Molecular Targets of Active Anticancer Compounds Derived from Marine Sources

Abstract
1. Introduction
2. Molecular Targets of Marine-Derived Anticancer Candidates
2.1. Targeting the Kinases Related to Cell Survival and Proliferation Signaling Pathway
2.1.1. Protein Kinase C (PKC)
2.1.2. Insulin-Like Growth Factor-1 Receptor (IGF-1R)
2.1.3. Cyclin-Dependent Kinases (CDKs)
2.1.4. Glycogen Synthase Kinase-3 Beta (GSK-3β)
2.1.5. Multi-Target Inhibitors of Receptor Tyrosine Kinases
2.2. Targeting Transcription Factors Related to Cancer Gene Expression
2.3. Targeting Histone Deacetylases Related to Epigenetic Regulation of Cancer
2.4. Targeting Proteasome and Deubiquitylating Enzymes Related to Oncoprotein Degradation
2.5. Targeting the Heat Shock Protein (Hsp90) Related to Cancer Oncoprotein Maturity
2.6. Targeting P-gp, Patched, and PXR Related to the Cancer Multidrug Resistance
2.7. Compounds Targeting Other Cancer Related Molecules
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ruiz-Torres, V.; Encinar, J.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An Updated Review on Marine Anticancer Compounds: The Use of Virtual Screening for the Discovery of Small-Molecule Cancer Drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pomponi, S.A. The bioprocess–technological potential of the sea. J. Biotechnol. 1999, 70, 5–13. [Google Scholar] [CrossRef]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. 3.06-Nature as Source of Medicines; Novel Drugs from Nature; Screening for Antitumor Activity. In Comprehensive Natural ProductsⅡ; Elsevier: Oxford, UK, 2010; pp. 135–175. [Google Scholar]
- Mayer, A.M.S.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2012–2013: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-inflammatory, Antiprotozoal, Antituberculosis, and Antiviral activities; Affecting the Immune and Nervous Systems, and Other Miscellaneous Mechanisms of Action. Mar. Drugs 2017, 15, 273. [Google Scholar]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
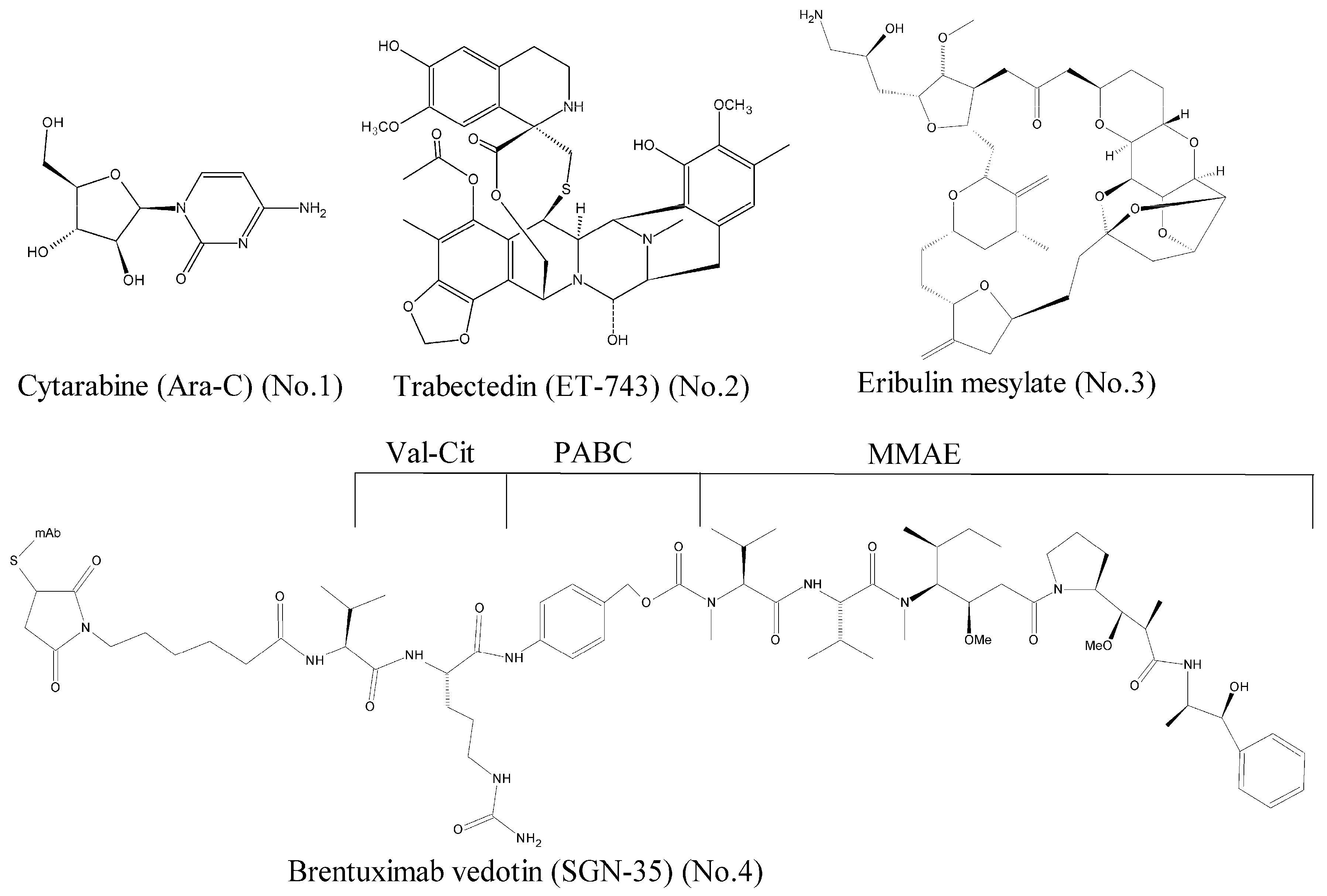
- Hunnisett-Dritz, D. Successful importation of cytarabine into the United States during a critical national drug shortage. Am. J. Health Syst. Pharm. 2012, 69, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Hickford, S.J.H.; Blunt, J.W.; Munro, M.H.G. Antitumour polyether macrolides: Four new halichondrins from the New Zealand deep-water marine sponge Lissodendoryx sp. Bioorg. Med. Chem. 2009, 17, 2199–2203. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase inhibitors from marine sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [PubMed]
- Sako, T.; Tauber, A.I.; Jeng, A.Y.; Yuspa, S.H.; Blumberg, P.M. Contrasting actions of staurosporine, a protein kinase C inhibitor, on human neutrophils and primary mouse epidermal cells. Cancer Res. 1988, 48, 4646–4650. [Google Scholar] [PubMed]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef] [PubMed]
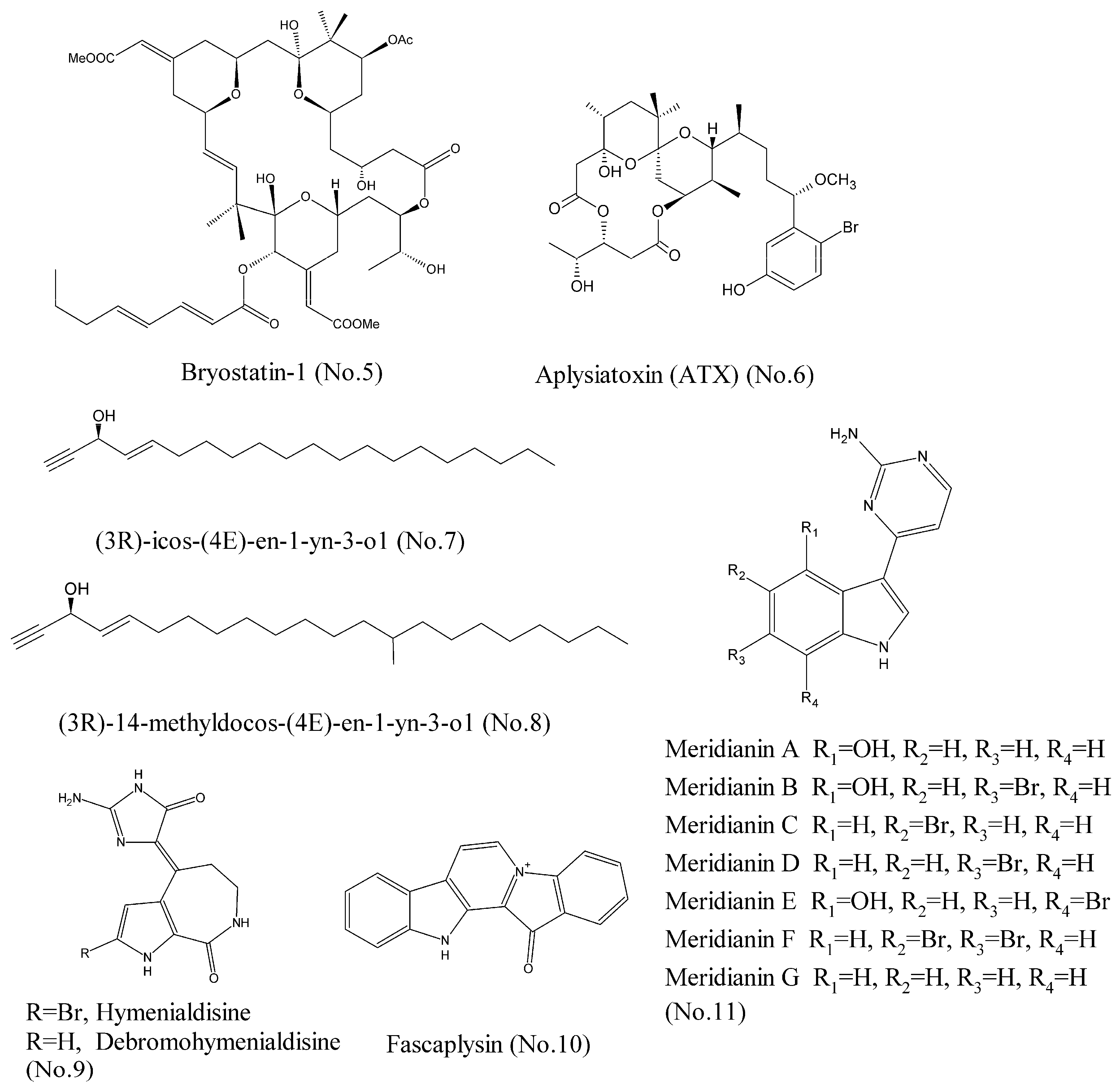
