy-8,11-dimethyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (11)
To a solution of lactam 17 (3.17 g, 12.0 mmol) in DMF (250 mL) was slowly added NaH (60% oil dispersion, 580 mg, 15.0 mmol, 1.2 eq.) over 10 min at 0 °C. The reaction mixture was stirred for 30 min at 0 °C, after which BnBr (1.70 mL, 15.0 mmol, 1.2 eq.) was added dropwise over 25 min. The reaction mixture was stirred for 1 h at 25 °C. The reaction mixture was diluted with H2O (300 mL) and extracted with CHCl3 (3 × 200 mL). The combined extracts were washed with brine (200 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 99:1) to afford compound 11 (3.81 g, 89%) as a colorless amorphous. [α] −108.2 (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.41–7.31 (5H, m, 10-O-Bn-H), 6.70 (1H, s, 7-H), 6.18 (1H, brs, 3-N-H), 5.18 (1H, d, J = 11.6 Hz, 10-OCH2Ph), 5.08 (1H, d, J = 11.6 Hz, 10-OCH2Ph), 3.92 (1H, d, J = 4.7 Hz, 1-H), 3.82 (3H, s, 9-OCH3), 3.81 (1H, dd, J = 10.3, 4.7 Hz 2-H), 3.52 (1H, d, J = 6.6 Hz, 5-H), 3.17 (1H, ddd, J = 10.3, 3.9, 1.0 Hz, 2-H), 3.13 (1H, dd, J = 17.3, 6.6 Hz, 6-H), 2.76 (1H, d, J = 17.3 Hz, 6-H), 2.31 (3H, s, 11-N-CH3), 2.25 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 172.2 (s, C-4), 149.4 (s, C-9), 148.3 (s, C-10), 137.6 (s, C-1’), 131.5 (s, C-8), 128.6 (d, C-3’, C-5’), 128.4 (d, C-6a), 128.1 (d, C-4’), 128.0 (d, C-2’, C-6’), 126.2 (s, C-10a), 125.8 (d, C-7), 74.2 (t, 10-OCH2Ph), 60.1 (q, 9-OCH3), 59.0 (d, C-5), 50.5 (d, C-1), 46.3 (t, C-2), 39.8 (q, 11-N-CH3), 27.4 (t, C-6), 15.8 (q, 8-CH3); IR (KBr) 3169, 3028, 2936, 1678, 1337, 1310, 1055, 702 cm−1; EIMS m/z (%) 352 (M+, 38), 295 (23), 294 (100), 261 (14), 204 (46), 203 (61), 174 (10), 91 (11); HREIMS m/z 352.1785 (M+, calcd for C21H24N2O3 352.1787).
3.1.10. Synthesis of (1R,5S)-10-(Benzyloxy)-9-methoxy-8,11-dimethyl-3-(2,4,5-trimethoxy-3-methylbenzyl)-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (23)
To a solution of NaH (60% oil dispersion, 80.3 mg, 2.00 mmol) in THF (10 mL) was added a solution of lactam 11 (705 mg, 2.00 mmol) in THF (10 mL) at 0 °C. The reaction mixture was stirred for 30 min at 0 °C, after which a solution of bromide 22 (550 mg, 2.00 mmol) in THF (10 mL) was added at 25 °C. The reaction mixture was stirred for 19 h at 25 °C. The reaction mixture was diluted with H2O (100 mL) and extracted with CHCl3 (3 × 100 mL). The combined extracts were washed with brine (150 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 49:1) to afford compound 23 (602 mg, 55%) as a yellow gummy solid and starting material 11 (139 mg, 20% recovery). [α] −55.6 (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.35–7.29 (3H, m, 10-O-Bn-H), 7.25–7.22 (2H, m, 10-O-Bn-H), 6.72 (1H, s, 7-H), 5.91 (1H, s, 6’-H), 5.06 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 5.00 (1H, d, J = 15.1 Hz, 3-N-CH2Ar), 4.66 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 4.14 (1H, d, J = 15.1 Hz, 3-N-CH2Ar), 3.93 (1H, brd, J = 4.8 Hz, 1-H), 3.71 (3H, s, 9-OCH3), 3.69-3.65 (2H, m, 2-H, 5-H), 3.67 (3H, s, 4’-OCH3), 3.57 (3H, s, 2’-OCH3), 3.35 (3H, s, 5’-OCH3), 3.18 (1H, dd, J = 17.2, 6.4 Hz, 6-H), 2.96 (1H, dd, J = 11.9 Hz, 2-H), 2.88 (1H, d, J = 17.2 Hz, 6-H), 2.32 (3H, s, 11-N-CH3), 2.25 (3H, s, 8-CH3), 2.13 (3H, s, 3’-CH3); 13C-NMR (100 MHz, CDCl3) δ 170.6 (s, C-4), 150.6 (s, C-2’), 149.4 (s, C-9), 149.4 (s, C-5’), 148.6 (s, C-10), 146.8 (s, C-4’), 137.3 (s, Bn), 131.4 (s, C-8), 128.5 (s, C-6a), 128.5 (d, Bn), 128.4 (d, Bn), 128.1 (d, Bn), 126.4 (s, C-10a), 125.4 (d, C-7), 125.1 (s, C-3’), 124.2 (s, C-1’), 107.8 (d, C-6’), 74.1 (t, 10-OCH2Ph), 60.9 (q, 2’-OCH3), 60.1 (q, 4’-OCH3), 59.9 (q, 9-OCH3), 59.3 (d, C-5), 55.1 (q, 5’-OCH3), 51.4 (d, C-1), 50.5 (t, C-2), 42.6 (t, 3-N-CH2Ar), 39.7 (q, 11-N-CH3), 27.5 (t, C-6), 15.7 (q, 8-CH3), 9.4 (q, 3’-CH3); IR (CHCl3) 3024, 2943, 2467, 1641, 1452, 1339, 1244, 1061 cm−1; EIMS m/z (%) 547 (11), 546 (M+, 32), 351 (11), 295 (25), 294 (100), 204 (27), 203 (21), 195 (18); HREIMS m/z 546.2731 (M+, calcd for C32H38N2O6 546.2730).
