A Sterol from Soft Coral Induces Apoptosis and Autophagy in MCF-7 Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
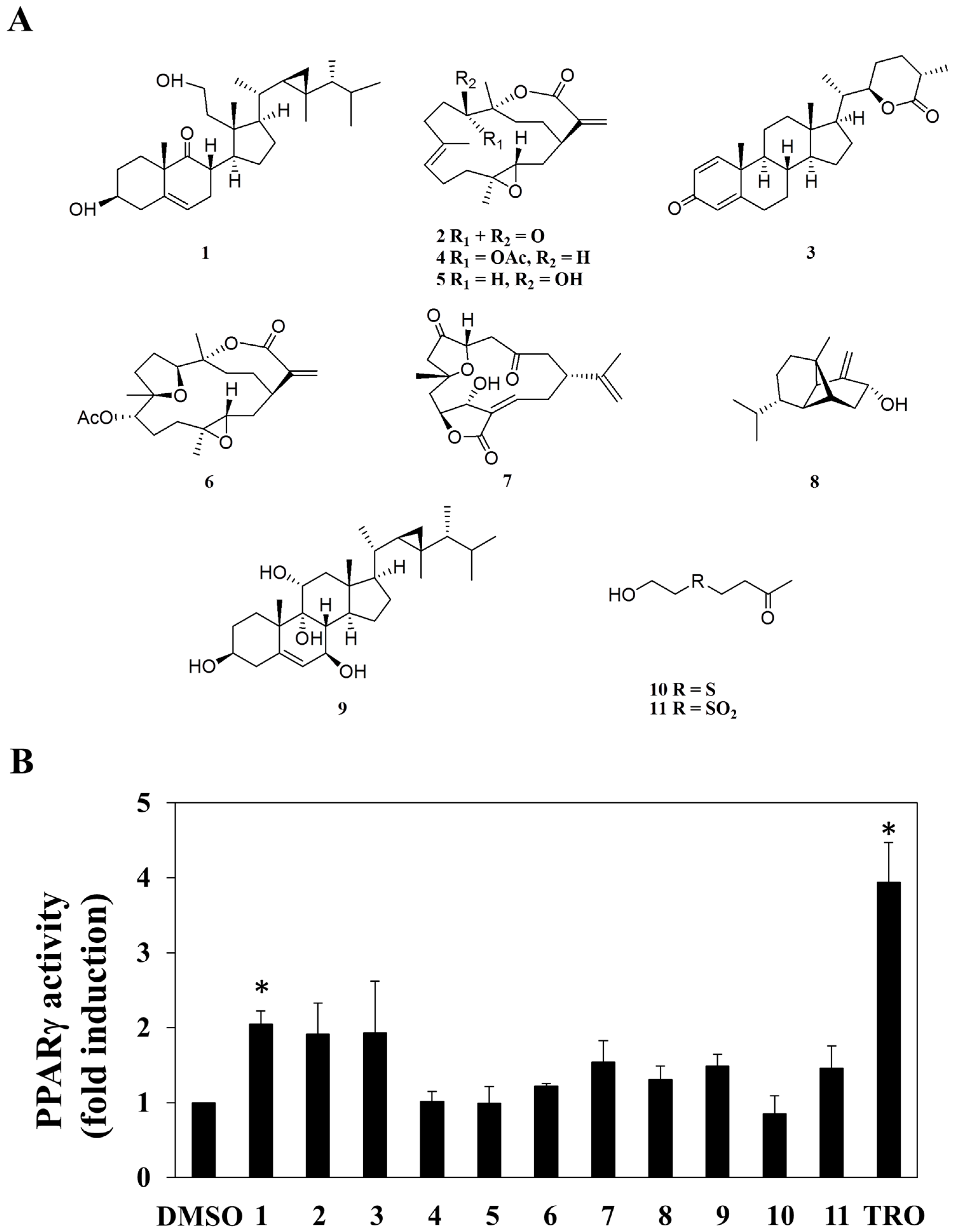
2.1. Screening for PPARγ Activators from a Small Marine Compound Library
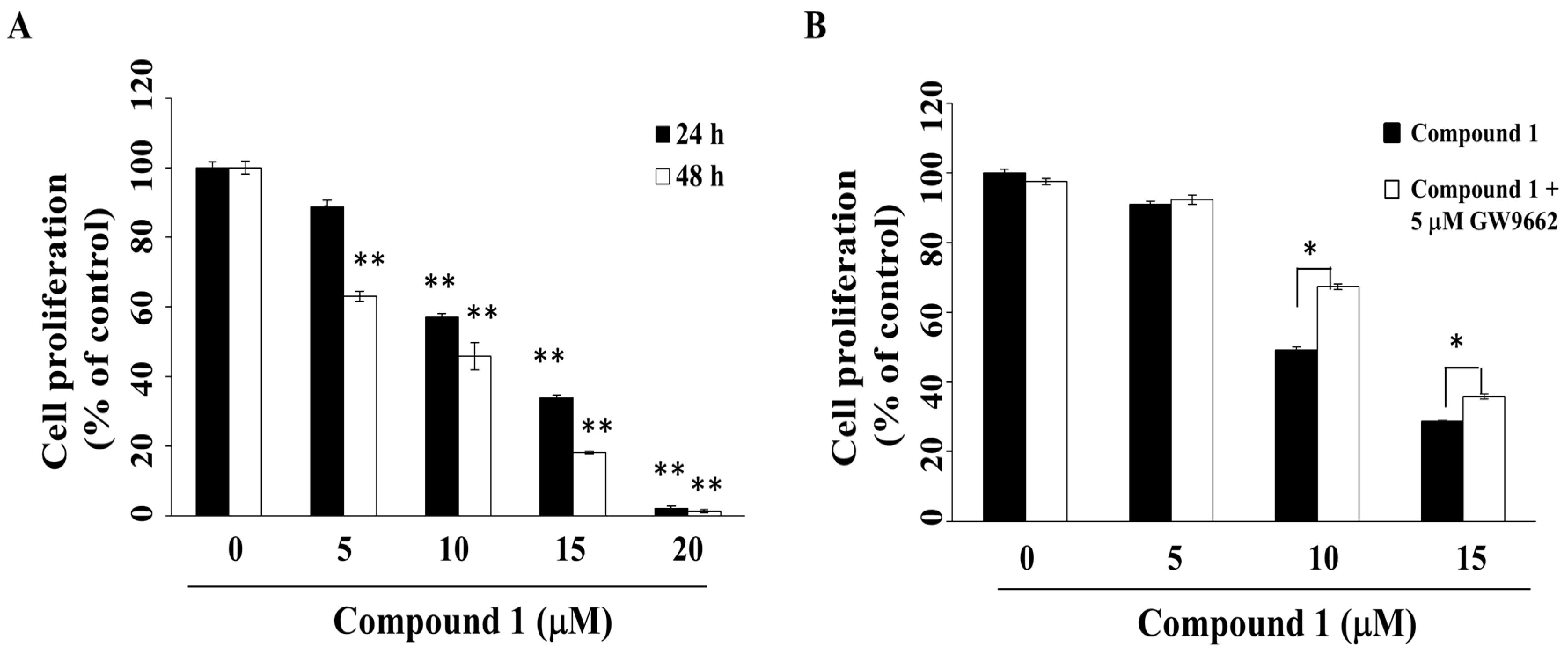
2.2. Compound 1 Inhibits Cell Growth in Part through PPARγ Activation
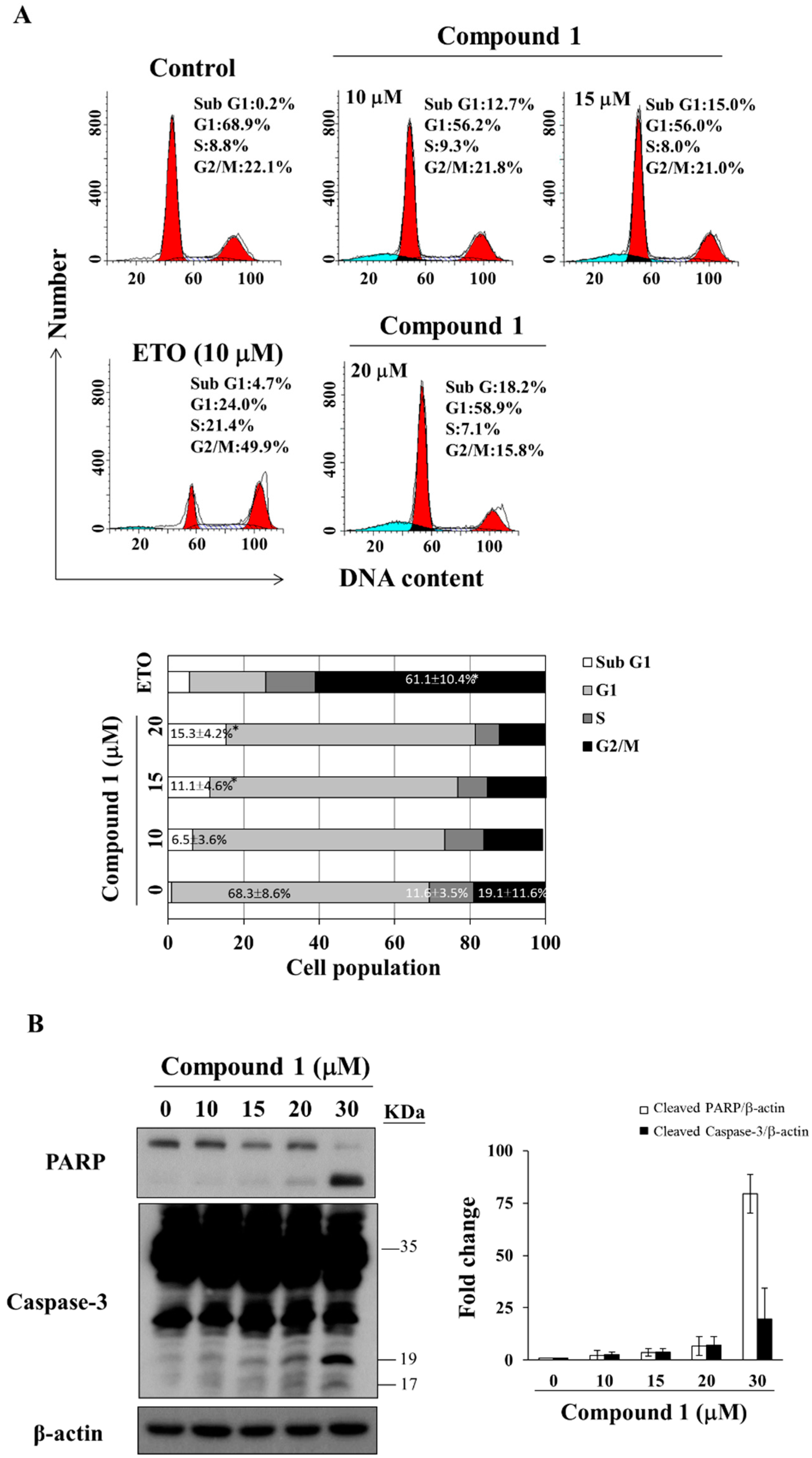
2.3. Compound 1 Induces Caspase-Dependent Apoptosis
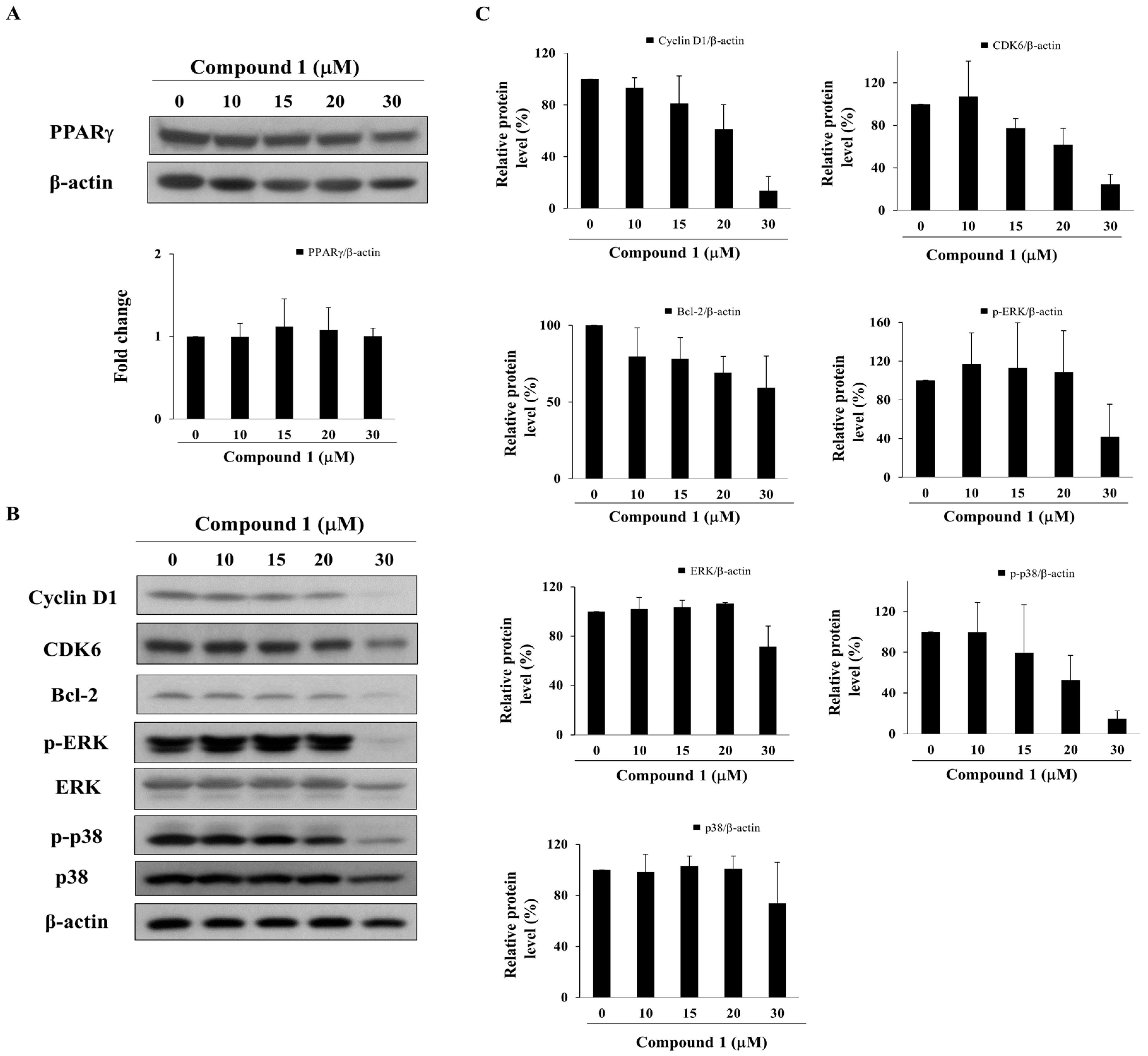
2.4. Compound 1 Upregulates the Expression of PPARγ Target Gene Products
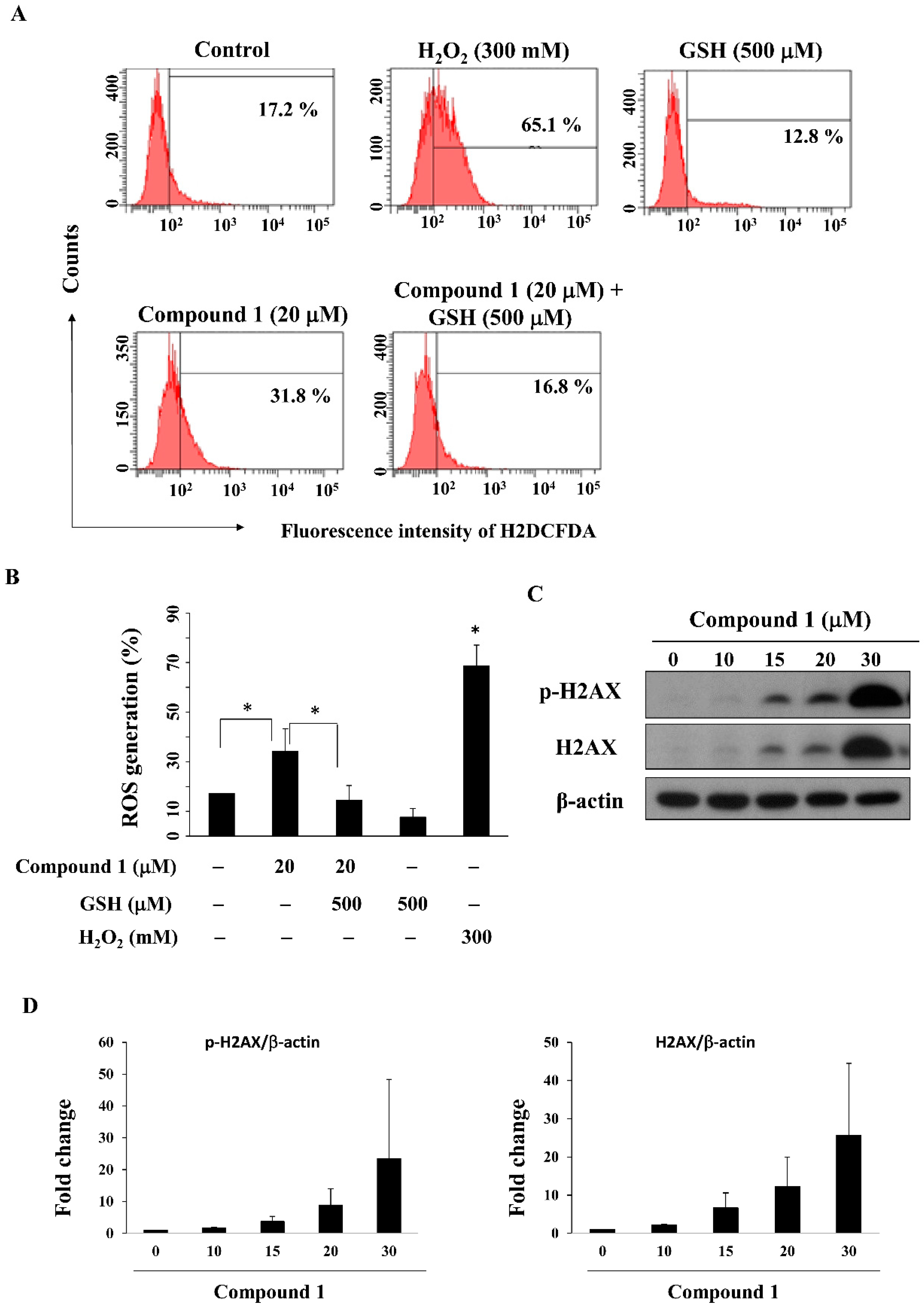
2.5. Compound 1 Increases Reactive Oxygen Species (ROS) Generation
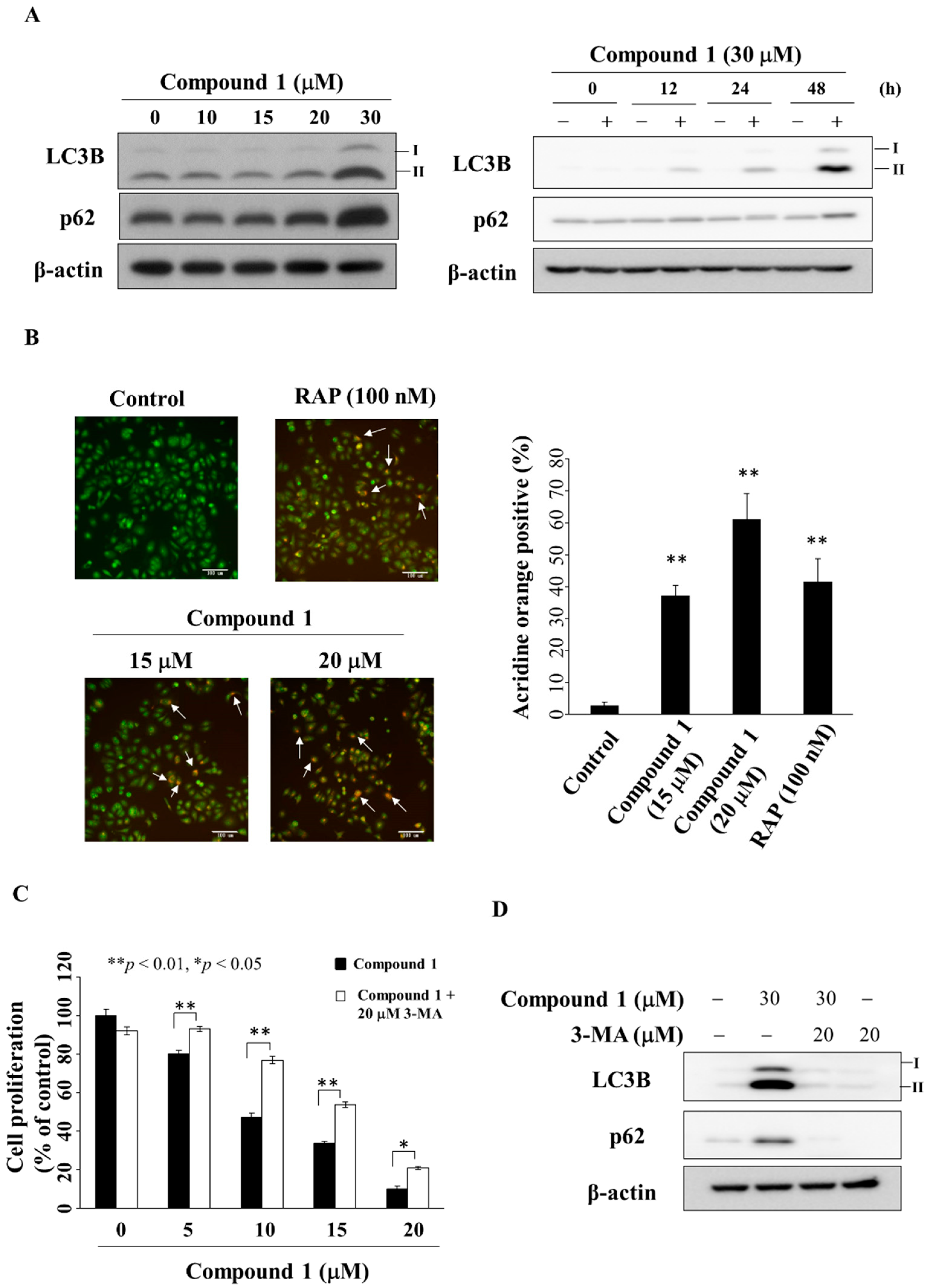
2.6. Compound 1 Induces Autophagy
3. Experimental Section
3.1. Marine Compound Library
3.2. Chemicals and Reagents
3.3. Cell Culture
3.4. Cell Viability
3.5. Flow Cytometry Analysis
3.6. Detection of Autophagosomes by Staining with Acridine Orange (AO)
3.7. Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Makarem, N.; Bandera, E.V.; Lin, Y.; Jacques, P.F.; Hayes, R.B.; Parekh, N. Carbohydrate nutrition and risk of adiposity-related cancers: Results from the Framingham Offspring cohort (1991–2013). Br. J. Nutr. 2017, 117, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Blunder, M.; Liu, X.; Malainer, C.; Blazevic, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor γ (PPARγ): A review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Phillip Koeffler, H. Peroxisome proliferator-activated receptor γ and cancers. Clin. Cancer Res. 2003, 9, 1–9. [Google Scholar]
- Yu, H.N.; Lee, Y.R.; Noh, E.M.; Lee, K.S.; Kim, J.S.; Song, E.K.; Han, M.K.; Lee, Y.C.; Kwon, K.B.; Lee, S.J.; et al. Induction of G(1) phase arrest and apoptosis in MDA-MB-231 breast cancer cells by troglitazone, a synthetic peroxisome proliferator-activated receptor γ (PPARγ) ligand. Cell Biol. Int. 2008, 32, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, Y.; Sano, H.; Kawahito, Y.; Mukai, S.; Yamada, R.; Kohno, M.; Inoue, K.; Hla, T.; Kondo, M. