Bacillamidins A–G from a Marine-Derived Bacillus pumilus

 ,
,  and
and
Abstract
1. Introduction
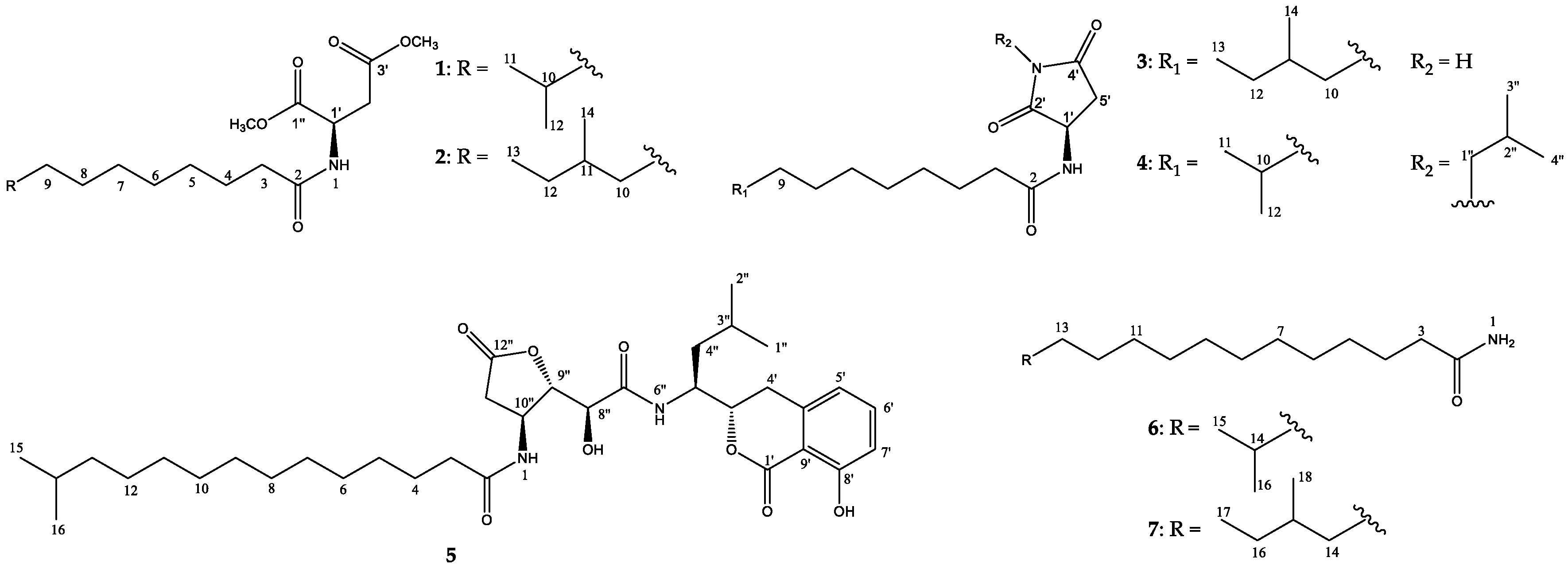
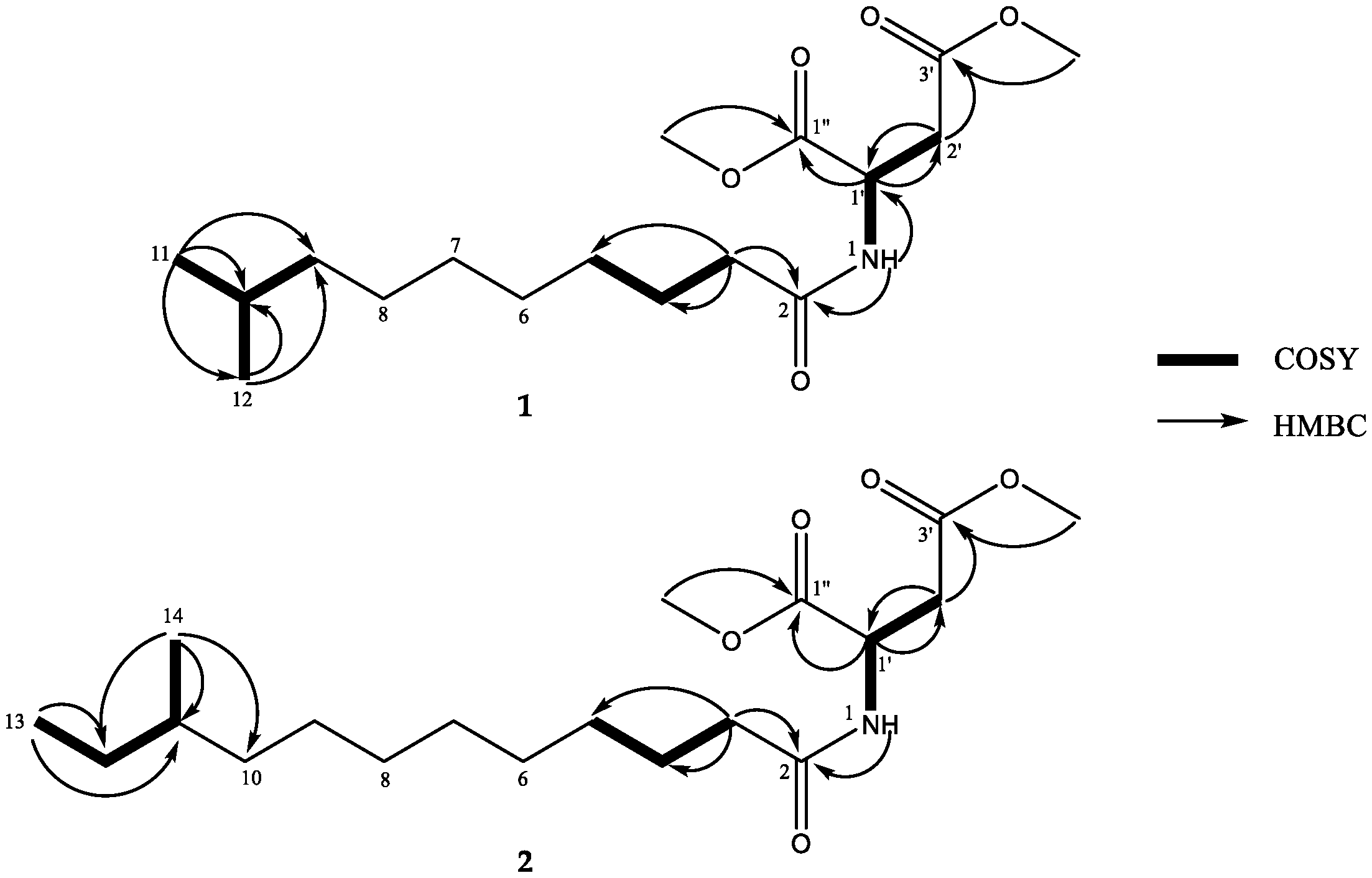
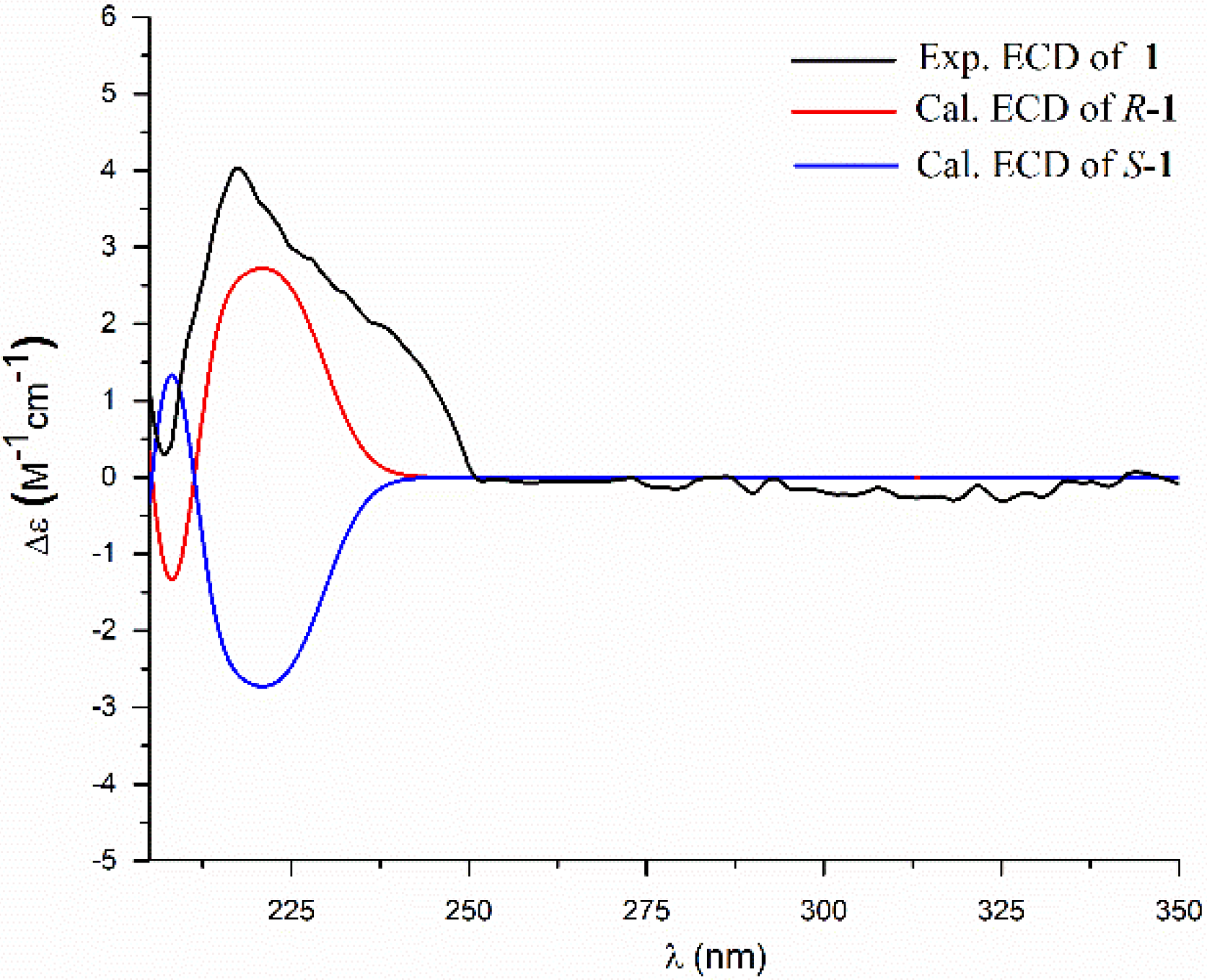
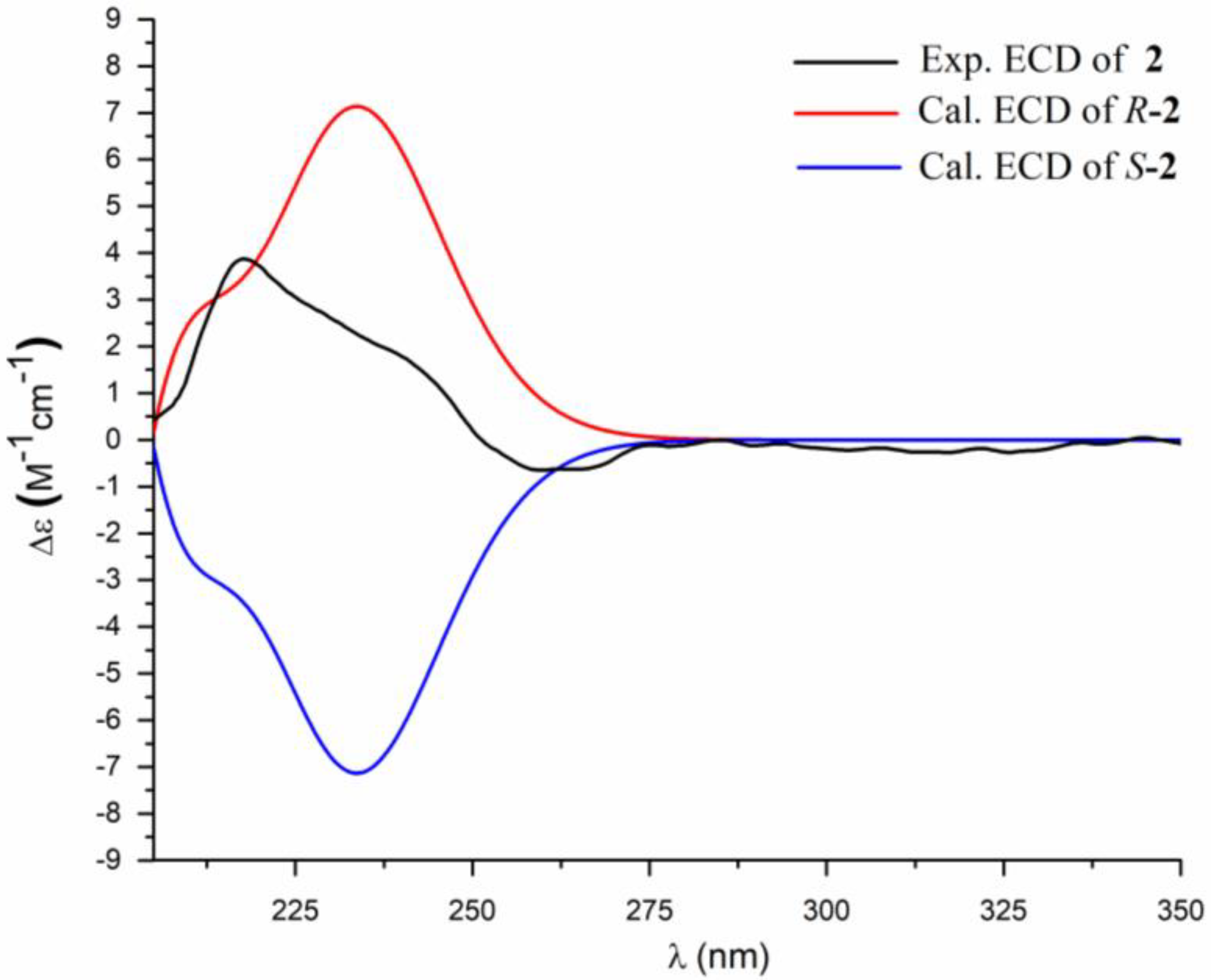
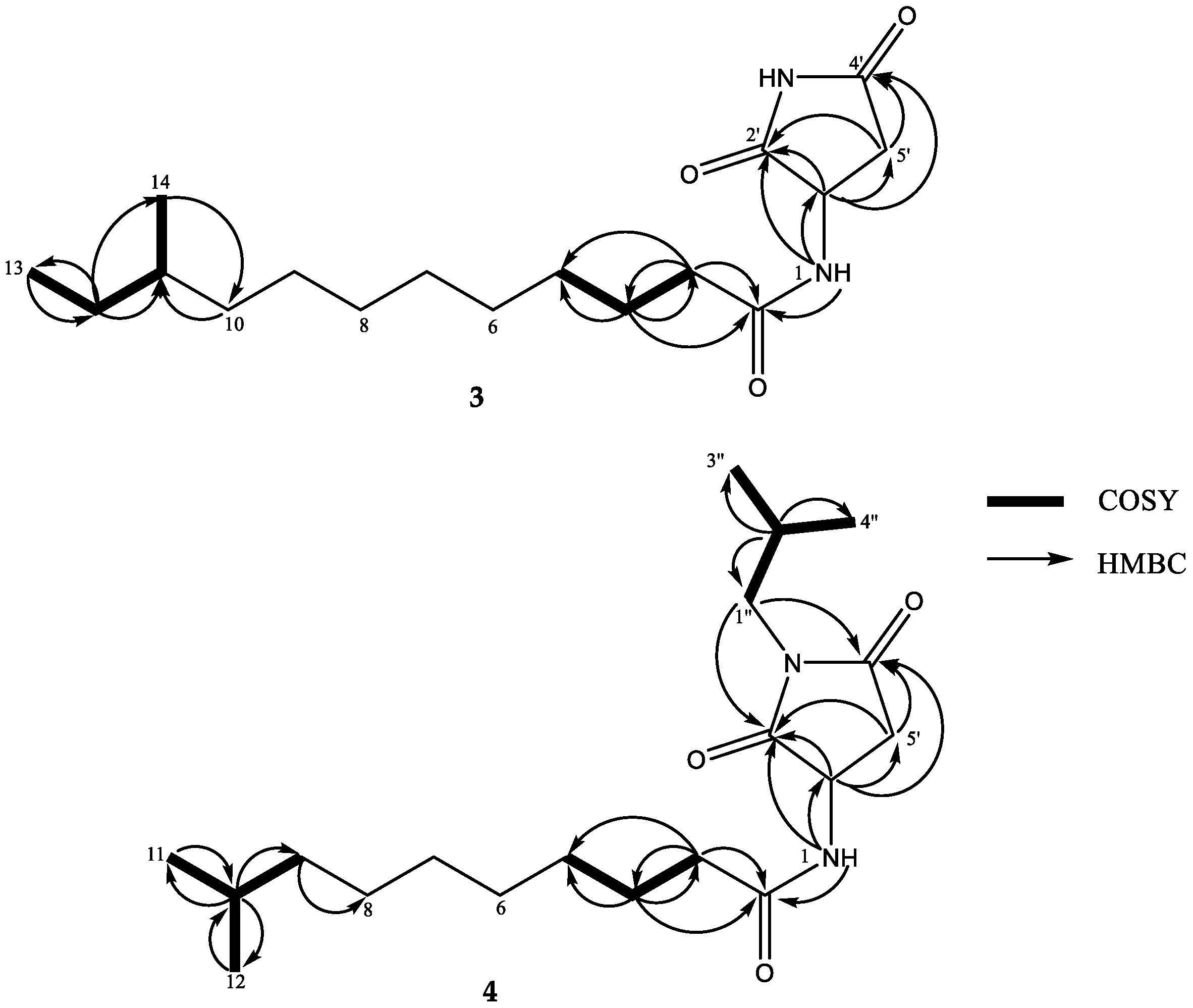
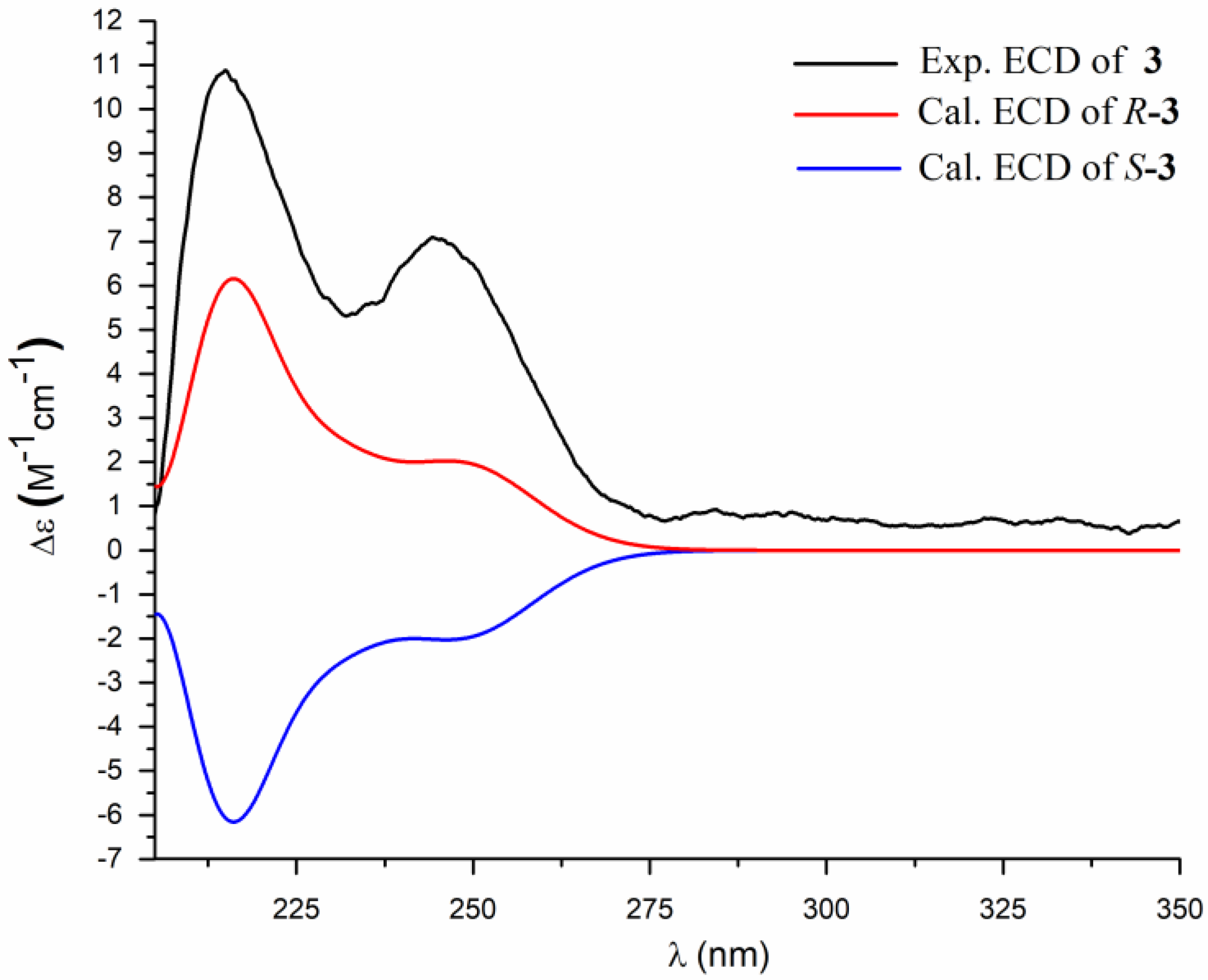
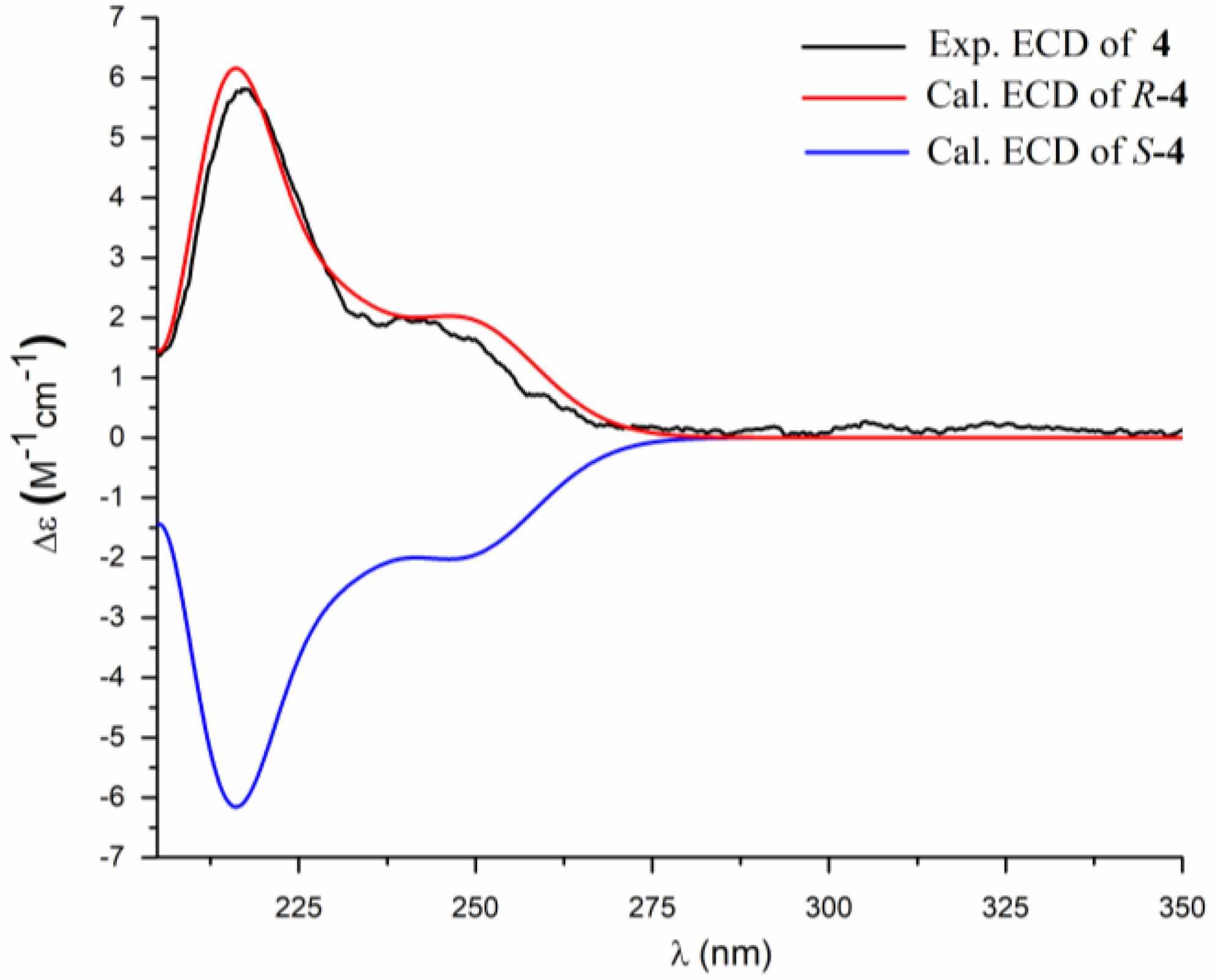
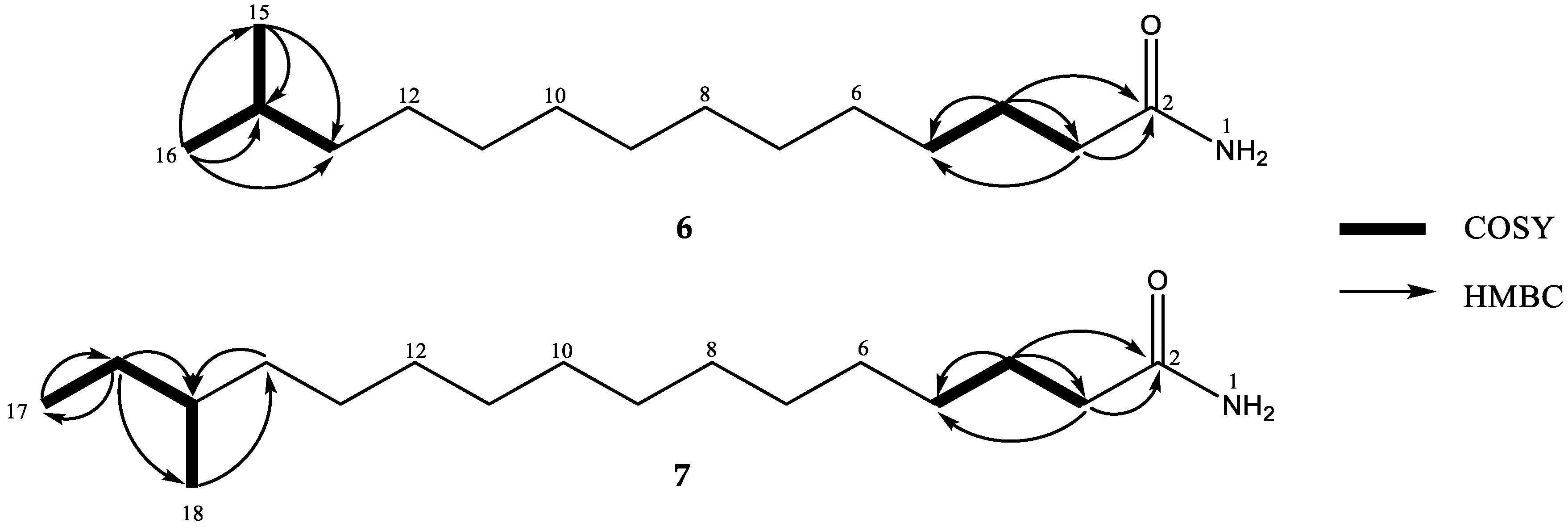
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Strain and Cultivation
3.3. Extraction and Isolation
3.4. ECD Calculation Methods
3.5. Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Joint, I.; Muhling, M.; Querellou, J. Culturing marine bacteria—An essential prerequisite for biodiscovery. Microb. Biotechnol. 2010, 3, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Rampelotto, P.H. Resistance of microorganisms to extreme environmental conditions and its contribution to astrobiology. Sustainability 2010, 2, 1602–1623. [Google Scholar] [CrossRef]
- Schinke, C.; Martins, T.; Queiroz, S.C.N.; Melo, I.S.; Reyes, F.G.R. Antibacterial compounds from marine bacteria, 2010–2015. J. Nat. Prod. 2017, 80, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Mondol, M.A.M.; Shin, H.J.; Islam, M.T. Diversity of secondary metabolites from marine Bacillus species: Chemistry and biological activity. Mar. Drugs 2013, 11, 2846–2872. [Google Scholar] [CrossRef] [PubMed]
- Devi, P.; Wahidullah, S.; Rodrigues, C.; Souza, L.D. The sponge-associated bacterium Bacillus licheniformis SAB1: A source of antimicrobial compounds. Mar. Drugs 2010, 8, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Naughton, L.M.; Montánchez, I.; Dobson, A.D.W.; Rai, D.K. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Hogberg, L.D.; Heddini, A.; Cars, O. The global need for effective antibiotics: Challenges and recent advances. Trends Pharmacol. Sci. 2010, 31, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Daoust, J. Isolation and Structure Elucidation of Bioactive Marine Natural Products. Ph.D. Thesis, The University of British Columbia, Vancouver, BC, Canada, 2011; doi:10.14288/1.0062104. [Google Scholar]
- Nord, L.I.; Vaag, P.; Duus, J. Quantification of organic and amino acids in beer by 1H NMR spectroscopy. Anal. Chem. 2004, 76, 4790–4798. [Google Scholar] [CrossRef]
- Jin, T.Y.; Shen, T.; Zhou, M.X.; Li, A.L.; Feng, D.; Zheng, B.; Gong, J.; Sun, J.W.; Li, L.Y.; Xiang, L. Chemical constituents from Portulaca oleracea and their bioactivities. J Chin. Pharm. Sci. 2016, 25, 898–905. [Google Scholar] [CrossRef]
- Iton, J.; Omoto, S.; Nishizawa, N.; Kodama, Y.; Lnouye, S. Chemical structures of amicoumacins produced by Bacillus pumilus. Agric. Biol. Chem. 1982, 46, 2659–2665. [Google Scholar] [CrossRef]
- Bai, J.; Liu, D.; Yu, S.; Proksch, P.; Lin, W. Amicoumacins from the marine-derived bacterium Bacillus sp. with the inhibition of NO production. Tetrahedron Lett. 2014, 55, 6286–6291. [Google Scholar] [CrossRef]
- Shimojima, Y.; Hayashi, H.; Ooka, T.; Shibukawa, W.; Iitaka, Y. Studies on AI-77s, microbial products with pharmacological activity structures and the chemical nature of AI-77s. Tetrahedron Lett. 1982, 23, 5435–5438. [Google Scholar] [CrossRef]
- Azumi, M.; Ogawa, K.; Fujita, T.; Takeshita, M.; Yoshida, R.; Furumai, T.; Igarashi, Y. Bacilosarcins A and B, novel bioactive isocoumarins with unusual heterocyclic cores from the marine-derived bacterium Bacillus subtilis. Tetrahedron 2008, 64, 6420–6425. [Google Scholar] [CrossRef]