3.1.11. Synthesis of (1R,4R,5S)-10-(Benzyloxy)-9-methoxy-8,11-dimethyl-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (24)
To a solution of lactam 23 (50.0 mg, 92.0 µmol) in THF (3.0 mL) at 0 °C was slowly added LiAlH2(OEt)2 (1.0 mol/L in CH2Cl2, 1.10 mL, 1.10 mmol, 12 eq.) over 10 min. The reaction mixture was stirred at 0 °C for 3 h. The reaction mixture was quenched with AcOH (100 µL, 1.90 mmol, 20.8 eq.), followed by the addition of KCN (35.8 mg, 549 µmol, 6.0 eq.) in H2O (2.0 mL), and stirring was continued for 14 h at 25 °C. The reaction mixture was neutralized with 5% NaHCO3 solution and diluted with saturated Rochell’s salt aq., and the mixture was stirred for 1 h. The reaction mixture was extracted with CHCl3 (3 × 30 mL). The combined extracts were washed with brine (40 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 24 (38.2 mg, 75%) as a colorless gummy solid. [α] −23.1 (c 1.3, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.34–7.27 (5H, m, 10-O-Bn-H), 6.59 (1H, s, 7-H), 6.33 (1H, s, 6’-H), 5.07 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 4.83 (1H, d, J = 11.3 Hz, 10-OCH2Ph), 3.93 (1H, brs, 1-H), 3.81 (3H, s, 9-OCH3), 3.76 (1H, brs, 4-H), 3.71 (3H, s, 4’-OCH3), 3.58 (1H, d, J = 13.5 Hz, 3-N-CH2Ar), 3.54 (3H, s, 5’-OCH3), 3.46 (1H, d, J = 13.5 Hz, 3-N-CH2Ar), 3.34 (3H, s, 2’-OCH3), 3.26 (1H, brd, J = 7.7 Hz, 5-H), 3.03 (1H, dd, J = 17.9, 7.7 Hz, 6-H), 2.84 (1H, dd, J = 10.4, 3.0 Hz, 2-H), 2.56 (1H, d, J = 10.4 Hz, 2-H), 2.35 (1H, d, J = 17.9 Hz, 6-H), 2.23 (3H, s, 8-CH3), 2.15 (3H, s, 11-N-CH3), 2.13 (3H, s, 3’-CH3); 13C-NMR (100 MHz, CDCl3) δ 150.9 (s, C-2’), 148.8 (s, C-9), 148.7 (s, C-5’), 148.2 (s, C-10), 146.8 (s, C-4’), 137.2 (s, Bn), 129.9 (s, C-6a), 129.8 (s, C-8), 128.3 (d, Bn), 128.2 (d, Bn), 127.9 (d, Bn), 126.7 (s, C-10a), 125.4 (s, C-3’), 124.4 (s, C-1’), 124.0 (d, C-7), 116.5 (s, 4-CN), 109.7 (d, C-6’), 74.2 (t, 10-OCH2Ph), 60.7 (q, 2’-OCH3), 59.9 (q, 4’-OCH3), 59.8 (q, 9-OCH3), 59.0 (d, C-4), 55.2 (d, C-5), 55.1 (q, 5’-OCH3), 53.7 (t, C-2), 53.1 (t, 3-N-CH2Ar), 52.6 (d, C-1), 41.0 (q, 11-N-CH3), 24.9 (t, C-6), 15.5 (q, 8-CH3), 9.2 (q, 3’-CH3); IR (CHCl3) 3015, 2938, 2226, 1485, 1321, 1227, 1088, 1011 cm−1; EIMS m/z (%) 557 (M+, 1), 295 (25), 294 (100), 204 (13), 203 (16); HREIMS m/z 557.2893 (M+, calcd for C33H39N3O5 557.2890).
