Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508


Abstract
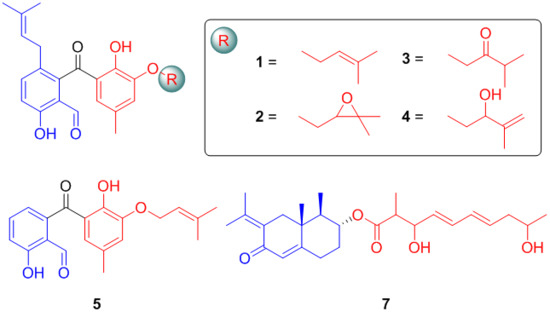
1. Introduction
2. Results and Discussion
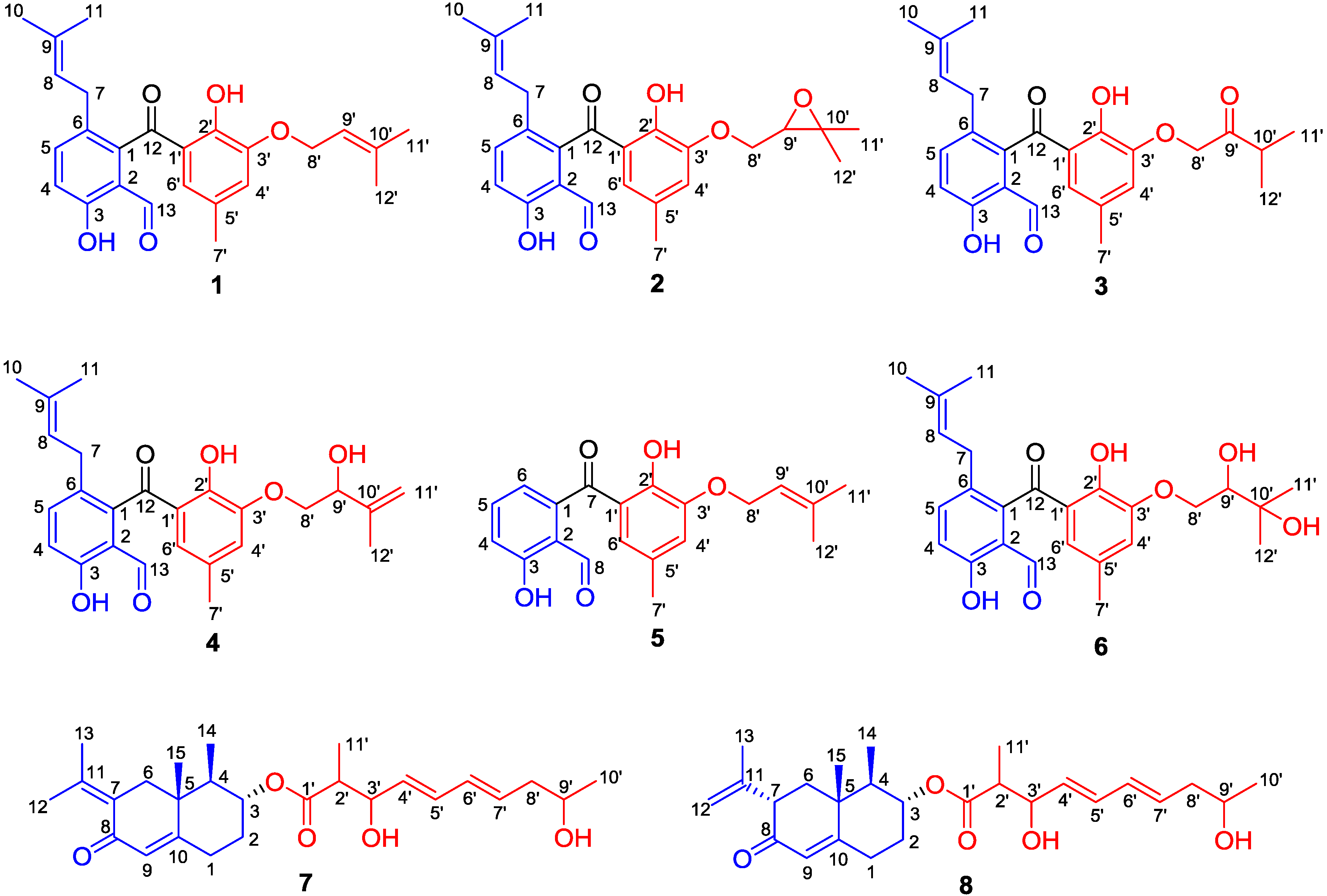
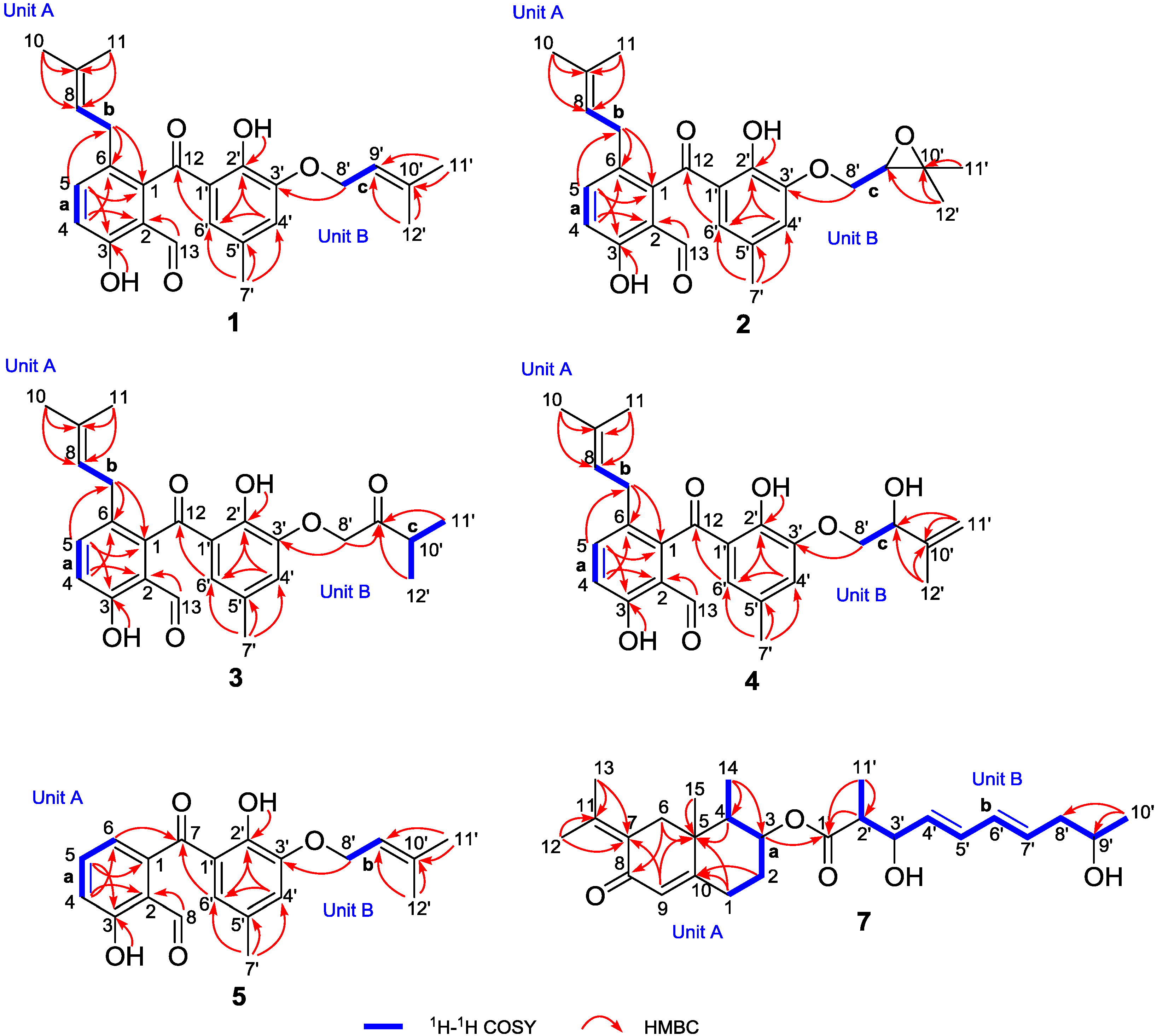
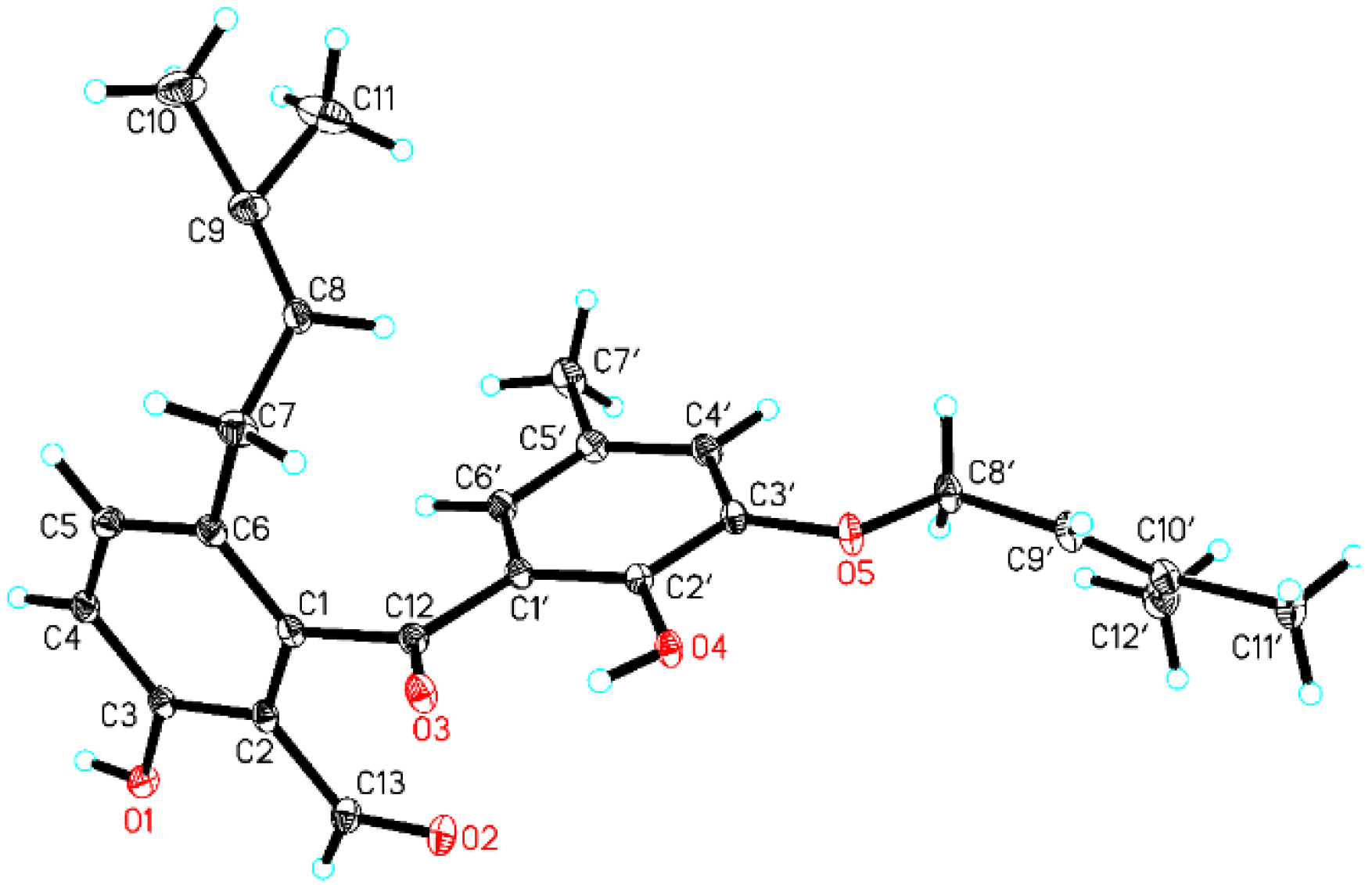
2.1. Structure Elucidation
2.2. Cytotoxicity Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction and Isolation
3.4. X-Ray Analysis of Tenellone D (1)
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.D.; Li, X.M.; Li, X.; Xu, G.M.; Liu, Y.; Wang, B.G. Aspewentins D–H, 20-nor-isopimarane derivatives from the deep sea sediment-derived fungus Aspergillus wentii SD-310. J. Nat. Prod. 2016, 79, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, L.; Hao, J.J.; Wang, L.P.; Zhu, W.M. α-glucosidase inhibitors from the marine-derived fungus Aspergillus flavipes HN4-13. J. Nat. Prod. 2016, 79, 2977–2981. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine fungi: A source of potential anticancer compounds. Front. Microbiol. 2018, 8, 2536. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.J.; Bae, S.Y.; Won, T.H.; You, M.J.; Kim, S.H.; Oh, D.C.; Lee, S.K.; Oh, K.B.; Shin, J. Asperphenins A and B, lipopeptidyl benzophenones from a marine-derived Aspergillus sp. fungus. Org. Lett. 2017, 19, 2066–2069. [Google Scholar] [CrossRef] [PubMed]
- Soldatou, S.; Baker, B.J. Cold-water marine natural products, 2006 to 2016. Nat. Prod. Rep. 2017, 34, 585–626. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: 2017 updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Xue, Y.R.; Liu, C.H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhai, H.J.; Zhu, K.K.; Yu, J.H.; Zhang, Y.Y.; Wang, Y.Y.; Jiang, C.S.; Zhang, X.Y.; Zhang, Y.; Zhang, H. Bioactive pyridone alkaloids from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Mar. Drugs 2018, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.J.; Cheng, W.; Heydari, H.; Wang, B.; Zhu, K.; Konuklugil, B.; Lin, W.H. Sorbicillinoid-based metabolites from a sponge-derived fungus Trichoderma saturnisporum. Mar. Drugs 2018, 16, 226. [Google Scholar] [CrossRef] [PubMed]
- Daletos, G.; Ebrahim, W.; Ancheeva, E.; El-Neketi, M.; Song, W.G.; Lin, W.H.; Proksch, P. Natural products from deep-sea-derived fungi a new source of novel bioactive compounds? Curr. Med. Chem. 2018, 25, 186–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, H.; Gigant, B.; Yu, Y.M.; Wu, Y.P.; Chen, X.Z.; Lai, Q.H.; Yang, Z.Y.; Chen, Q.; Yang, J.L. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.X.; Wu, Y.; Xie, S.S.; Sun, W.G.; Guo, Y.; Li, X.N.; Liu, J.J.; Li, H.; Wang, J.P.; Luo, Z.W.; et al. Phomopsterones A and B, two functionalized ergostane-type steroids from the endophytic fungus Phomopsis sp. TJ507A. Org. Lett. 2017, 19, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Raju, R.; Salim, A.A.; Khalil, Z.G.; Capon, R.J. Cytochalasins from an Australian marine sediment-derived Phomopsis sp. (CMB-M0042F): Acid-mediated intramolecular cycloadditions enhance chemical diversity. J. Org. Chem. 2017, 82, 9704–9709. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Krohn, K.; Ahmed, I.; Draeger, S.; Schulz, B.; Di Pietro, S.; Pescitelli, G. Phomopsinones A–D: Four new pyrenocines from endophytic fungus Phomopsis sp. Eur. J. Org. Chem. 2012, 2012, 1783–1789. [Google Scholar] [CrossRef]