- Takahashi, M.; Osawa, K.; Ueda, J.; Tsuai, C.T. On the chemical structure of a new acid amide, N-(13-methyltetradecyl)-acetamide, from the fruits of Cpsicum annuum L (author’s transl). Yakugaku Zasshi J. Pharm. Soc. Jpn. 1977, 97, 758–761. [Google Scholar] [CrossRef]
- Milbum, A.H.; Truter, E.V. The components of wool wax. Part II. Synthesis of the acids and alcohols of the iso- and the (+)-anteiso-series. J. Chem. Soc. 1954, 3344–3351. [Google Scholar] [CrossRef]
- Williams, D.E.; Dalisay, D.; Chen, J.; Polishchuck, E.A.; Patrick, G.N.; Ko, M.; Av-Gay, Y.; Li, H.; Magarvey, N.; Andersen, R.J. Aminorifamycins and sporalactams produced in culture by a Micromonospora sp. isolated from a northeastern-Pacific marine sediment are potent antibiotics. Org. Lett. 2017, 19, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Y.; Wang, R.; Deng, L.Q.; Zhang, X.L.; Min, C. A new isoflavanone from Ficus tikoua Bur. J. Nat. Res. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protocol. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (J, Hz) | δC, Type | δH (J, Hz) | δC, Type | |
| 1-NH | 8.31, d (7.6) | 8.42, d (7.6) | ||
| 2 | 172.2, CO | 172.2, CO | ||