3.1.12. Synthesis of (1R,4R,5S)-10-Hydroxy-9-methoxy-8,11-dimethyl-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (25)
To a solution of 24 (115 mg, 206 µmol) and pentamethylbenzene (306 mg, 2.06 mmol, 10 eq.) in CH2Cl2 (30 mL) was added BCl3 (1.0 mol/L in CH2Cl2, 1.00 mL, 1.00 mmol, 5 eq.) over 10 min at −78 °C and the mixture was stirred for 2 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and quenched with saturated NaHCO3 solution at 0 °C. The mixture was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were dried over Na2SO4 and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 25 (80.7 mg, 84%) as a colorless amorphous. [α] −35.9 (c 0.9, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 6.38 (1H, s, 6’-H), 6.37 (1H, s, 7-H), 5.75 (1H, brs, 10-OH), 4.10 (1H, brs, 1-H), 3.77 (1H, brs, 4-H), 3.76 (3H, s, 9-OCH3), 3.74 (3H, s, 4’-OCH3), 3.61 (1H, d, J = 13.5 Hz, 3-N-CH2Ar), 3.57 (3H, s, 5’-OCH3), 3.55 (1H, d, J = 13.5 Hz, 3-N-CH2Ar), 3.38 (3H, s, 2’-OCH3), 3.32 (1H, brd, J = 7.8 Hz, 5-H), 3.08 (1H, dd, J = 18.1, 7.8 Hz, 6-H), 2.99 (1H, dd, J = 11.0, 3.0 Hz, 2-H), 2.80 (1H, d, J = 11.0 Hz, 2-H), 2.38 (3H, s, 11N -CH3), 2.35 (1H, d, J = 18.1 Hz, 6-H), 2.22 (3H, s, 8-CH3), 2.13 (3H, s, 3’-CH3); 13C-NMR (100 MHz, CDCl3) δ 151.0 (s, C-2’), 148.9 (s, C-5’), 146.8 (s, C-4’), 145.5 (s, C-10), 142.8 (s, C-9), 130.5 (s, C-6a), 127.9 (s, C-8), 125.5 (s, C-3’), 124.5 (s, C-1’), 120.2 (d, C-7), 119.6 (s, C-10a), 116.7 (s, 4-CN), 109.7 (d, C-6’), 60.9 (q, 9-OCH3), 60.6 (q, 4’-OCH3), 60.1 (q, 2’-OCH3), 58.7 (d, C-4), 55.3 (q, 5’-OCH3), 55.3 (d, C-5), 53.4 (t, C-2), 53.3 (t, 3-N-CH2Ar), 52.4 (d, C-1), 41.4 (q, 11-N-CH3), 25.0 (t, C-6), 15.5 (q, 8-CH3), 9.3 (q, 3’-CH3); IR (CHCl3) 3534, 3015, 2940, 2226, 1487, 1331, 1227, 1088, 1011 cm−1; EIMS m/z (%) 467 (M+, 1), 441 (13), 440 (48), 247 (16), 246 (12), 245 (57), 205 (19), 204 (100), 195 (20); HREIMS m/z 467.2421 (M+, calcd for C26H33N3O5 467.2420).
3.1.13. Synthesis of (1R,4R,5S)-9-Methoxy-8,11-dimethyl-7,10-dioxo-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,2,3,4,5,6,7,10-octahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (6d)
To a solution of phenol 25 (16.6 mg, 35.5 µmol) in THF (1 mL) was added salcomine (11.5 mg, 35.5 µmol, 1.0 eq.) at 25 °C, and the reaction mixture was stirred for 3 h under O2 atmosphere. The reaction mixture was filtered through a cellulose pad and washed with EtOAc. The filtrate was concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 1:1) to afford compound 6d (13.5 mg, 79%) as a yellow amorphous. [α] +104.7 (c 0.5, CHCl3); 1H-NMR (400 MHz, CDCl3); δ 6.52 (1H, s, 6’-H), 4.01 (3H, s, 9-OCH3), 3.88 (1H, brs, 1-H), 3.76 (3H, s, 4’-OCH3), 3.69 (1H, brs, 4-H), 3.66 (3H, s, 5’-OCH3), 3.61 (2H, s, 3-N-CH2Ar), 3.56 (3H, s, 2’-OCH3), 3.31 (1H, brd, J = 7.3 Hz, 5-H), 3.00 (1H, dd, J = 11.3, 3.2 Hz, 2-H), 2.69 (1H, dd, J = 20.7, 7.3 Hz, 6-H), 2.62 (1H, d, J = 11.3 Hz, 2-H), 2.35 (3H, s, 11-N-CH3), 2.16 (1H, d, J = 20.7 Hz, 6-H), 2.15 (3H, s, 3’-CH3), 1.94 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 186.8 (s, C-7), 182.2 (s, C-10), 155.4 (s, C-9), 151.4 (s, C-2’), 149.2 (s, C-5’), 147.5 (s, C-4’), 140.9 (s, C-6a), 137.6 (s, C-10a), 128.3 (s, C-8), 126.0 (s, C-3’), 123.8 (s, C-1’), 116.1 (s, 4-CN), 109.9 (d, C-6’), 61.1 (q, 2’-OCH3), 61.0 (q, 9-OCH3), 60.2 (q, 4’-OCH3), 57.9 (d, C-4), 55.7 (q, 5’-OCH3), 54.5 (d, C-5), 53.2 (t, 3-N-CH2Ar), 51.9 (t, C-2), 51.4 (d, C-1), 41.5 (q, 11-N-CH3), 20.8 (t, C-6), 9.5 (q, 3’-CH3), 8.6 (q, 8-CH3); IR (CHCl3) 3015, 2941, 2228, 1653, 1308, 1236, 1088, 1009 cm−1; EI-MS m/z (%) 481 (M+, 9), 220 (11), 219 (15), 218 (45), 196 (14), 195 (100); HREIMS m/z 481.2212 (M+, calcd for C26H31N3O6 481.2213).
