Abstract
As an important glycosaminoglycan, keratan sulfate (KS) mainly exists in corneal and cartilage, possessing various biological activities. In this study, we purified KS from blue shark (Prionace glauca) cartilage and prepared KS oligosaccharides (KSO) through keratanase II-catalyzed hydrolysis. The structures of KS and KSO were characterized using multi-dimensional nuclear magnetic resonance (NMR) spectra and liquid chromatography-mass spectrometry (LC-MS). Shark cartilage KS was highly sulfated and modified with ~2.69% N-acetylneuraminic acid (NeuAc) through α(2,3)-linked to galactose. Additionally, KS exhibited binding affinity to Ricinus communis agglutinin I (RCA120) in a concentration-dependent manner, a highly toxic lectin from beans of the castor plant. Furthermore, KSO from dp2 to dp8 bound to RCA120 in the increasing trend while the binding affinity of dp8 was superior to polysaccharide. These results define novel structural features for KS from Prionace glauca cartilage and demonstrate the potential application on ricin-antidote exploitation.
1. Introduction
Glycosaminoglycans (GAGs) are anionic, linear polysaccharides consisting of repeating disaccharide units of hexuronic acid (d-glucuronic acid, GlcA, and/or its C5-epimer l-iduronic acid, IdoA) or hexose (d-galactose) and hexosamine (d-glucosamine, GlcN, or d-galactosamine, GalN). GAGs are categorized into four classes on the basis of variations in monosaccharide compositions, linkage types, and the degree and pattern of sulfated substitution: chondroitin sulfate (CS)/dermatan sulfate (DS), heparin (HP)/heparan sulfate (HS), hyaluronan (HA), and keratan sulfate (KS) [1,2].
KS is the only type of GAGs without uronic acid, and mainly composed of alternating repeating disaccharide unit of β (1,3)-linked galactose (Gal) and β (1,4)-linked N-acetylglucosamine (GlcNAc) with sulfation occurring at the C6 of both saccharide units [3,4,5]. In general, the degree of sulfation of KS varies in different tissues, such as cornea [6], cartilage [7], and brain [8]. In addition, minor but significant structural components, such as sialylation and fucosylation, are important modifications found in KS and essential for controlling the degradation of these molecules [9,10,11,12]. The N-acetylneuraminic acid (NeuAc) residues can be either α (2,3)- or α (2,6)-linked to Gal and typically occupy non-reducing terminal positions, while the fucose residues are α (1,3)-linked to GlcNAc [13].
Additionally, KS participates in diversely important biological processes in vivo. The high abundance of KS in cornea and cartilage is crucial in maintaining the proper hydration levels and keeps the cornea transparent [3,14]. In addition, KS in bones serves as a structural component endowed with cell binding properties [15]. KS proteoglycan with specific sulfation patterns, 6-O-sulfated-GlcNAc and non-sulfated Gal, is required in a critical period of visual cortex plasticity [16]. KS in microglia inhibits neural cell adhesion and the growth of spinal cord neurite in experimental autoimmune neuritis [17,18]. KS can also act as a diagnostic marker for carcinomas of the female genital tract [19]. Furthermore, KS oligosaccharides (KSO) also perform specific activities. KS disaccharide, Gal6Sβ1→4GlcNAc6S, inhibits interleukin 12 production by macrophages in murine Thy-1 type autoimmune disease [20]. Shirato et al. suggested KS disaccharide for treatment of airway inflammatory responses arising from bacterial infections [21]. KS disaccharide also prevents the progression of emphysema in murine models and is effective for treating chronic obstructive pulmonary disease [22].
Ricin is a potent cytotoxic glycoprotein derived from the beans of castor plant (Ricinus communis). It is composed of chain A and chain B, linked by a disulfide bond. Ricin A chain (32 kDa) is a ribosome-inactivating enzyme, while ricin B chain (34 kDa) is a galactose/N-acetylgalactosamine binding lectin [23]. The leading cause of death is that ricin can dissolve red blood cells and further cause the paralysis of cardiovascular and respiratory centers. Due to high toxicity and accessibility, ricin has been widely used as a terrorist weapon and for political assassination. It was reported that over 700 people have died of ricin poisoning. However, there is no antidote currently available for ricin poisoning. Thus, it is critical for the exploitation of antidote. Ricinus communis agglutinin I (RCA120) recognizes carbohydrate chains with non-reducing terminal β-d-galactose and Galβ1→4GlcNAc sequence [24]. Possessing the repeating structure of Galβ1→4GlcNAc, KS may be the candidate compound as an antidote.