| 3 | 2.08, t (7.6) | 34.9, CH2 | 2.08, t (7.6) | 34.9, CH2 |
| 4 | 1.48, m | 25.1, CH2 | 1.47, m | 25.1, CH2 |
| 5 | 1.23, m | 28.4, CH2 | 1.23, m | 28.4, CH2 |
| 6 | 1.23, m | 28.7, CH2 | 1.23, m | 28.7, CH2 |
| 7 | 1.23, m | 29.2, CH2 | 1.23, m | 28.9, CH2 |
| 8 | 1.23, m | 26.7, CH2 | 1.23, m | 29.3, CH2 |
| 9 | 1.13, m | 38.4, CH2 | 1.23, m | 26.4, CH2 |
| 10 | 1.48, m | 27.4, CH | 1.09, m; 1.27, m | 36.0, CH2 |
| 11 | 0.84, d (6.4) | 22.5, CH3 | 1.30, m | 33.7, CH |
| 12 | 0.84, d (6.4) | 22.5, CH3 | 1.10, m | 29.0, CH2 |
| 13 | 0.83, t (9.0) | 11.2, CH3 | ||
| 14 | 0.78, d (9.0) | 19.1, CH3 | ||
| 1′ | 4.60, ddd (9.6, 7.6, 6.0) | 48.4, CH | 4.60, ddd (9.6, 7.6, 6.0) | 48.4, CH |
| 2′ | 2.79, dd (16.4, 9.6) | 35.6, CH2 | 2.79, dd (16.4, 9.6) | 35.6, CH2 |
| 2.67, dd (16.4, 6.0) | 2.67, dd (16.4, 6.0) | |||
| 3′ | 171.2, CO | 171.3, CO | ||
| 3′-OCH3 | 3.61, s | 51.6, OCH3 | 3.61, s | 51.6, OCH3 |
| 1″ | 170.4, CO | 170.4, CO | ||
| 1″-OCH3 | 3.60, s | 52.0, OCH3 | 3.60, s | 52.1, OCH3 |
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δH (J, Hz) | δC, Type | δH (J, Hz) | δC, Type | |
| 1-NH | 8.42, d (7.6) | 8.51, d (7.6) | ||
| 2 | 172.5, CO | 172.6, CO | ||