- Xie, S.S.; Wu, Y.; Qiao, Y.B.; Guo, Y.; Wang, J.P.; Hu, Z.X.; Zhang, Q.; Li, X.N.; Huang, J.F.; Zhou, Q.; et al. Protoilludane, illudalane, and botryane sesquiterpenoids from the endophytic fungus Phomopsis sp. TJ507A. J. Nat. Prod. 2018, 81, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Gao, J.; Shen, Y.; Chu, Y.L.; Xu, Q.; Tan, R.X. Immunosuppressive diterpenes from Phomopsis sp. S12. Eur. J. Org. Chem. 2014, 2014, 5728–5734. [Google Scholar] [CrossRef]
- Li, L.Y.; Sattler, I.; Deng, Z.W.; Groth, I.; Walther, G.; Menzel, K.D.; Peschel, G.; Grabley, S.; Lin, W.H. A-seco-oleane-type triterpenes from Phomopsis sp. (strain HKI0458) isolated from the mangrove plant Hibiscus tiliaceus. Phytochemistry 2008, 69, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Bunyapaiboonsri, T.; Yoiprommarat, S.; Srikitikulchai, P.; Srichomthong, K.; Lumyong, S. Oblongolides from the endophytic fungus Phomopsis sp. BCC9789. J. Nat. Prod. 2010, 73, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.W.; Liu, H.X.; Sun, Z.H.; Chen, Y.C.; Tan, Y.Z.; Zhang, W.M. Secondary metabolites from the deep-sea derived fungus Acaromyces ingoldii FS121. Molecules 2016, 21, 371. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, Z.H.; Liu, Z.; Chen, Y.C.; Liu, H.X.; Li, H.H.; Zhang, W.M. Dichotocejpins A–C: New diketopiperazines from a deep-sea-derived fungus Dichotomomyces cejpii FS110. Mar. Drugs 2016, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Zhang, L.; Chen, Y.C.; Li, S.N.; Tan, G.H.; Sun, Z.H.; Pan, Q.L.; Ye, W.; Li, H.H.; Zhang, W.M. Cytotoxic pimarane-type diterpenes from the marine sediment-derived fungus Eutypella sp. FS46. Nat. Prod. Res. 2017, 31, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Zhang, L.; Chen, Y.C.; Sun, Z.H.; Pan, Q.L.; Li, H.H.; Zhang, W.M. Monoterpenes and sesquiterpenes from the marine sediment-derived fungus Eutypella scoparia FS46. J. Asian Nat. Prod. Res. 2017, 19, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Tan, H.B.; Chen, Y.C.; Li, S.N.; Huang, Z.L.; Guo, H.; Li, H.H.; Gao, X.X.; Liu, H.X.; Zhang, W.M. Lithocarpins A–D: Four tenellone-macrolide conjugated [4 + 2] hetero-adducts from the deep-sea derived fungus Phomopsis lithocarpus FS508. Org. Chem. Front. 2018, 5, 1792–1797. [Google Scholar] [CrossRef]
- Zhang, C.W.; Ondeyka, J.G.; Herath, K.B.; Guan, Z.Q.; Collado, J.; Platas, G.; Pelaez, F.; Leavitt, P.S.; Gurnett, A.; Nare, B.; et al. Tenellones A and B from a Diaporthe sp. Two highly substituted benzophenone inhibitors of parasite cGMP-dependent protein kinase activity. J. Nat. Prod. 2005, 68, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.D.; Jiang, Z.D.; Gallagher, R.T. Pine root (Deuteromycetes) fungal metabolites and analogs and derivatives thereof for anticancer agents. USA 5932613 A, 1999. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 141.0, C | 140.6, C | ||
| 2 | 117.4, C | 117.3, C | ||
| 3 | 160.8, C | 160.8, C | ||
| 4 | 7.06, d, (8.8) | 119.5, CH | 7.07, d, (8.7) | 119.6, CH |
| 5 | 7.45, d, (8.8) | 138.5, CH | 7.45, d, (8.7) | 138.7, CH |
| 6 | 129.9, C | 129.9, C | ||
| 7 | 3.12, s | 31.0, CH2 | 3.12, s | 31.0, CH2 |
| 8 | 5.05, m | 121.9, CH | 5.05, m | 121.7, CH |
| 9 | 134.1, C | 134.2, C | ||
| 10 | 1.47, s | 17.8, CH3 | 1.47, s | 17.8, CH3 |
| 11 | 1.58, s | 25.7, CH3 | 1.59, s | 25.7, CH3 |
| 12 | 203.1, C=O | 203.1, C=O | ||
| 13 | 9.71, s | 194.5, C=O | 9.71, s | 194.4, C=O |