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor γ agonists through induction of apoptosis. Biochem. Biophys. Res. Commun. 2000, 270, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Y.; Liu, Y.; Bi, Z.Y. Pioglitazone inhibits growth of human retinoblastoma cells via regulation of NF-κB inflammation signals. J. Recept. Signal Transduct. Res. 2017, 37, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Chen, C.Y.; Pinzone, J.J.; Ringel, M.D.; Chen, C.S. Beyond peroxisome proliferator-activated receptor γ signaling: The multi-facets of the antitumor effect of thiazolidinediones. Endocr.-Relat. Cancer 2006, 13, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yang, J.; Lee, S.L.; Kulp, S.K.; Chen, C.S. PPARγ-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009, 276, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Colin-Cassin, C.; Yao, X.; Cerella, C.; Chbicheb, S.; Kuntz, S.; Mazerbourg, S.; Boisbrun, M.; Chapleur, Y.; Diederich, M.; Flament, S.; et al. PPARγ-inactive Δ2-troglitazone independently triggers ER stress and apoptosis in breast cancer cells. Mol. Carcinog. 2015, 54, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Akinyeke, T.O.; Stewart, L.V. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARγ-independent mechanism. Cancer Biol. Ther. 2011, 11, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Feldman, P.L.; Lambert, M.H.; Henke, B.R. PPAR modulators and PPAR pan agonists for metabolic diseases: The next generation of drugs targeting peroxisome proliferator-activated receptors? Curr. Top. Med. Chem. 2008, 8, 728–749. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. Potential remains for PPAR-targeted drugs. Nat. Rev. Drug Discov. 