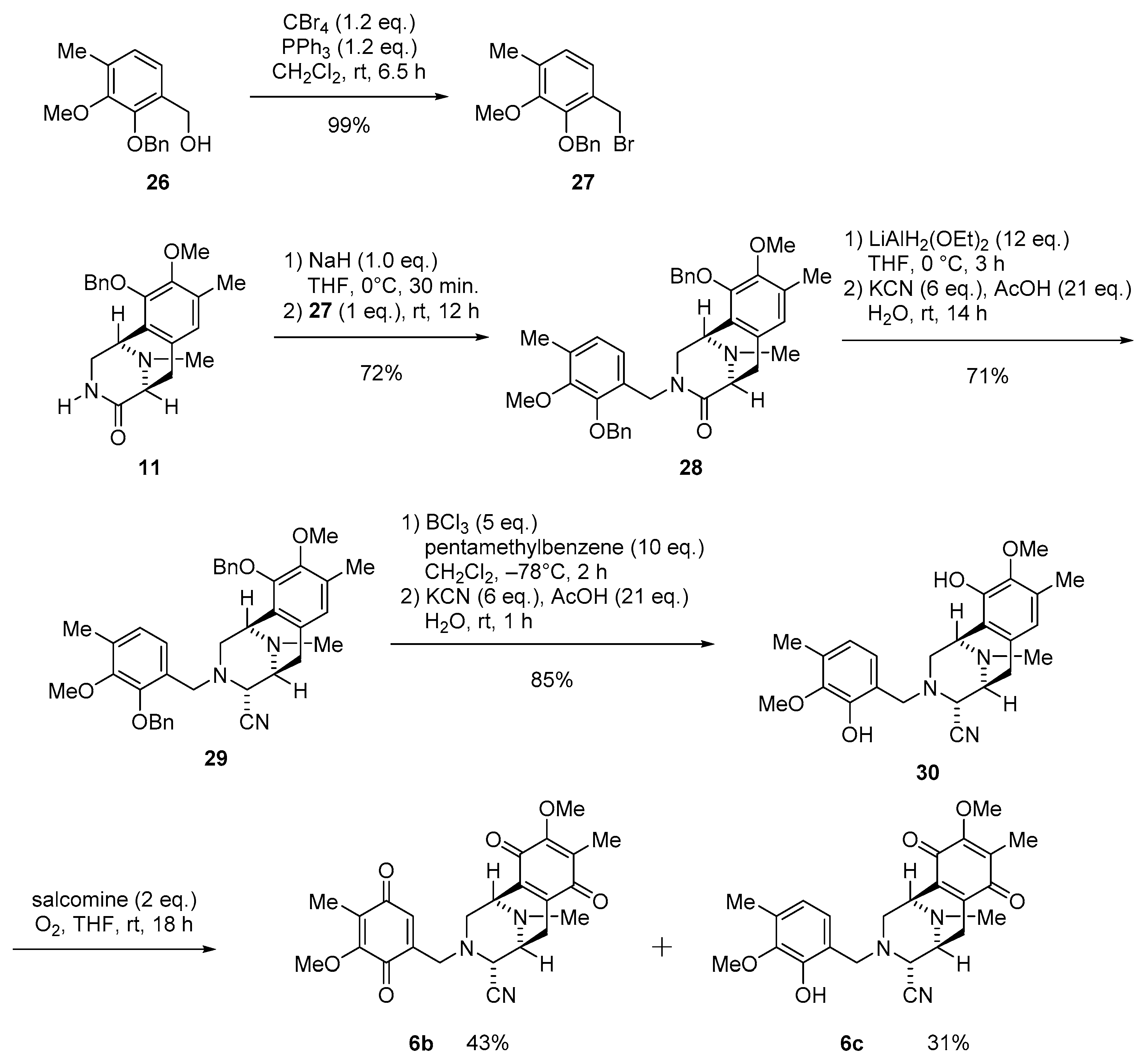
3.1.14. Synthesis of 2-(Benzyloxy)-1-(bromomethyl)-3-methoxy-4-methylbenzene (27)
To a solution of alcohol 26 (100 mg, 387 µmol) in CH2Cl2 (2 mL) was added PPh3 (125 mg, 465 µmol, 1.2 eq.) and CBr4 (162 mg, 465 µmol, 1.2 eq.) at 25 °C, and the reaction mixture was stirred for 6.5 h. The reaction mixture was diluted with H2O (10 mL) and extracted with CHCl3 (3 × 10 mL). The combined extracts were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 4:1) to afford compound 27 (123 mg, 99%) as a colorless oil. 1H-NMR (400 MHz, CDCl3) δ 7.54–7.31 (5H, m, Bn-H), 7.03 (1H, d, J = 7.8 Hz, 6-H), 6.93 (1H, d, J = 7.8 Hz, 5-H), 5.12 (2H, s, 2-OCH2Ph), 4.56 (2H, s, 1-CH2Br), 3.86 (3H, s, 3-OCH3), 2.30 (3H, s, 4-CH3); 13C-NMR (100 MHz, CDCl3) δ 151.7 (s, C-3), 150.1 (s, C-2), 137.4 (s, Bn), 133.6 (s, C-4), 129.9 (s, C-1), 128.5 (d, Bn), 128.4 (d, Bn), 128.2 (d, Bn), 126.1 (d, C-5), 125.2 (d, C-6), 75.2 (t, 2-OCH2Ph), 60.2 (q, 3-OCH3), 41.4 (t, 1-CH2Br), 15.9 (q, 4-CH3); IR (CHCl3) 3034, 3012, 2936, 1462, 1414, 1278, 1227, 1069 cm−1; EIMS m/z (%) : 322 (1), 320 (M+, 1), 241 (10), 151 (11), 150 (100), 149 (19), 91 (50); HREIMS m/z 320.0413 (M+, calcd for C16H17BrO2 320.0412).