| 1’ | 121.0, C | 121.3, C | ||
| 2’ | 151.7, C | 151.6, C | ||
| 3’ | 148.0, C | 147.7, C | ||
| 4’ | 6.93, d, (1.9) | 121.8, CH | 7.03, s | 123.7, CH |
| 5’ | 128.6, C | 129.1, C | ||
| 6’ | 6.47, d, (1.9) | 123.6, CH | 6.56, s | 125.1, CH |
| 7’ | 2.17, s | 21.1, CH3 | 2.19, s | 21.0, CH3 |
| 8α’ | 4.62, d, (5.7) | 66.3, CH2 | 4.12, dd, (9.5, 7.8) | 71.8, CH2 |
| 8β’ | 4.43, dd, (9.5, 2.8) | |||
| 9’ | 5.55, m | 119.4, CH | 4.07, dd, (7.8, 2.8) | 76.6, CH |
| 10’ | 138.7, C | 71.1, C | ||
| 11’ | 1.76, s | 18.4, CH3 | 1.69, s | 29.8, CH3 |
| 12’ | 1.80, s | 26.0, CH3 | 1.71, s | 28.4, CH3 |
| 3-OH | 11.51, s | 11.50, s | ||
| 2’-OH | 12.09, s | 12.13, s | ||
| No. | 3 | 4 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 140.6, C | 141.7, C | ||
| 2 | 117.3, C | 119.2, C | ||
| 3 | 160.8, C | 161.1, C | ||
| 4 | 7.08, d, (8.7) | 119.6, CH | 7.11, d, (8.6) | 119.6, CH |
| 5 | 7.46, d, (8.7) | 138.7, CH | 7.55, d, (8.6) | 139.2, CH |
| 6 | 129.9, C | 130.5, C | ||
| 7 | 3.13, s | 31.0, CH2 | 3.14, d, (7.2) | 31.4, CH2 |
| 8 | 5.06, m | 121.7, CH | 5.08, m | 122.3, CH |
| 9 | 134.2, C | 133.8, C | ||
| 10 | 1.48, s | 17.8, CH3 | 1.46, s | 17.6, CH3 |
| 11 | 1.59, s | 25.7, CH3 | 1.55, s | 25.7, CH3 |
| 12 | 203.2, C=O | 203.6, C=O | ||
| 13 | 9.72, s | 194.4, C=O | 9.98, s | 194.1, C=O |
| 1’ | 121.5, C | 123.2, C | ||
| 2’ | 151.5, C | 152.1, C | ||
| 3’ | 147.0, C | 148.8, C | ||
| 4’ | 6.90, d, (1.9) | 123.4, CH | 7.18, d, (2.0) | 123.0, CH |
| 5’ | 128.8, C | 129.2, C | ||
| 6’ | 6.57, s | 125.4, CH | 6.69, s | 124.6, CH |
| 7’ | 2.16, s | 21.0, CH3 | 2.16, s | 20.7, CH3 |
| 8α’ | 4.79, s | 72.9, CH2 | 4.01, dd, (9.8, 7.5) | 74.3, CH2 |