2010, 9, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.H.; Yeh, C.L.; Yeh, S.L.; Lin, E.S.; Wang, L.Y.; Wang, Y.H. Quercetin metabolites inhibit MMP-2 expression in A549 lung cancer cells by PPAR-γ associated mechanisms. J. Nutr. Biochem. 2016, 33, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Qu, J.; Yang, R.; Ge, M.X.; Mei, Y.; Zhou, B.T.; Qu, Q. Phytochemicals mediate the expression and activity of OCTN2 as activators of the PPARγ/RXRα pathway. Front. Pharmacol. 2016, 7, 189. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Ota, I.; Masui, T.; Itaya-Hironaka, A.; Shobatake, R.; Okamoto, H.; Takasawa, S.; Kitahara, T. Effect of resveratrol on cancer progression through the REG III expression pathway in head and neck cancer cells. Int. J. Oncol. 2016, 49, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Chen, L.; Kong, D.; Zhang, X.; Lu, C.; Lu, Y.; Zheng, S. Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J. Cell. Mol. Med. 2014, 18, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.R.; Huang, C.Y.; Chen, B.W.; Tsai, Y.Y.; Shih, S.P.; Hwang, T.L.; Dai, C.F.; Wang, S.Y.; Sheu, J.H. New bioactive steroids from the soft coral Klyxum flaccidum. RSC Adv. 2015, 5, 12546–12554. [Google Scholar] [CrossRef]
- Hsieh, P.W.; Chang, F.R.; McPhail, A.T.; Lee, K.H.; Wu, Y.C. New cembranolide analogues from the Formosan soft coral Sinularia flexibilis and their cytotoxicity. Nat. Prod. Res. 2003, 17, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.H.; Chou, K.J.; Wen, Z.H.; Wang, G.H.; Wu, Y.C.; Dai, C.F.; Sheu, J.H. Paraminabeolides A−F, cytotoxic and anti-inflammatory marine withanolides from the soft coral Paraminabea acronocephala. J. Nat. Prod. 2011, 74, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Chiu, S.J.; Tsai, Y.T.; Chang, C.M.; Wang, J.Y.; Wang, E.T.; Hou, M.F.; Huang, C.Y.; Sheu, J.H.; Chang, W.C. A soft coral natural product, 11-episinulariolide acetate, inhibits gene expression of cyclooxygenase-2 and interleukin-8 through attenuation of calcium signaling. Molecules 2013, 18, 7023–7034. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.J.; Tseng, Y.J.; Huang, C.Y.; Wen, Z.H.; Dai, C.F.; Sheu, J.H. Cytotoxic and anti-inflammatory diterpenoids from the Dongsha atoll soft coral Sinularia flexibilis. Tetrahedron 2012, 68, 244–249. [Google Scholar] [CrossRef]
- Tseng, Y.J.; Ahmed, A.F.; Dai, C.F.; Chiang, M.Y.; Sheu, J.H. Sinulochmodins A-C, three novel terpenoids from the soft coral Sinularia lochmodes. Org. Lett. 2005, 7, 3813–3816. [Google Scholar] [CrossRef] [PubMed]
- Jean, Y.H.; Chen, W.F.; Duh, C.Y.; Huang, S.Y.; Hsu, C.H.; Lin, C.S.; Sung, C.S.; Chen, I.M.; Wen, Z.H. Inducible nitric oxide synthase and cyclooxygenase-2 participate in anti-inflammatory and analgesic effects of the natural marine compound lemnalol from Formosan soft coral Lemnalia cervicorni. Eur. J. Pharmacol. 2008, 578, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.R.; Huang, C.Y.; Tsai, Y.Y.; Lin, Y.S.; Hwang, T.L.; Su, J.H.; Sung, P.J.; Dai, C.F.; Sheu, J.H. New cytotoxic and anti-inflammatory steroids from the soft coral Klyxum flaccidum. Bioorg. Med. Chem. Lett. 2016, 26, 3253–3257. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.H.; Chao, C.H.; Wu, M.H.; Sheu, J.H. A neuroprotective sulfone of marine origin and the in vivo anti-inflammatory activity of an analogue. Eur. J. Med. Chem. 2010, 45, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Bai, L.Y.; Lin, W.Y. Identification of a triterpenoid as a novel PPARγ activator derived from Formosan plants. Phytother. Res. 2017, 31, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, R.; Luo, B.; Yang, X.; Qiu, L.; Xiong, J.; Jiang, M.; Liu, Y.; Zhang, Z.; Wu, Y. Macrophage peroxisome proliferator-activated receptor γ deficiency delays skin wound healing through impairing apoptotic cell clearance in mice. Cell Death Dis. 2015, 6, e1597. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, L.H.; Huang, B.; Wang, R.Y.; Yuan, S.X.; Tao, Q.F.; Xu, Y.; Sun, H.Y.; Lin, C.; Zhou, W.P. Pioglitazone, a PPARγ agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol. Carcinog. 2015, 54, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Zhang, Y.; Chen, C.; Li, Y.Q.; Ma, C.; Wang, Z.J. Pioglitazone inhibits advanced glycation end product-induced matrix metalloproteinases and apoptosis by suppressing the activation of MAPK and NF-κB. Apoptosis 2016, 21, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Hasegawa, A.; Yamamori, M.; Okamura, N. In vitro and in vivo cytotoxicity of troglitazone in pancreatic cancer. J. Exp. Clin. Cancer Res. 2017, 36, 91. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Murch, O.; Thiemermann, C. Peroxisome proliferator-activated receptor γ antagonists GW9662 and T0070907 reduce the protective effects of lipopolysaccharide preconditioning against organ failure caused by endotoxemia. Crit. Care Med. 2006, 34, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Facompre, M.; Wattez, N.; Kluza, J.; Lansiaux, A.; Bailly, C. Relationship between cell cycle changes and variations of the mitochondrial membrane potential induced by etoposide. Mol. Cell Biol. Res. Commun. 2000, 4, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 2007, 282, 37006–37015. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Watanabe, K.; Date, M.; Daito, M.; Ohura, K. Possible involvement of p38 in mechanisms underlying acceleration of proliferation by 15-deoxy-Δ12,14-prostaglandin J2 and the precursors in leukemia cell line THP-1. J. Pharmacol. Sci. 2004, 94, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Osman, I.; Segar, L. Pioglitazone, a PPARγ agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/p70S6K and ERK signaling. Biochem. Pharmacol. 