3.1.15. Synthesis of (1R,5S)-10-(Benzyloxy)-3-(2-(benzyloxy)-3-methoxy-4-methylbenzyl)-9-methoxy-8,11-dimethyl-2,3,5,6-tetrahydro-1,5-epiminobenzo[d]azocin-4(1H)-one (28)
To a solution of NaH (60% oil dispersion, 5.70 mg, 142 µmol, 1.0 eq.) in THF (10 mL) was added a solution of lactam 11 (50.0 mg, 142 µmol) in THF (0.7 mL) at 0 °C. The reaction mixture was stirred for 30 min at 0 °C, after which a solution of bromide 27 (45.6 mg, 142 µmol, 1.0 eq.) in THF (0.7 mL) was added at 25 °C. The reaction mixture was stirred for 12 h at 25 °C. The reaction mixture was diluted with H2O (20 mL) and extracted with CHCl3 (3 × 20 mL). The combined extracts were washed with brine (40 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (CHCl3−MeOH = 49:1) to afford compound 28 (60.8 mg, 72%) as a colorless oil. [α] −67.0 (c 2.2, CHCl3); 1H-NMR (400 MHz, CDCl3); δ 7.37–7.25 (10H, m, 10-O-Bn-H, 2’-O-Bn-H), 6.71 (1H, s, 7-H), 6.50 (1H, d, J = 7.8 Hz, 5’-H), 5.96 (1H, d, J = 7.8 Hz, 6’-H), 4.98 (1H, d, J = 11.4 Hz, 10-OCH2Ph), 4.88 (2H, s, 2’-OCH2Ph), 4.84 (1H, d, J = 11.4 Hz, 10-OCH2Ph), 4.76 (1H, d, J = 15.5 Hz, 3-N-CH2Ar), 4.12 (1H, d, J = 15.5 Hz, 3-N-CH2Ar), 3.90 (1H, brd, J = 4.6 Hz, 1-H), 3.76 (3H, s, 3’-OCH3), 3.72 (3H, s, 9-OCH3), 3.63 (1H, d, J = 6.2 Hz, 5-H), 3.59 (1H, dd, J = 11.8, 4.6 Hz, 2-H), 3.14 (1H, dd, J = 16.9, 6.2 Hz, 6-H), 2.87 (1H, d, J = 11.8 Hz, 2-H), 2.82 (1H, d, J = 16.9 Hz, 6-H), 2.31 (3H, s, 11-N-CH3), 2.28 (3H, s, 8-CH3), 2.17 (3H, s, 4’-CH3); 13C-NMR (100 MHz, CDCl3) δ 170.3 (s, C-4), 151.2 (s, C-3’), 149.8 (s, C-2’), 149.4 (s, C-9), 148.4 (s, C-10), 137.4 (s×2, Bn), 131.2 (s, C-8), 130.9 (s, C-4’), 128.6 (d, Bn), 128.4 (d×2, Bn), 128.4 (s, C-6a), 128.2 (d, Bn), 128.0 (d, Bn), 127.9 (d, Bn), 126.3 (s, C-10a), 125.7 (d, C-7), 125.7 (d, C-5’), 121.9 (d, C-6’), 74.6 (t, 2’-OCH2Ph), 74.1 (t, 10-OCH2Ph), 60.0 (q, 3’-OCH3), 59.9 (q, 9-OCH3), 59.4 (d, C-5), 51.5 (d, C-1), 50.7 (t, C-2), 43.2 (t, 3-N-CH2Ar), 39.7 (q, 11-N-CH3), 27.4 (t, C-6), 15.7 (q, 8-CH3), 15.6 (q, 4’-CH3); IR (CHCl3) 3013, 2938, 2467, 1641, 1449, 1337, 1273, 1061 cm−1; EIMS m/z (%) 593 (17), 592 (M+, 40), 295 (26), 294 (100), 204 (29), 203 (20), 91 (10); HREIMS m/z 592.2934 (M+, calcd for C37H40N2O5 592.2937).