- Khan, T.K.; Nelson, T.J. Protein kinase C activator bryostatin-1 modulates proteasome function. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Nakagawa, Y.; Near, K.E.; Staveness, D. Computer-guided design, synthesis, and protein kinase C affinity of a new salicylate-based class of bryostatin analogs. Org. Lett. 2014, 16, 5136–5139. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Yanagita, R.C. Synthesis and biological activities of simplified analogs of the natural PKC ligands, bryostatin-1 and aplysiatoxin. Chem. Rec. 2014, 14, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Ashida, Y.; Yanagita, R.C.; Takahashi, C.; Kawanami, Y.; Irie, K. Binding mode prediction of aplysiatoxin, a potent agonist of protein kinase C, through molecular simulation and structure-activity study on simplified analogs of the receptor-recognition domain. Bioorg. Med. Chem. 2016, 24, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Amin, H.; Rah, B.; Goswami, A. Targeting EGFR and IGF 1R: A promising combination therapy for metastatic cancer. Front. Biosci. (Sch. Ed.) 2013, 5, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Novello, S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat. Rev. 2012, 38, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Fidler, M.J.; Shersher, D.D.; Borgia, J.A.; Bonomi, P. Targeting the insulin-like growth factor receptor pathway in lung cancer: Problems and pitfalls. Ther. Adv. Med. Oncol. 2012, 4, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Zovko, A.; Novak, M.; Hååg, P.; Kovalerchick, D.; Holmlund, T.; Färnegårdh, K.; Ilan, M.; Carmeli, S.; Lewensohn, R.; Viktorsson, K. Compounds from the marine sponge Cribrochalina vasculum offer a way to target IGF-1R mediated signaling in tumor cells. Oncotarget 2016, 7, 50258–50276. [Google Scholar] [CrossRef] [PubMed]
- Zovko, A.; Viktorsson, K.; Hååg, P.; Kovalerchick, D.; Färnegårdh, K.; Alimonti, A.; Ilan, M.; Carmeli, S.; Lewensohn, R. Marine sponge Cribrochalina vasculum compounds activate intrinsic apoptotic signaling and inhibit growth factor signaling cascades in non–small cell lung carcinoma. Mol. Cancer Ther. 2014, 13, 2941–2954. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yan, X.-J.; Chen, H.-M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2006, 59, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Sauleau, P.; Retailleau, P.; Nogues, S.; Carletti, I.; Marcourt, L.; Raux, R.; Mourabit, A.A.; Debitus, C. Dihydrohymenialdisines, new pyrrole-2-aminoimidazole alkaloids from the marine sponge Cymbastela cantharella. Tetrahedron Lett. 2011, 52, 2676–2678. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, R.A. Meridianins: Marine-derived potent kinase inhibitors. Mini Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-Aided Drug Design Applied to Marine Drug Discovery: Meridianins as Alzheimer’s Disease Therapeutic Agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
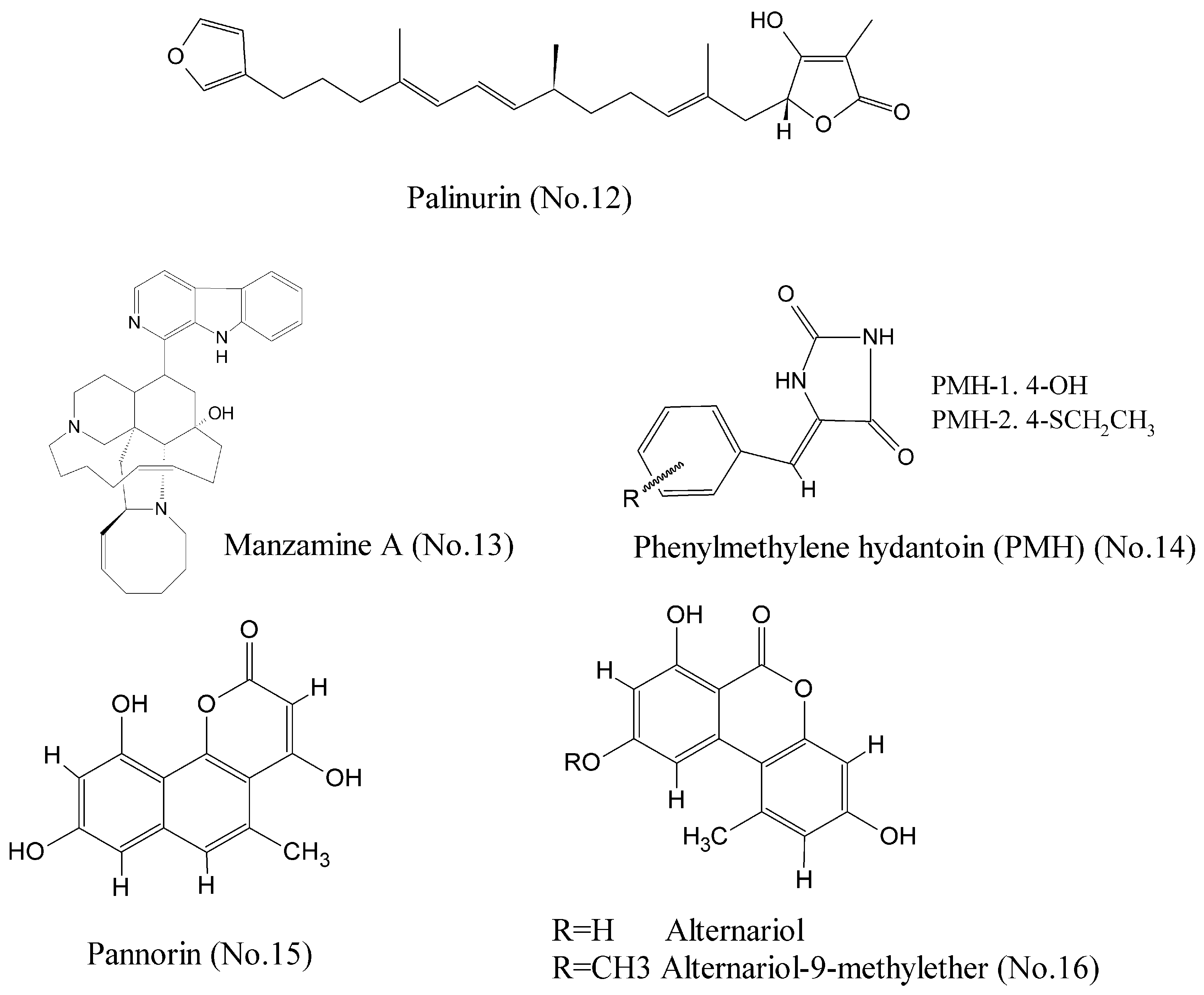