2016, 101, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Ahn, J.H.; Cheon, H.G. Apoptotic action of peroxisome proliferator-activated receptor-gamma activation in human non small-cell lung cancer is mediated via proline oxidase-induced reactive oxygen species formation. Mol. Pharmacol. 2007, 72, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Kollipara, R.K.; Singh, D.K.; Sudderth, J.; Hu, Z.; Nguyen, H.; Wang, S.; Humphries, C.G.; Carstens, R.; Huffman, K.E.; et al. Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell MeTable 2014, 20, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010, 704, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yang, X.; Chen, T.; Xi, Z.; Jiang, X. The PPARγ agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014, 21, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, Z.Y.; Simon, H.U. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin. Cancer Biol. 2013, 23, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Yang, Y.; Xu, K.; Koike, T.; Zheng, X. Transport of autophagosomes in neurites of PC12 cells during serum deprivation. Autophagy 2008, 4, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Chu, P.C.; Lin, W.Y.; Chiu, S.J.; Weng, J.R. A triterpenoid from wild bitter gourd inhibits breast cancer cells. Sci. Rep. 2016, 6, 22419. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Chiu, S.J.; Chu, P.C.; Weng, J.R. FTY720 induces autophagyassociated apoptosis in human oral squamous carcinoma cells, in part, through a reactive oxygen species/mcl-1-dependent mechanism. Sci. Rep. 2017, 7, 5600. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Avila-Roman, J.; Talero, E.; de Los Reyes, C.; Garcia-Maurino, S.; Motilva, V. Microalgae-derived oxylipins decrease inflammatory mediators by regulating the subcellular location of NFκB and PPAR-γ. Pharmacol. Res. 2017, 128, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, B.K.; Yang, H.H.; Sung, P.J.; Weng, C.F. Excavatolide B inhibits nonsmall cell lung cancer proliferation by altering peroxisome proliferator activated receptor gamma expression and PTEN/AKT/NF-κB expression. Environ. Toxicol. 