3.1.16. Synthesis of (1R,4R,5S)-10-(Benzyloxy)-3-(2-(benzyloxy)-3-methoxy-4-methylbenzyl)-9-methoxy-8,11-dimethyl-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (29)
To a solution of lactam 28 (85.6 mg, 144 µmol) in THF (4.5 mL) at 0 °C was slowly added LiAlH2(OEt)2 (1.0 mol/L in CH2Cl2, 1.70 mL, 1.70 mmol, 12 eq.) over 10 min. The reaction mixture was stirred at 0 °C for 3 h. The reaction mixture was quenched with AcOH (170 µL, 3.00 mmol, 20.8 eq.), followed by the addition of KCN (57.8 mg, 866 µmol, 6.0 eq.) in H2O (2.0 mL), and stirring was continued for 14 h at 25 °C. The reaction mixture was neutralized with 5% NaHCO3 solution and diluted with saturated Rochell’s salt aq., and the mixture was stirred for 1 h. The reaction mixture was extracted with CHCl3 (3 × 30 mL). The combined extracts were washed with brine (40 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 29 (61.5 mg, 71%) as a colorless gummy solid. [α] −31.2 (c 0.8, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.43–7.25 (10H, m, 10-O-Bn-H, 2’-O-Bn-H), 6.70 (1H, d, J = 7.8 Hz, 5’-H), 6.53 (1H, d, J = 7.8 Hz, 6’-H), 6.46 (1H, s, 7-H), 5.03 (1H, d, J = 11.2 Hz, 10-OCH2Ph), 4.72 (1H, d, J = 11.2 Hz, 10-OCH2Ph), 4.59 (1H, d, J = 10.6 Hz 2’-OCH2Ph), 4.54 (1H, d, J = 10.6 Hz 2’-OCH2Ph), 3.95 (1H, brs, 1-H), 3.71 (3H, s, 9-OCH3), 3.70 (1H, s, 4-H), 3.68 (3H, s, 3’-OCH3), 3.58 (1H, d, J = 13.1 Hz, 3-N-CH2Ar), 3.45 (1H, d, J = 13.1 Hz, 3-N-CH2Ar), 3.23 (1H, brd, J = 7.8 Hz, 5-H), 2.95 (1H, dd, J = 16.7, 7.8 Hz, 6-H), 2.79 (1H, dd, J = 11.0, 3.0 Hz, 2-H), 2.56 (1H, d, J = 11.0 Hz, 2-H), 2.27 (1H, d, J = 16.7 Hz, 6-H), 2.20 (3H, s, 4’-CH3), 2.14 (3H, s, 11-N-CH3), 2.12 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 151.8 (s, C-3’), 150.6 (s, C-2’), 148.9 (s, C-9), 148.4 (s, C-10), 137.9 (s, Bn), 137.5 (s, Bn), 131.8 (s, C-4’), 130.1 (s, C-8), 129.9 (s, C-6a), 128.7 (s, C-1’), 128.5 (d, Bn), 128.3 (d, Bn), 128.2 (d, Bn), 128.1 (d, Bn), 127.9 (d, Bn), 127.7 (d, Bn), 126.5 (s, C-10a), 125.5 (d, C-5’), 124.9 (d, C-6’), 124.4 (d, C-7), 116.7 (s, 4-CN), 75.0 (t, 2’-OCH2Ph), 74.3 (t, 10-OCH2Ph), 60.1 (q, 3’-OCH3), 60.0 (q, 9-OCH3), 59.4 (d, C-4), 55.4 (d, C-5), 53.8 (t, 3-N-CH2Ar), 53.7 (t, C-2), 52.7 (d, C-1), 41.2 (q, 11-N-CH3), 25.0 (t, C-6), 15.7 (q, 8-CH3), 15.7 (q, 4’-CH3); IR (CHCl3) 3015, 2930, 2226, 1454, 1321, 1076, 1028, 700 cm−1; EI-MS m/z (%) 603 (M+, 1), 337 (11), 295 (24), 294 (100), 204 (13), 203 (18), 91 (14); HREIMS m/z 603.3099 (M+, calcd for C38H41N3O4 603.3097).
3.1.17. Synthesis of (1R,4R,5S)-10-Hydroxy-3-(2-hydroxy-3-methoxy-4-methylbenzyl)-9-methoxy-8,11-dimethyl-1,2,3,4,5,6-hexahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (30)
To a solution of 29 (47.8 mg, 79.2 µmol) and pentamethylbenzene (117 mg, 792 µmol, 10 eq.) in CH2Cl2 (13 mL) was added BCl3 (1.0 mol/L in CH2Cl2, 400 µL, 400 µmol, 5.0 eq.) over 17 min at −78 °C and the mixture was stirred for 2 h. The reaction mixture was diluted with CH2Cl2 (10 mL) and quenched with saturated NaHCO3 solution (20 mL) at 0 °C. The mixture was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were dried over Na2SO4 and concentrated in vacuo to give a residue. To a solution of the obtained residue (168 mg) in THF (5 mL), AcOH (100 µL, 1.66 mmol, 21 eq.) was added. The reaction mixture was stirred for 5 min, after which KCN (31.0 mg, 475 µmol, 6 eq.) in H2O (5.0 mL) was added. The reaction mixture was stirred for 1 h at 25 °C. The reaction mixture was neutralized with 5% NaHCO3 and diluted with saturated Rochell’s salt aq., and the mixture was stirred for 1 h. The reaction mixture was extracted with CHCl3 (3 × 20 mL). The combined extracts were washed with brine (50 mL), dried over Na2SO4, and concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 30 (28.5 mg, 85%) as a colorless gummy solid. [α] −46.5 (c 0.2, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.53 (1H, brs, 2’-OH), 6.67 (1H, d, J = 7.8 Hz, 6’-H), 6.58 (1H, d, J = 7.8 Hz, 5’-H), 6.52 (1H, s, 7-H), 5.63 (1H, s, 10-OH), 4.16 (1H, brs, 1-H), 3.80 (1H, brs, 4-H), 3.78 (3H, s, 9-OCH3), 3.74 (1H, d, J = 13.7 Hz, 3-N-CH2Ar), 3.68 (1H, d, J = 13.7 Hz, 3-N-CH2Ar), 3.67 (3H, s, 3’-OCH3), 3.37 (1H, brd, J = 7.0 Hz, 5-H), 3.14 (1H, dd, J = 19.2, 7.0 Hz, 6-H), 3.01 (1H, dd, J = 10.8, 2.7 Hz, 2-H), 2.84 (1H, d, J = 10.8 Hz, 2-H), 2.44 (1H, d, J = 19.2 Hz, 6-H), 2.41 (3H, s, 11-N-CH3), 2.29 (3H, s, 8-CH3), 2.19 (3H, s, 4’-CH3); 13C-NMR (100 MHz, CDCl3) δ 149.3 (s, C-2’), 146.2 (s, C-3’), 145.4 (s, C-10), 143.3 (s, C-9), 132.0 (s, C-4’), 129.1 (s, C-8), 129.1 (s, C-6a), 123.9 (s, C-6’), 121.4 (d, C-7), 121.3 (d, C-5’), 118.8 (s, C-1’), 118.0 (s, C-10a), 115.3 (s, 4-CN), 60.8 (q, 9-OCH3), 59.6 (q, 3’-OCH3), 57.9 (d, C-4), 57.8 (t, 3-N-CH2Ar), 55.1 (d, C-5), 53.3 (t, C-2), 52.2 (d, C-1), 41.5 (q, 11-N-CH3), 24.7 (t, C-6), 15.9 (q, 8-CH3), 15.9 (q, 4’-CH3); IR (CHCl3) 3532, 3007, 2928, 2232, 1464, 1418, 1242, 1227, 1074 cm−1; FABMS m/z 424 [M + H]+; HRFABMS m/z 424.2234 ([M + H]+, calcd for C24H30N3O4 424.2236).