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; del Monte-Millán, M.; Medina, M.; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.A.; Johnson, J.D.; Linley, P.A.; Gunasekera, S.E.; Wright, A.E. A novel activity from an old compound: Manzamine a reduces the metastatic potential of AsPC-1 pancreatic cancer cells and sensitizes them to trail-induced apoptosis. Investig. New Drugs 2011, 29, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Sallam, A.A.; Mohyeldin, M.M.; Foudah, A.I.; Akl, M.R.; Nazzal, S.; Meyer, S.A.; Liu, Y.-Y.; El Sayed, K.A. Marine natural products-inspired phenylmethylene hydantoins with potent in vitro and in vivo antitumor activities via suppression of brk and fak signaling. Org. Biomol. Chem. 2014, 12, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Wiese, J.; Imhoff, J.; Gulder, T.; Labes, A.; Schmaljohann, R. Marine Fungi as Producers of Benzocoumarins, a New Class of Inhibitors of Glycogen-Synthase-Kinase 3β. Mar. Drugs 2016, 14, 200. [Google Scholar] [CrossRef] [PubMed]
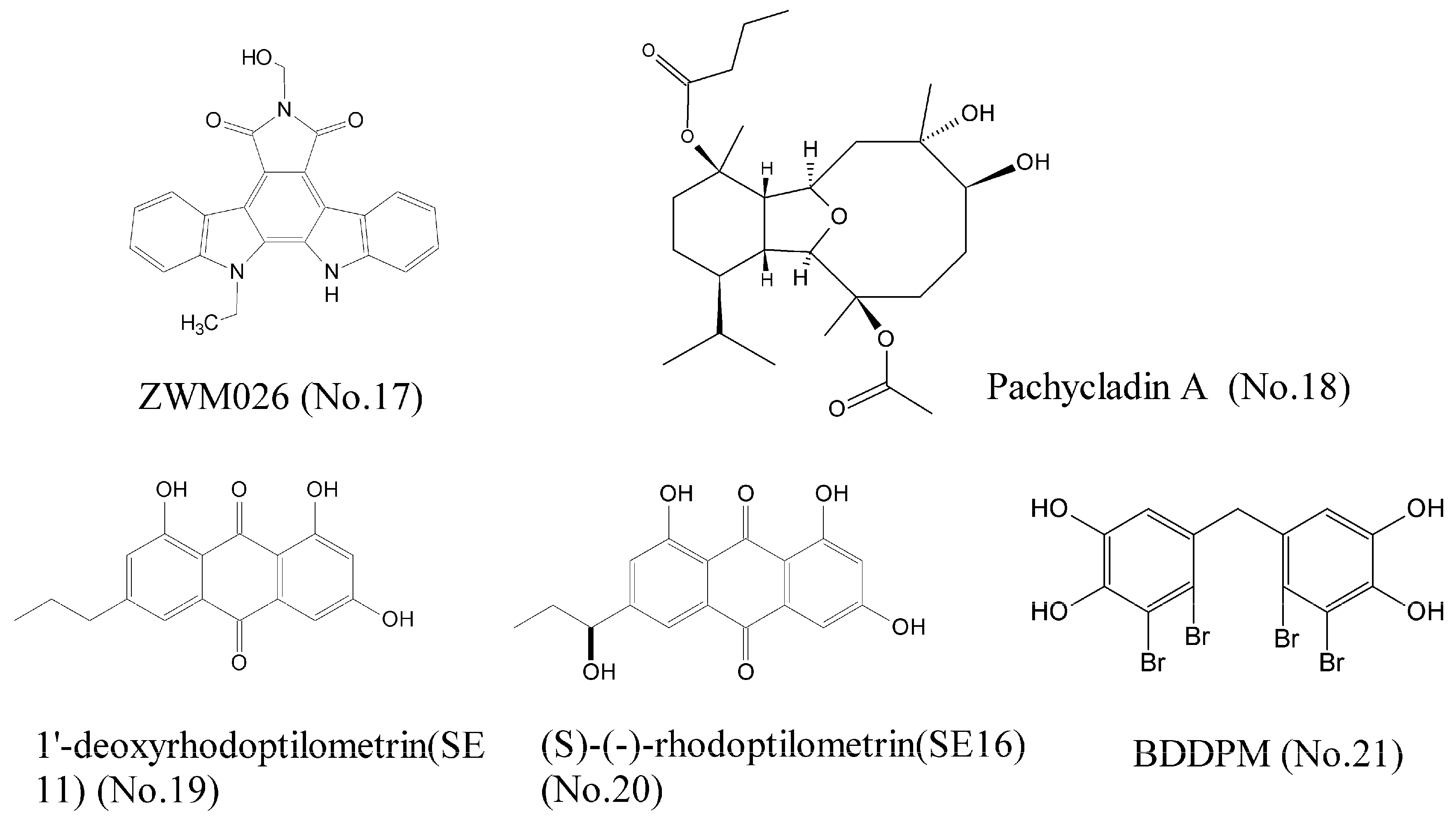
- Song, X.; Qi, X.; Wang, Q.; Zhu, W.; Li, J. A novel multi-target inhibitor harboring selectivity of inhibiting egfr T790M sparing wild-type EGFR. Am. J. Cancer Res. 2017, 7, 1884–1898. [Google Scholar] [PubMed]
- Mohyeldin, M.M.; Akl, M.R.; Siddique, A.B.; Hassan, H.M.; El Sayed, K.A. The marine-derived pachycladin diterpenoids as novel inhibitors of wild-type and mutant EGFR. Biochem. Pharmacol. 2017, 126, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, W.; Ebada, S.S.; Bergermann, A.; Chovolou, Y.; Totzke, F.; Kubbutat, M.H.G.; Lin, W.; Proksch, P. Cytotoxic effects of the anthraquinone derivatives 1′-deoxyrhodoptilometrin and (S)-(-)-rhodoptilometrin isolated from the marine echinoderm Comanthus sp. Arch. Toxicol. 2017, 91, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.-J.; Jiang, B.; Wu, N.; Li, X.; Liu, S.; Luo, J.; Shi, D. Anti-Angiogenic Properties of BDDPM, a Bromophenol from Marine Red Alga Rhodomela confervoides, with Multi Receptor Tyrosine Kinase Inhibition Effects. Int. J. Mol. Sci. 2015, 16, 13548–13560. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Luo, J.; Jiang, B.; Wang, L.; Wang, S.; Wang, C.; Fu, C.; Li, J.; Shi, D. Marine bromophenol bis (2,3-dibromo-4,5-dihydroxy-phenyl)-methane inhibits the proliferation, migration, and invasion of hepatocellular carcinoma cells via modulating β1-integrin/FAK signaling. Mar. Drugs 2015, 13, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Shin, H.-W.; Chun, Y.-S.; Leutou, A.S.; Son, B.W.; Park, J.-W. Diacetoxyscirpenol as a new anticancer agent to target hypoxia-inducible factor 1. Oncotarget 2016, 7, 62107–62122. [Google Scholar] [CrossRef] [PubMed]
- Goey, A.K.L.; Chau, C.H.; Sissung, T.M.; Cook, K.M.; Venzon, D.J.; Castro, A.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; McMahon, J.B.; et al. Screening and Biological Effects of Marine Pyrroloiminoquinone Alkaloids: Potential Inhibitors of the HIF-1α/p300 Interaction. J. Nat. Prod. 2016, 79, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Deb, S.; Muñoz, R.M.; Subler, M.A.; Deb, S.P. The tumor suppressor p53 and the oncoprotein simian virus 40 T antigen bind to overlapping domains on the MDM2 protein. Mol. Cell. Biol. 1993, 13, 6849–6857. [Google Scholar] [CrossRef] [PubMed]