| 3 | 2.08, t (7.6) | 34.9, CH2 | 2.08, t (7.6) | 34.8, CH2 |
| 4 | 1.23, m | 25.0, CH2 | 1.48, m | 25.0, CH2 |
| 5 | 1.23, m | 28.5, CH2 | 1.23, m | 28.4, CH2 |
| 6 | 1.23, m | 28.8, CH2 | 1.23, m | 28.8, CH2 |
| 7 | 1.23, m | 29.0, CH2 | 1.23, m | 29.2, CH2 |
| 8 | 1.23, m | 29.3, CH2 | 1.23, m | 26.7, CH2 |
| 9 | 1.23, m | 26.5, CH2 | 1.13, m | 38.4, CH2 |
| 10 | 1.09, m; 1.27, m | 36.2, CH2 | 1.48, m | 27.4, CH |
| 11 | 1.30, m | 33.7, CH | 0.83, d (6.6) | 22.5, CH3 |
| 12 | 1.10, m | 29.0, CH2 | 0.83, d (6.6) | 22.5, CH3 |
| 13 | 0.83, t (9.0) | 11.2, CH3 | ||
| 14 | 0.78, d (9.0) | 19.1, CH3 | ||
| 1′ | 4.35, ddd (9.6, 7.6, 6.0) | 49.5, CH | 4.35, ddd (9.6, 7.6, 6.0) | 48.4, CH |
| 2′ | 177.7, CO | 176.5, CO | ||
| 3′-NH | 11.19, s | |||
| 4′ | 176.4, CO | 175.2, CO | ||
| 5′ | 2.84, dd (16.4, 9.6) | 36.0, CH2 | 2.91, dd (16.4, 9.6) | 34.8, CH2 |
| 2.53, dd (16.4, 6.0) | 2.53, dd (16.4, 6.0) | |||
| 1″ | 3.19, d (7.2) | 45.4, CH2 | ||
| 2″ | 1.90, m | 26.7, CH | ||
| 3″ | 0.84, d (6.6) | 20.0, CH3 | ||
| 4″ | 0.84, d (6.6) | 20.0, CH3 | ||
| Position | 5 | ||||
|---|---|---|---|---|---|
| δH (J, Hz) | δC, Type | Position | δH (J, Hz) | δC, Type | |