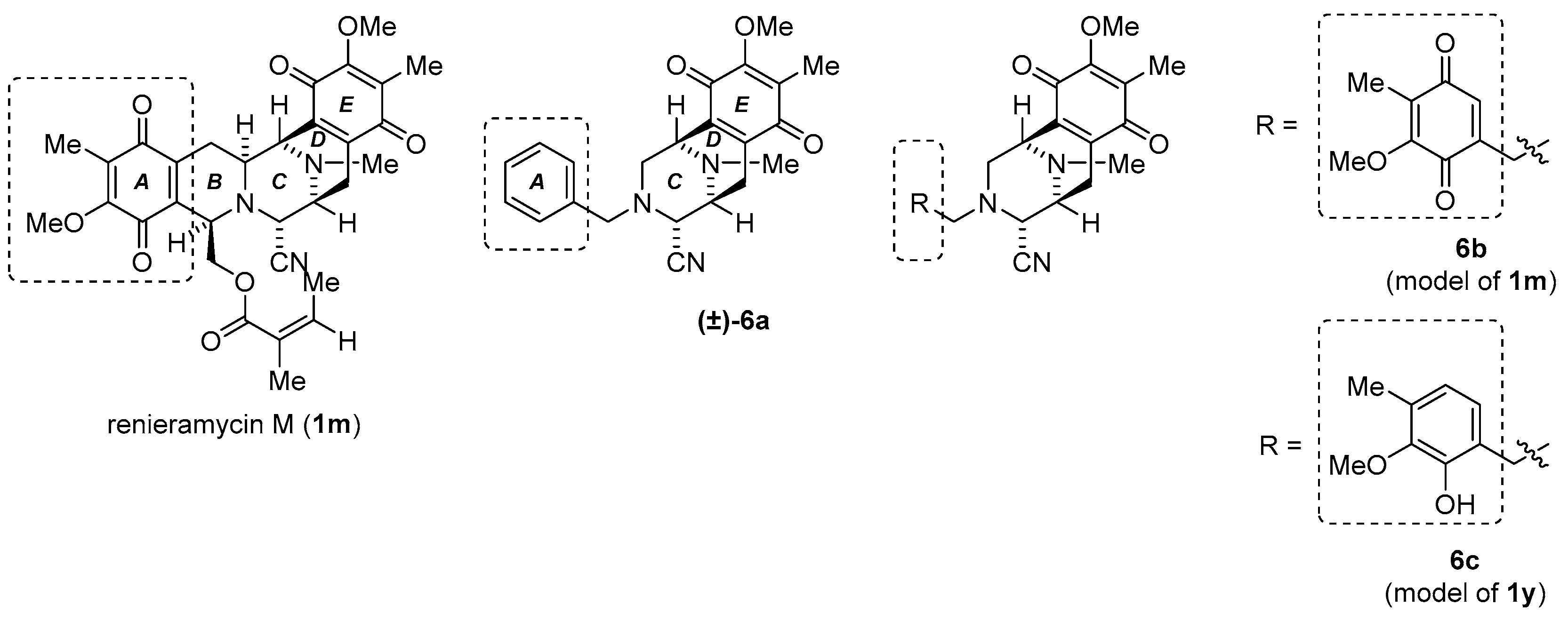
3.1.18. Synthesis of (1R,4R,5S)-9-Methoxy-3-((5-methoxy-4-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)methyl)-8,11-dimethyl-7,10-dioxo-1,2,3,4,5,6,7,10-octahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (6b) and (1R,4R,5S)-3-(2-hydroxy-3-methoxy-4-methylbenzyl)-9-methoxy-8,11-dimethyl-7,10-dioxo-1,2,3,4,5,6,7,10-octahydro-1,5-epiminobenzo[d]azocine-4-carbonitrile (6c)
To a solution of phenol 30 (17.3 mg, 40.8 µmol) in THF (1.5 mL) was added salcomine (27.6 mg, 81.6 µmol, 2.0 eq.) at rt, and the reaction mixture was stirred for 18 h under O2 atmosphere. The reaction mixture was filtered through a cellulose pad and washed with EtOAc. The filtrate was concentrated in vacuo to give a residue. The residue was purified by SiO2 flash column chromatography (n-Hex.−EtOAc = 2:1) to afford compound 6b (8.0 mg, 43%) as a yellow oil, and 6c (5.6 mg, 31%) as a yellow oil.