| 8β’ | 4.15, dd, (9.8, 3.9) | |||
| 9’ | 210.6, C=O | 4.47, dd, (7.5, 3.9) | 74.0, CH | |
| 10’ | 3.00, m | 37.2, CH | 146.0, C | |
| 11α’ | 1.20, s | 18.1, CH3 | 4.92, s | 112.2, CH2 |
| 11β’ | 5.12, m | |||
| 12’ | 1.21, s | 29.9, CH3 | 1.83, s | 19.0, CH3 |
| 3-OH | 11.51, s | |||
| 2’-OH | 12.10, s | |||
| No. | δH (J in Hz) | δC | No. | δH (J in Hz) | δC |
|---|---|---|---|---|---|
| 1 | 142.5, C | 4’ | 6.96, s | 121.7, CH | |
| 2 | 117.9, C | 5’ | 128.3, C | ||
| 3 | 162.9, C | 6’ | 6.69, m | 124.2, CH | |
| 4 | 7.16, d, (8.5) | 120.5, CH | 7’ | 2.12, s | 21.2, CH3 |
| 5 | 7.59, dd, (8.5, 7.3) | 136.1, CH | 8’ | 4.62, d, (7.0) | 66.3, CH2 |
| 6 | 6.98, d, (7.3) | 120.0, CH | 9’ | 5.55, m | 119.5, CH |
| 7 | 201.0, C=O | 10’ | 138.8, C | ||
| 8 | 9.91, s | 195.0, C=O | 11 | 1.76, s | 18.4, CH3 |
| 1’ | 119.9, C | 12’ | 1.80, s | 26.0, CH3 | |
| 2’ | 152.3, C | 3-OH | 11.74, s | ||
| 3’ | 148.1, C | 2’-OH | 11.93, s |
| No. | δH (J in Hz) | δC | No. | δH (J in Hz) | δC |
|---|---|---|---|---|---|
| 1α | 2.34, m | 30.2, CH2 | 13 | 1.85, s | 22.3, CH3 |
| 1β | 2.45, m | 14 | 0.98, d, (6.7) | 10.8, CH3 | |
| 2α | 1.46, m | 31.7, CH2 | 15 | 1.03, s | 17.3, CH3 |
| 2β | 2.16, overlapped | 1’ | 175.2, C=O | ||
| 3 | 4.87, td, (11.2, 4.4) | 74.2, CH | 2’ | 2.56, m | 46.0, CH |
| 4 | 1.67, dt, (11.2, 6.7) | 46.1, CH | 3’ | 4.24, m | 74.5, CH |
| 5 | 42.3, C | 4’ | 5.59, dd, (15.0, 7.0) | 131.5, CH | |
| 6α | 2.16, overlapped | 41.2, CH2 | 5’ | 6.27, dd, (15.0, 10.4) | 132.5, CH |
| 6β | 2.91, d, (13.7) | 6’ | 6.12, dd, (15.0, 10.4) | 132.5, CH | |
| 7 | 127.2, C | 7’ | 5.72, dd, (15.0, 7.0) | 131.5, CH | |
| 8 | 191.7, C=O | 8’ | 2.26, m | 42.7, CH2 | |