2017, 32, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Kim, B.H.; Kim, Y.I.; Kim, K.Y.; Hwangbo, Y.; Jang, J.Y.; Dong, S.H.; Kim, H.J.; Chang, Y.W.; Chang, R. The peroxisome proliferator-activated receptor γ ligands, pioglitazone and 15-deoxy-Δ(12,14)-prostaglandin J(2), have antineoplastic effects against hepatitis B virus-associated hepatocellular carcinoma cells. Int. J. Oncol. 2010, 36, 223–231. [Google Scholar] [PubMed]
- Yang, C.C.; Wang, Y.C.; Wei, S.; Lin, L.F.; Chen, C.S.; Lee, C.C.; Lin, C.C.; Chen, C.S. Peroxisome proliferator-activated receptor γ-independent suppression of androgen receptor expression by troglitazone mechanism and pharmacologic exploitation. Cancer Res. 2007, 67, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Kruk, J.; Aboul-Enein, H.Y. Reactive oxygen and nitrogen species in carcinogenesis: Implications of oxidative stress on the progression and development of several cancer types. Mini-Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, G.J.; Yi, S.S.; Heo, S.H.; Park, C.R.; Nam, H.S.; Cho, M.K.; Lee, S.H. Cisplatin and resveratrol induce apoptosis and autophagy following oxidative stress in malignant mesothelioma cells. Food Chem. Toxicol. 2016, 97, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.C.; Hsiao, C.D.; Chen, W.M.; Wen, Y.S.; Lin, Y.C.; Chang, T.W.; Yao, F.Y.; Hung, S.C.; Wang, J.Y.; Chiu, J.H.; et al. Cytotoxic effects of 15d-PGJ2 against osteosarcoma through ROS-mediated AKT and cell cycle inhibition. Oncotarget 2014, 5, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Han, E.J.; Im, C.N.; Park, S.H.; Moon, E.Y.; Hong, S.H. Combined treatment with peroxisome proliferator-activated receptor (PPAR) γ ligands and γ radiation induces apoptosis by PPAR γ-independent up-regulation of reactive oxygen species-induced deoxyribonucleic acid damage signals in non-small cell lung cancer cells. Int. J. Radiat. Oncol. 2013, 85, e239–e248. [Google Scholar]