6b: [α] −70.3 (c 0.3, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 6.27 (1H, t, J = 1.8 Hz, 2’-H), 4.00 (3H, s, 9-OCH3), 3.90 (3H, s, 5’-OCH3), 3.86 (1H, brs, 1-H), 3.74 (1H, brd, J = 1.8 Hz, 4-H), 3.49 (1H, d, J = 16.5, 1.8 Hz, 3-N-CH2Ar), 3.39 (1H, d, J = 16.5, 1.8 Hz, 3-N-CH2Ar), 3.34 (1H, brd, J = 7.3 Hz, 5-H), 2.98 (1H, dd, J = 11.1, 3.0 Hz, 2-H), 2.75 (1H, dd, J = 20.7, 7.3 Hz, 6-H), 2.54 (1H, d, J = 11.1 Hz, 2-H), 2.34 (3H, s, 11-N-CH3), 2.20 (1H, d, J = 20.7 Hz, 6-H), 1.98 (3H, s, 8-CH3), 1.89 (3H, s, 4’-CH3); 13C-NMR (100 MHz, CDCl3) δ 187.4 (s, C-3’), 186.7 (s, C-7), 182.5 (s, C-6’), 182.2 (s, C-10), 155.9 (s, C-5’), 155.5 (s, C-9), 141.3 (s, C-1’), 140.9 (s, C-6a), 136.9 (s, C-10a), 132.9 (d, C-2’), 129.1 (s, C-8), 129.0 (s, C-4’), 116.0 (s, 4-CN), 61.1 (q, 9-OCH3), 60.7 (q, 5’-OCH3), 59.3 (d, 4-C), 54.7 (d, 5-C), 52.3 (t, 3-N-CH2Ar), 51.2 (d, 1-C), 50.8 (t, 2-C), 41.4 (q, 11-N-CH3), 20.8 (t, 6-C), 8.7 (q, 8-CH3), 8.5 (q, 4’-CH3); IR (CHCl3) 3017, 2945, 2359, 2230, 1655, 1612, 1308, 1234, 1153 cm−1; EIMS m/z (%) 451 (M+, 6), 261 (18), 260 (37), 233 (11), 232 (25), 220 (12), 219 (43), 218 (100), 204 (26), 190 (11), 176 (13), 166 (19), 83 (10); HREIMS m/z 451.1740 (M+, calcd for C24H25N3O6 451.1743).
6c: [α] +95.3 (c 0.2, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.92 (1H, brs, 2’-OH), 6.68 (1H, d, J = 7.8 Hz, 6’-H), 6.60 (1H, d, J = 7.8 Hz, 5’-H), 4.00 (3H, s, 9-OCH3), 3.92 (1H, brs, 1-H), 3.78 (1H, d, J = 14.0 Hz, 3-N-CH2Ar), 3.75 (1H, brs, 4-H), 3.73 (1H, d, J = 14.0 Hz, 3-N-CH2Ar), 3.70 (3H, s, 3’-OCH3), 3.38 (1H, brd, J = 7.4 Hz, 5-H), 2.99 (1H, dd, J = 11.7, 3.2 Hz, 2-H), 2.79 (1H, dd, J = 20.8, 7.4 Hz, 6-H), 2.70 (1H, d, J = 11.7 Hz, 2-H), 2.36 (3H, s, 11-N-CH3), 2.23 (1H, d, J = 20.8 Hz, 6-H), 2.20 (3H, s, 4’-CH3), 2.00 (3H, s, 8-CH3); 13C-NMR (100 MHz, CDCl3) δ 186.2 (s, C-7), 182.1 (s, C-10), 155.4 (s, C-9), 149.0 (s, C-2’), 146.1 (s, C-3’), 140.8 (s, C-6a), 136.9 (s, C-10a), 132.2 (s, C-4’), 129.2 (s, C-8), 124.0 (d, C-6’), 121.6 (d, C-5’), 118.2 (s, C-1’), 114.9 (s, 4-CN), 61.0 (q, 9-OCH3), 59.8 (q, 3’-OCH3), 57.5 (d, C-4), 57.2 (t, 3-N-CH2Ar), 54.3 (d, C-5), 51.3 (t, C-2), 51.1 (d, C-1), 41.5 (q, 11-N-CH3), 20.7 (t, C-6), 15.9 (q, 4’-CH3), 8.8 (q, 8-CH3); IR (CHCl3) 3524, 3022, 2945, 2853, 2359, 2234, 1653, 1614, 1308, 1236, 1152 cm−1; EIMS m/z (%) : 437 (M+, 5), 411 (24), 410 (100), 261 (19), 260 (80), 259 (12), 245 (12), 234 (24), 233 (20), 232 (43), 231 (14), 220 (21), 219 (49), 218 (98), 217 (12), 204 (26), 203 (13), 202 (15), 192 (19), 190 (12), 176 (13), 151 (14), 150 (33), 149 (17), 91 (13), 77 (12). HREIMS m/z 437.1956 (M+, calcd for C24H27N3O5 437.1951).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}