- Malloy, K.L.; Choi, H.; Fiorilla, C.; Valeriote, F.A.; Matainaho, T.; Gerwick, W.H. Hoiamide D, a marine cyanobacteria-derived inhibitor of p53/MDM2 interaction. Bioorg. Med. Chem. Lett. 2012, 22, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Pande, V.; Sharma, R.K.; Inoue, J.-I.; Otsuka, M.; Ramos, M.J. A molecular modeling study of inhibitors of nuclear factor kappa-B (p50)—DNA binding. J. Comput. Aided Mol. Des. 2003, 17, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Folmer, F.; Jaspars, M.; Solano, G.; Cristofanon, S.; Henry, E.; Tabudravu, J.; Black, K.; Green, D.H.; Küpper, F.C.; Aalbersberg, W.; et al. The inhibition of TNF-α-induced NF-κB activation by marine natural products. Biochem. Pharmacol. 2009, 78, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Issa, J.-P.J. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol. Cell. Biol. 2003, 23, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Benedetti, R.; Pérez-Rodríguez, S.; Nebbioso, A.; García-Rodríguez, J.; Carafa, V.; Stuhldreier, M.; Conte, M.; Rodríguez-Barrios, F.; Stunnenberg, H.G.; et al. Indole-derived psammaplin A analogues as epigenetic modulators with multiple inhibitory activities. J. Med. Chem. 2012, 55, 9467–9491. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.J.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-Y.; Wang, J.-D.; Wang, X.; Liu, H.-C.; Zhang, M.-M.; Liu, Y.-C.; Zhang, C.-H.; Su, Y.; Shen, Y.-Y.; Guo, Y.-W.; et al. Marine-derived chromopeptide A, a novel class Ⅰ HDAC inhibitor, suppresses human prostate cancer cell proliferation and migration. Acta Pharmacol. Sin. 2017, 38, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.-P.; Lee, M.-G.; El-Shazly, M.; Juan, Y.-S.; Wen, Z.-H.; Du, Y.-C.; Su, J.-H.; Sung, P.-J.; Chen, Y.-C.; Yang, J.-C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Matsunaga, K.; Saito, M.; Hagiya, S.; Furukawa, K.-I.; Nakamura, H.; Ohizumi, Y. Halenaquinone, a novel phosphatidylinositol 3-kinase inhibitor from a marine sponge, induces apoptosis in pc12 cells. Eur. J. Pharmacol. 2001, 413, 37–45. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Sloss, C.M.; Wang, F.; Liu, R.; Xia, L.; Houston, M.; Ljungman, D.; Palladino, M.A.; Cusack, J.C. Proteasome inhibition activates epidermal growth factor receptor (EGFR) and EGFR-independent mitogenic kinase signaling pathways in pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 5116–5123. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S.; Eustáquio, A.S.; McGlinchey, R.P. Advances in and applications of proteasome inhibitors. Curr. Opin. Chem. Biol. 2008, 12, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Wu, Q.-H.; Rowley, D.C.; Al-Kareef, A.M.Q.; Wang, H. Anticancer agent-based marine natural products and related compounds. J. Asian Nat. Prod. Res. 2015, 17, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide natural products: Potent 20 S proteasome inhibitors as promising cancer chemotherapeutics. Angew. Chem. Int. Ed. Engl. 2010, 49, 9346–9367. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Lam, K.S. Generating a generation of proteasome inhibitors: From microbial fermentation to total synthesis of salinosporamide a (marizomib) and other salinosporamides. Mar. Drugs 2010, 8, 835–880. [Google Scholar] [CrossRef] [PubMed]