| 9 | 5.77, d, (1.8) | 126.9, CH | 9’ | 3.86, m | 67.5, CH |
| 10 | 164.9, C | 10’ | 1.20, d (6.2) | 23.1, CH3 | |
| 11 | 143.7, C | 11’ | 1.18, d (7.2) | 14.3, CH3 | |
| 12 | 2.10, s | 22.8, CH3 |
| Compounds | IC50 (μM) a | |||
|---|---|---|---|---|
| HepG-2 | MCF-7 | SF-268 | A549 | |
| 1 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 |
| 4 | 88.6 ± 3.1 | 85.7 ± 7.4 | 67.7 ± 3.1 | >100 |
| 5 | 16.0 ± 0.1 | 25.1 ± 1.1 | 23.0 ± 0.9 | 17.6 ± 0.3 |
| 6 | >100 | >100 | >100 | >100 |
| 7 | 90.9 ± 2.0 | 81.1 ± 2.8 | 92.5 ± 4.3 | 59.2 ± 2.1 |
| 8 | 26.2 ± 0.8 | 29.6 ± 4.6 | 28.8 ± 0.2 | 25.5 ± 0.4 |
| cisplatin | 2.4 ± 0.1 | 3.2 ± 0.1 | 3.3 ± 0.3 | 1.6 ± 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.-L.; Liu, H.-X.; Chen, Y.-C.; Tan, H.-B.; Guo, H.; Xu, L.-Q.; Li, S.-N.; Huang, Z.-L.; Li, H.-H.; Gao, X.-X.; et al. Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Mar. Drugs 2018, 16, 329. https://doi.org/10.3390/md16090329
Xu J-L, Liu H-X, Chen Y-C, Tan H-B, Guo H, Xu L-Q, Li S-N, Huang Z-L, Li H-H, Gao X-X, et al. Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Marine Drugs. 2018; 16(9):329. https://doi.org/10.3390/md16090329
Chicago/Turabian StyleXu, Jian-Lin, Hong-Xin Liu, Yu-Chan Chen, Hai-Bo Tan, Heng Guo, Li-Qiong Xu, Sai-Ni Li, Zi-Lei Huang, Hao-Hua Li, Xiao-Xia Gao, and et al. 2018. "Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508" Marine Drugs 16, no. 9: 329. https://doi.org/10.3390/md16090329
APA StyleXu, J.-L., Liu, H.-X., Chen, Y.-C., Tan, H.-B., Guo, H., Xu, L.-Q., Li, S.-N., Huang, Z.-L., Li, H.-H., Gao, X.-X., & Zhang, W.-M. (2018). Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Marine Drugs, 16(9), 329. https://doi.org/10.3390/md16090329