| 1-NH | 8.34, d (6.8) | 1″ | 0.80, d (6.4) | 21.3, CH3 | |
| 2 | 171.9, CO | 2″ | 0.88, d (6.4) | 23.3, CH3 | |
| 3 | 1.90, t (7.6) | 35.0, CH2 | 3″ | 1.56, m | 24.0, CH |
| 4 | 1.48, m | 24.9, CH2 | 4″ | 1.32, m | 38.4, CH2 |
| 5-10 | 1.25, m | 28.5–29.2 | 1.68, m | ||
| 11 | 1.25, m | 29.3, CH2 | 5″ | 4.19, dddd (3.6, 7.6, 10.4, 13.6) | 48.3, CH |
| 12 | 1.25, m | 26.7, CH2 | 6″-NH | 7.94, d (9.6) | |
| 13 | 1.13, m | 38.4, CH2 | 7″ | 169.8, CO | |
| 14 | 1.49, m | 27.4, CH | 8″ | 4.27, dd (6.0, 2.4) | 71.6, CH |
| 15 | 0.83, d, (6.6) | 22.5, CH3 | 8″-OH | 6.18, d (6.0) | |
| 16 | 0.83, d, (6.6) | 22.5, CH3 | 9″ | 4.61, dd (2.4, 2.0) | 85.7, CH |
| 1′ | 168.9, CO | 10″ | 4.32, m | 46.1, CH | |
| 3′ | 4.69, ddd (10.4, 8.0, 2.8) | 80.8, CH | 11″ | 2.16, dd (18.0, 2.2) | 36.0, CH2 |
| 4′ | 2.87, m | 29.0, CH2 | 2.85, m | ||
| 5′ | 6.82, d (7.6) | 118.5, CH | 12″ | 175.6, CO | |
| 6′ | 7.48, dd (8.4, 7.6) | 136.2, CH | |||
| 7′ | 6.84, d (8.4) | 115.2, CH | |||
| 8′ | 160.8, C | ||||
| 8″-OH | 10.79, s | ||||
| 9′ | 108.3, C | ||||
| 10′ | 140.4, C | ||||
| Compounds | P. aeruginosa PA-01 | A. baumannii ATCC19606 | E. coli BW25113 |