- Gullo, V.P.; McAlpine, J.; Lam, K.S.; Baker, D.; Petersen, F. Drug discovery from natural products. J. Ind. Microbiol. Biotechnol. 2006, 33, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.S.; Gerwick, W.H. The carmaphycins: New proteasome inhibitors exhibiting an α,β-epoxyketone warhead from a marine cyanobacterium. ChemBioChem 2012, 13, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Bi, C.; Schmitt, S.M.; Li, X.I.N.; Fan, Y.; Zhang, N.A.N.; Dou, Q.P. Metal-based 2,3-indolinedione derivatives as proteasome inhibitors and inducers of apoptosis in human cancer cells. Int. J. Mol. Med. 2014, 34, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Turcu, F.E.R.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, K.; Anchoori, R.; Iizuka, Y.; Meints, J.; MacNeill, L.; Vogel, R.I.; Orlowski, R.Z.; Lee, M.K.; Roden, R.B.S.; Bazzaro, M. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses. Clin. Cancer Res. 2014, 20, 3174–3186. [Google Scholar] [CrossRef] [PubMed]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Suresh Kumar, K.G. The multifaceted roles of USP7: New therapeutic opportunities. Cell Biochem. Biophys. 2011, 60, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Miyazaki, M.; Kodrasov, M.P.; Rotinsulu, H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Yokosawa, H.; Nicholson, B.; Tsukamoto, S. Spongiacidin C, a pyrrole alkaloid from the marine sponge stylissa massa, functions as a USP7 inhibitor. Bioorg. Med. Chem. Lett. 2013, 23, 3884–3886. [Google Scholar] [CrossRef] [PubMed]
- Afifi, A.H.; Kagiyama, I.; El-Desoky, A.H.; Kato, H.; Mangindaan, R.E.P.; de Voogd, N.J.; Ammar, N.M.; Hifnawy, M.S.; Tsukamoto, S.; Sulawesins, A.-C. Furanosesterterpene Tetronic Acids That Inhibit USP7, from a Psammocinia sp. Marine Sponge. J. Nat. Prod. 2017, 80, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhao, Z.; Qi, X.; Tang, S.; Wang, Q.; Zhu, T.; Gu, Q.; Liu, M.; Li, J. Identification of epipolythiodioxopiperazines HDN-1 and chaetocin as novel inhibitor of heat shock protein 90. Oncotarget 2015, 6, 5263–5274. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-H.; Liu, Y.-C.; Su, J.-H.; El-Shazly, M.; Wu, C.-F.; Du, Y.-C.; Hsu, Y.-M.; Yang, J.-C.; Weng, M.-K.; Chou, C.-H.; et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase Ⅱ and HSP90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, P.; Lovchik, M.A.; Li, Y.; Tang, L.; Chen, Z.; Zeng, R.; Ma, D.; Yuan, J.; Yu, Q. Cyclodepsipeptide toxin promotes the degradation of HSP90 client proteins through chaperone-mediated autophagy. J. Cell Biol. 2009, 185, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Lopez, D.; Martinez-Luis, S. Marine natural products as breast cancer resistance protein inhibitors. Mar. Drugs 2015, 13, 2010–2029. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; El Sayed, K.; Chen, Z.-S.; Guo, H. Current status on marine products with reversal effect on cancer multidrug resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.-P.; Khanfar, M.A.; Kuang, Y.; Dai, C.-L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of p-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Tanaka, H.; Nagayoshi, Y.; Nakashima, H.; Tsutsumi, K.; Ohtsuka, T.; Takahata, S.; Tanaka, M.; Okada, H. Targeting the hedgehog signaling pathway with interacting peptides to Patched-1. J. Gastroenterol. 2012, 47, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, L.; Tribalat, M.-A.; Sauvard, L.; Cazareth, J.; Lalli, E.; Broutin, I.; Thomas, O.P.; Mus-Veteau, I. Natural paniceins from mediterranean sponge inhibit the multidrug resistance activity of Patched and increase chemotherapy efficiency on melanoma cells. Oncotarget 2015, 6, 22282–22297. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Hodnik, Ž.; Peterlin Mašič, L.; Tomašić, T.; Smodiš, D.; D’Amore, C.; Fiorucci, S.; Kikelj, D. Bazedoxifene-scaffold-based mimetics of solomonsterols A and B as novel pregnane X receptor antagonists. J. Med. Chem. 2014, 57, 4819–4833. [Google Scholar] [CrossRef] [PubMed]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; Fiorucci, S.; et al. 4-Methylenesterols from Theonella swinhoei sponge are natural pregnane-X-receptor agonists and farnesoid-X-receptor antagonists that modulate innate immunity. Steroids 2012, 77, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ogawa, H.; Ohno, O.; Yamori, T.; Suenaga, K.; Toyoshima, C. Biselyngbyasides, cytotoxic marine macrolides, are novel and potent inhibitors of the Ca2+ pumps with a unique mode of binding. FEBS Lett. 2015, 589, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, J.; Kedzior, M.; Guimarães, L.; Ross, A.B.; Peters, T.L.; Ambrose, A.J.; Schmidlin, C.J.; Zhang, D.D.; Costa-Lotufo, L.V.; Rodríguez, A.D.; et al. ATP-competitive, marine derived natural products that target the DEAD box helicase, eIF4A. Bioorg. Med. Chem. Lett. 2017, 27, 4082–4085. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Kalinovsky, A.I.; Shubina, L.K.; Bokemeyer, C.; Stonik, V.A.; Honecker, F. Mycalamide a shows cytotoxic properties and prevents egf-induced neoplastic transformation through inhibition of nuclear factors. Mar. Drugs 2012, 10, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Gürel, G.; Blaha, G.; Steitz, T.A.; Moore, P.B. Structures of triacetyloleandomycin and mycalamide a bind to the large ribosomal subunit of haloarcula marismortui. Antimicrob. Agents Chemother. 2009, 53, 5010–5014. [Google Scholar] [CrossRef] [PubMed]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Pörzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound Name | Marine Organism | Chemical Class | Molecular Target | Cancer Type/Cell lines | Refs. |
|---|---|---|---|---|---|---|