- Li, C.J.; Liao, W.T.; Wu, M.Y.; Chu, P.Y. New insights into the role of autophagy in tumor immune microenvironment. Int. J. Mol. Sci. 2017, 18, 1566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubín-de-Celis, S.; Zou, Z.J.; Fernandez, A.F.; Ci, B.; Kim, M.; Xiao, G.H.; Xie, Y.; Levine, B. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4176–4181. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Penault-Llorca, F.; Senovilla, L.; Dalban, C.; Enot, D.; Locher, C.; Prada, N.; Poirier-Colame, V.; Chaba, K.; Arnould, L.; et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy 2015, 11, 1878–1890. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, J.-R.; Chiu, C.-F.; Hu, J.-L.; Feng, C.-H.; Huang, C.-Y.; Bai, L.-Y.; Sheu, J.-H. A Sterol from Soft Coral Induces Apoptosis and Autophagy in MCF-7 Breast Cancer Cells. Mar. Drugs 2018, 16, 238. https://doi.org/10.3390/md16070238
Weng J-R, Chiu C-F, Hu J-L, Feng C-H, Huang C-Y, Bai L-Y, Sheu J-H. A Sterol from Soft Coral Induces Apoptosis and Autophagy in MCF-7 Breast Cancer Cells. Marine Drugs. 2018; 16(7):238. https://doi.org/10.3390/md16070238
Chicago/Turabian StyleWeng, Jing-Ru, Chang-Fang Chiu, Jing-Lan Hu, Chia-Hsien Feng, Chiung-Yao Huang, Li-Yuan Bai, and Jyh-Horng Sheu. 2018. "A Sterol from Soft Coral Induces Apoptosis and Autophagy in MCF-7 Breast Cancer Cells" Marine Drugs 16, no. 7: 238. https://doi.org/10.3390/md16070238
APA StyleWeng, J.-R., Chiu, C.-F., Hu, J.-L., Feng, C.-H., Huang, C.-Y., Bai, L.-Y., & Sheu, J.-H. (2018). A Sterol from Soft Coral Induces Apoptosis and Autophagy in MCF-7 Breast Cancer Cells. Marine Drugs, 16(7), 238. https://doi.org/10.3390/md16070238