|---|---|---|---|
| 1 | 64 | 58 | >128 |
| 2 | 64 | 64 | >128 |
| 3 | 64 | 64 | >128 |
| 4 | 64 | 58 | >128 |
| 5 | >128 | >128 | >128 |
| 6 | >128 | >128 | >128 |
| 7 | >128 | >128 | >128 |
| ofloxacin | 8 | 16 | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.-Y.; Hu, Y.-J.; Meng, F.-C.; Qu, S.-Y.; Wang, R.; Andersen, R.J.; Liao, Z.-H.; Chen, M. Bacillamidins A–G from a Marine-Derived Bacillus pumilus. Mar. Drugs 2018, 16, 326. https://doi.org/10.3390/md16090326
Zhou S-Y, Hu Y-J, Meng F-C, Qu S-Y, Wang R, Andersen RJ, Liao Z-H, Chen M. Bacillamidins A–G from a Marine-Derived Bacillus pumilus. Marine Drugs. 2018; 16(9):326. https://doi.org/10.3390/md16090326
Chicago/Turabian StyleZhou, Si-Yu, Yi-Jie Hu, Fan-Cheng Meng, Shen-Yue Qu, Rui Wang, Raymond J. Andersen, Zhi-Hua Liao, and Min Chen. 2018. "Bacillamidins A–G from a Marine-Derived Bacillus pumilus" Marine Drugs 16, no. 9: 326. https://doi.org/10.3390/md16090326
APA StyleZhou, S.-Y., Hu, Y.-J., Meng, F.-C., Qu, S.-Y., Wang, R., Andersen, R. J., Liao, Z.-H., & Chen, M. (2018). Bacillamidins A–G from a Marine-Derived Bacillus pumilus. Marine Drugs, 16(9), 326. https://doi.org/10.3390/md16090326