| 5 | Bryostatin-1 | bryozoan | oxygenated macrolide | PKC activator | sarcoma, melanoma, ovaria, cervical, neck and head carcinoma, esophageal, gastric, pancreatic, renal cell carcinoma, leukemia cells | [16,17,18] |
| 6 | Aplysiatoxin (ATX) | sea hare and cyanobacteria | polyacetate | PKC activator | Leukemia cell, breast cancer cell | [19] |
| 7,8 | (3R)-icos-(4E)-en-1-yn-3-ol and (3R)-14-methyldocos-(4E)-en-1-yn-3-ol | marine sponge | acetylene alcohols | IGF-1Rβ | NSCLC cells | [23,24] |
| 9 | Hymenialdisine and Debromohymenialdisine | marine sponge | pyrrole-2-aminoimidazole alkaloids | CDK1, CDK2, CDK5, | colon carcinoma cell lines LoVo and Caco-2 | [26,27] |
| 10 | Fascaplysin | marine sponge | carboline class alkaloid | CDK4 | osteosarcoma U2OS, colon carcinoma cell HCT116 | [25,28] |
| 11 | Meridianin A-G | marine tunicate | indole alkaloids | CDK1, CDK5 | / | [29,30] |
| 12 | Palinurin | marine sponge | linear furanosesquiterpene | GSK-3β | human neuroblastoma cells SH-SY5Y | [32] |
| 13 | Manzamine A | marine sponge | alkaloid | GSK-3β | pancreatic cancer cell | [33,34] |
| 14 | PMH-1 and PMH-2 | marine sponge | cyclic imide hydantoins | GSK-3β | prostate cancer cell | [35] |
| 15 | Pannorin | marine fungi | oxygenated benzocoumarin core | GSK-3β | / | [36] |
| 16 | Alternariol, and Alternariol-9-methylether | marine fungi | oxygenated benzocoumarin core | GSK-3β | / | [36] |
| 17 | ZWM026 | mangrove | indolocarbazoles | EGFR-T790M, ErbB2, ErbB3, ErbB4, and RET | lung cancer cells | [37] |
| 18 | Pachycladin A | Red Sea soft coral | diterpenoids | EGFR and PKC | breast cancer cell lines, cervical cancer HeLa cells | [38] |
| 19,20 | 1’-deoxyrhodoptilometrin (SE11) and (S)-(−)-rhodoptilometrin (SE16) | marine echinoderm | anthraquinone | IGF-1R, FAK, EGFR, ErbB2, and ErbB4 | glioma and colon carcinoma | [39] |
| 21 | BDDPM | marine red alga | bromophenol | FGFR2,3,VEGFR2,PDGFRα,PKB/Akt,eNOS | hepatoma carcinoma cell | [40,41] |
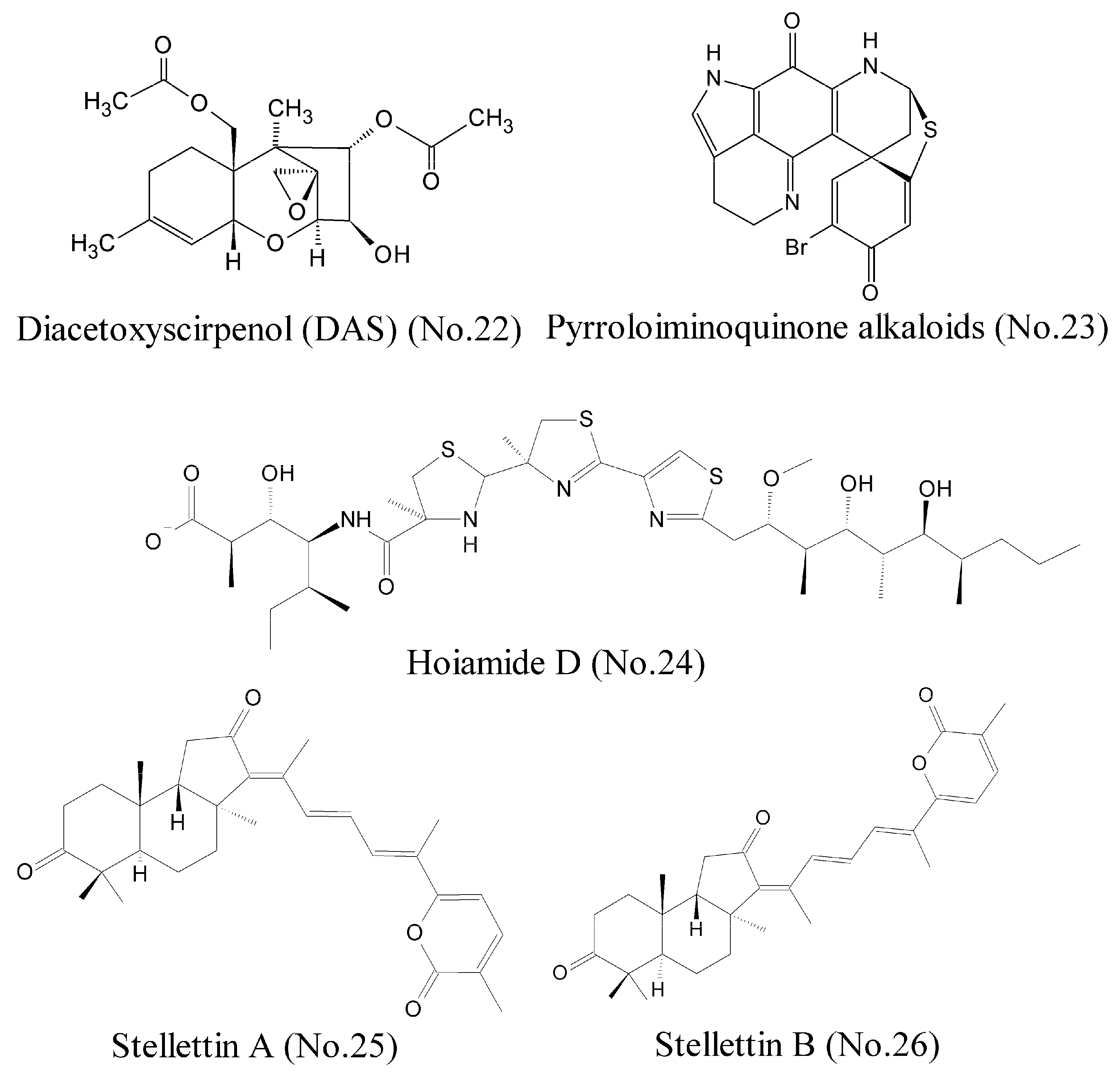
| 22 | Diacetoxyscirpenol (DAS) | marine red alga bacterium | enol | HIF-1α | lung cancer cell lines A549 | [43] |
| 23 | Pyrroloiminoquinone alkaloids | marine sponge | alkaloids | HIF-1α/p300 | colon and prostatic carcinoma | [44] |
| 24 | Hoiamide D | marine cyanobacteria | polyketide | p53/MDM2 | lung cell lines H460 | [46] |
| 25,26 | Stellettin A and Stellettin B | marine sponge | triterpenoids | p50/p65 | Leukemia cell line K562 | [48] |
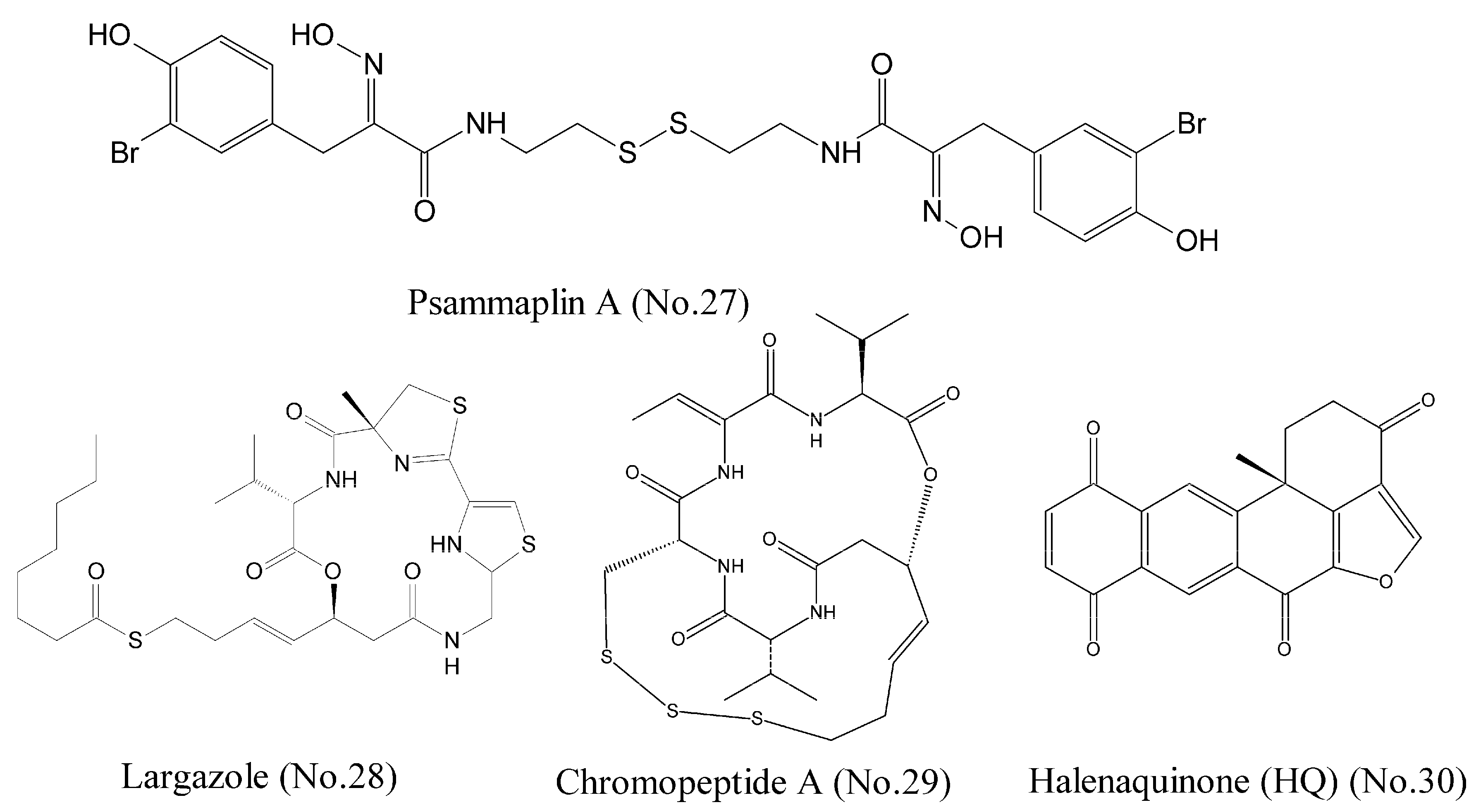
| 27 | Psammaplin A | marine sponge | indole | HDAC1 | lung, breast cancer cell lines | [50,51] |
| 28 | Largazole | marine cyanobacterium | cyclic depsipeptide | HDAC1 | colon cancer cell lines HCT116 | [52] |
| 29 | Chromopeptide A | marine bacterium | depsipeptide | HDAC1,2,3,8 | prostate cancer cell lines PC3 | [53] |
| 30 | Halenaquinone (HQ) | marine sponge | polycyclic quinone-type | HDACs | Molt 4, K562, MDA-MB-231, and DLD-1 cell lines | [54,55] |
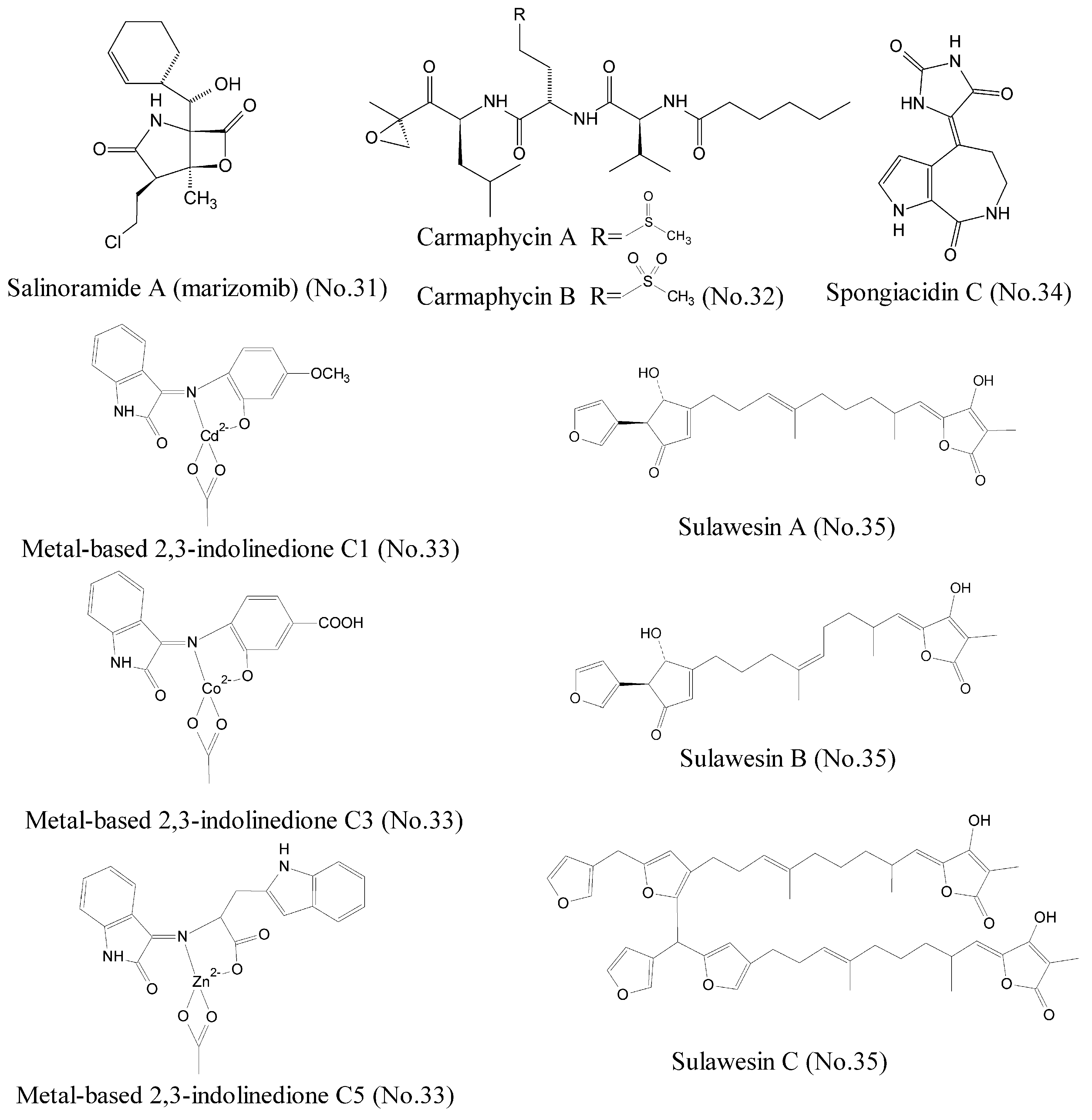
| 31 | Salinosporamide A | marine actinomycete bacteria | γ-lactam-β-lactone bicyclic core | 20S proteasome | melanoma, pancreatic carcinoma, or NSCLC | [59,60,61,62] |
| 32 | Carmaphycin A and carmaphycin B | marine cyanobacteria | leucine-derived α,β-epoxyketone | proteasome | lung and colon cancer cell lines | [63] |
| 33 | Metal-based 2, 3-indolinedione | marine organisms | metal-based complexes with derivatives of 2,3-indolinedione | 26S proteasome | breast cancer cell lines MDA-MB-231 and prostate cancer cell lines LNCaP and PC-3 | [64] |
| 34 | Spongiacidin C | marine sponge | pyrrole alkaloid | USP7 | / | [69] |
| 35 | Sulawesins A–C | marine sponge | furanosesterterpene tetronic acids | USP7 | / | [70] |
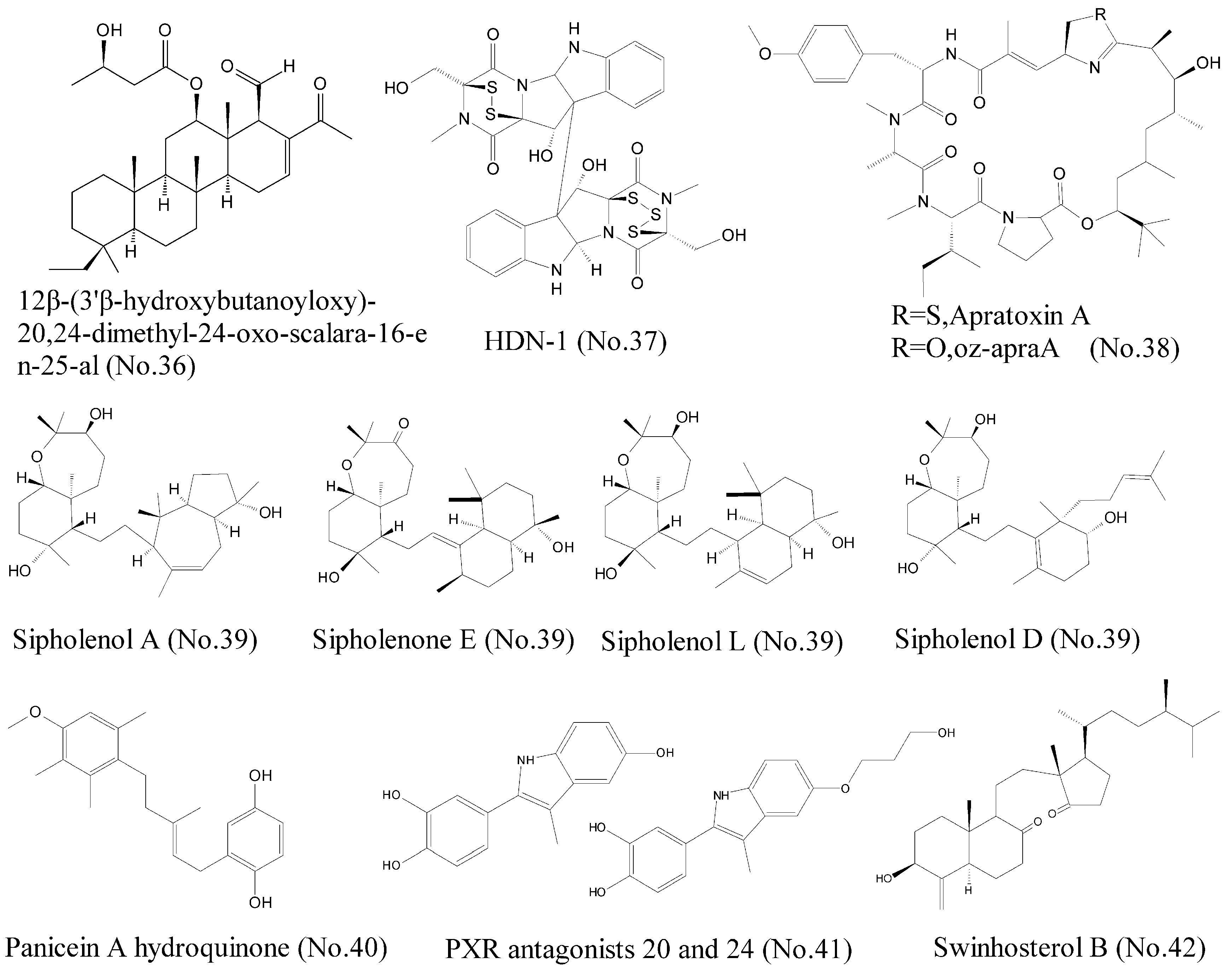
| 36 | 12β-(3′β-hydroxybutanoyloxy)-20, 24-dimethyl-24-oxo-scalara-16-en-25-al | marine sponge | sesterterpenoids | Hsp90 | Leukemia cell lines | [72] |
| 37 | HDN-1 | antarctic fungus | epipolythiopiperazine-2, 5-diones (ETPs) | Hsp90 | lung cancer cell lines | [71] |
| 38 | Apratoxin A (oz-apraA) | marine cyanobacterium | cyclodepsipeptide | Hsp90 | A549, MDA-MB-453, HEK293, SKoV3, and H4 cells | [73] |
| 39 | Sipholane triterpenoids | marine sponge | perhydrobenzoxepine ring and a bicyclodecane system | P-gp | human oral epidermoid carcinoma cell line KB-C2 and KB-V1 | [75,76] |
| 40 | Panicein A hydroquinone | marine sponge | hydroquinone | Patched | melanoma cells | [79] |
| 41 | PXR antagonists 20 and 24 | sponges and echinoderms | Sulfated steroids | PXR agonist | HepG2 cells | [81] |
| 42 | Swinhosterol B | marine sponge | 4-methylenesterols | PXR agonist | HepG2 cell | [82] |
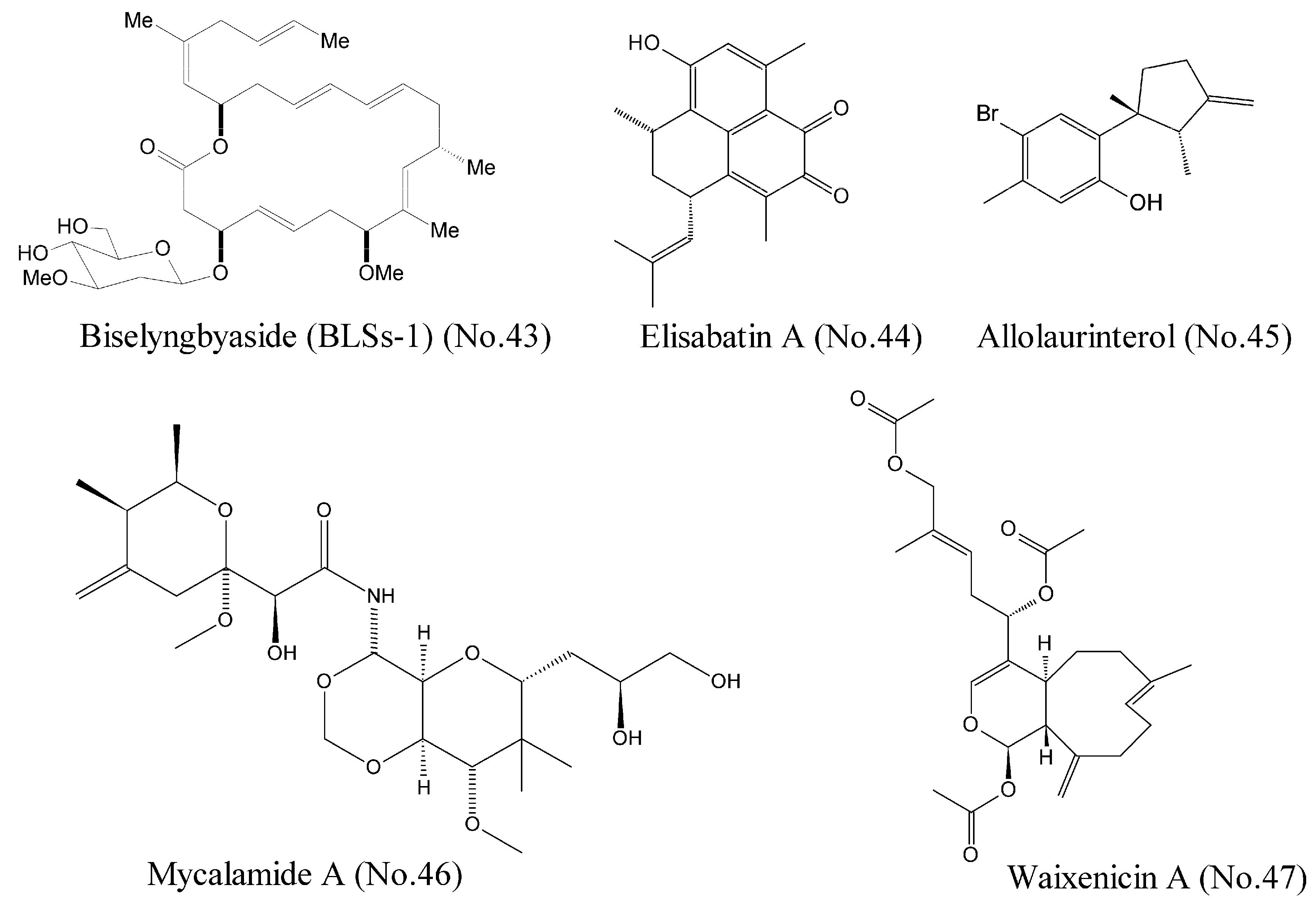
| 43 | Biselyngbyaside (BLSs-1) | marine cyanobacterium | macrolides | calcium channel | HeLa cells | [83] |
| 44 | Elisabatin A | Indian gorgonian octocoral | polyketone | eIF4A ATPase activity | A549 and MDA-MA-468 cell lines | [84] |
| 45 | Allolaurinterol | marine red alga | benzene derivative | eIF4A ATPase activity | A549 and MDA-MA-468 cell lines | [84] |
| 46 | Mycalamide A | marine sponge | lactones | protein synthesis inhibitor | JB6 Cl 41 P+, HeLa cell line | [85,86] |
| 47 | Waixenicin A | soft coral | polyketone | TRPM7 | Jurkat and RBL cells | [87] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Xiong, Y.; Qi, X.; Tang, W.; Dai, J.; Gu, Q.; Li, J. Molecular Targets of Active Anticancer Compounds Derived from Marine Sources. Mar. Drugs 2018, 16, 175. https://doi.org/10.3390/md16050175
Song X, Xiong Y, Qi X, Tang W, Dai J, Gu Q, Li J. Molecular Targets of Active Anticancer Compounds Derived from Marine Sources. Marine Drugs. 2018; 16(5):175. https://doi.org/10.3390/md16050175
Chicago/Turabian StyleSong, Xiaoping, Ying Xiong, Xin Qi, Wei Tang, Jiajia Dai, Qianqun Gu, and Jing Li. 2018. "Molecular Targets of Active Anticancer Compounds Derived from Marine Sources" Marine Drugs 16, no. 5: 175. https://doi.org/10.3390/md16050175
APA StyleSong, X., Xiong, Y., Qi, X., Tang, W., Dai, J., Gu, Q., & Li, J. (2018). Molecular Targets of Active Anticancer Compounds Derived from Marine Sources. Marine Drugs, 16(5), 175. https://doi.org/10.3390/md16050175