Based on these various physiological and pharmacological activities, the structural characterization of KS is extremely important for developing structure–activity relationships and diagnosing related diseases. Two hydrolytic enzymes, keratanase I and keratanase II, are useful in facilitating the structural determination of KS. Additionally, nuclear magnetic resonance (NMR) and liquid chromatography-mass spectrometry (LC-MS) are all powerful techniques for structural elucidation. Multi-dimensional NMR spectra have been applied to characterizing the capping segments, main chain repeating unit and linkage region of KS derived from bovine tracheal cartilage [25]. Due to the soft ionization mode, electrospray ionization tandem mass spectrometry (ESI-MS/MS) has been useful for sequencing KSO obtained from various biological samples [26,27].
In the present study, a highly sulfated KS was isolated from Prionace glauca cartilage. The structure of KS and KSO generated through enzymatic hydrolysis were elucidated by NMR and hydrophilic interaction liquid chromatography-Fourier transform mass spectrometry (HILIC-FTMS) analysis. The interaction of KS and KSO with RCA120 was also determined using SPR and MOE docking methods, providing a foundation for further pharmaceutical exploitation of Prionace glauca cartilage KS.
2. Results and Discussion
2.1. Isolation and Chemical Composition Analysis
The crude polysaccharide from Prionace glauca cartilage was generated through proteolysis as previously reported [28]. The polysaccharides were separated on QFF column in gradient elution (Figure S1A); and two charged uniform fractions were acquired. Based on the enzymatic hydrolysis characteristics of the two fractions towards GAGs enzyme, Peak I was inferred to be CS and comprised of a mixture of CSA, CSC, and CSD sequences, as determined through disaccharide composition analysis (Figure S2, Table S2). Peak II was degraded by neither chondroitinase nor heparinase. However, according to the results of monosaccharide composition and its low uronic acid content, we concluded Peak II to be KS (Table S1). Single and symmetric peaks on both RID and MALLS indicated that CS and KS were of high purity (Figure S1B,C). The molecular weight of KS from shark cartilage (Mw: 45,980) was much bigger than that from bovine corneal and chicken egg white (Mw: 14,300 and 36,800, respectively), which might be related to the unique marine environment, such as low temperature; high pressure and salt; and lack of oxygen, light, and nutrition [29,30].
2.2. NMR Spectroscopy
The structural features of KS were properly characterized through a combination of 1D 1H-NMR, DEPTQ NMR, and 2D 1H-1H COSY, 1H-13C HSQC, as well as 1H-13C HMBC. Major 1H- and 13C-chemical shifts identified from these spectra were assigned as Table 1. The NMR profiles of KS sample were roughly consistent with the reported structure of KS [31].

Table 1.
Major 1H and 13C chemical shifts (in ppm) data for shark cartilage KS.
In 1H-NMR spectrum, KS had a crowded region between 3.4 and 4.8 ppm, resulting in a severe signal overlap for the majority of the resonances. Two notable anomeric proton signals at 4.66 and 4.48 ppm were identical with H-1 of GlcNAc and Gal, respectively (Figure 1A). The anomeric carbons at 102.68 and 102.73 ppm were deduced by DEPTQ NMR and 1H-13C HSQC (Figure 1B,D).
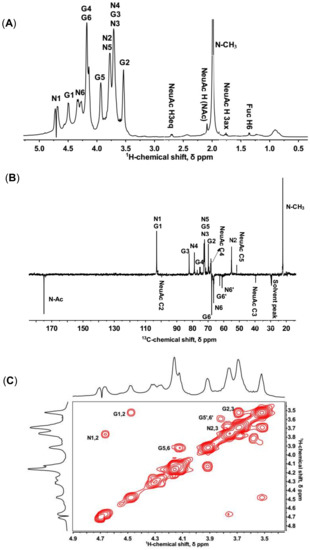
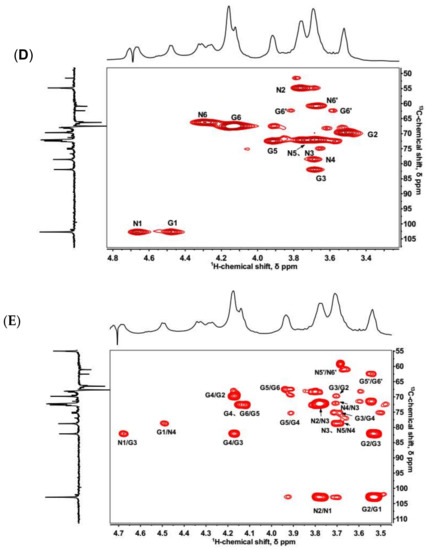
Figure 1.
1D and 2D NMR spectra of isolated KS. N and G stand for GlcNAc and Gal, respectively. (A) Full 1H-NMR spectrum; (B) DPETQ spectrum; (C) 1H-1H COSY spectrum. G1,2 refers to a cross-peak between G H-1 and G H-2, etc. G5′,6′ refers to a cross-peak between non-sulfated G H-5 and G H-6. (D) 1H-13C HSQC spectrum. G1 refers to a cross-peak between G H-1 and G C-1, etc. G6′ refers to a cross-peak between non-sulfated G H-6 and G C-6, etc. (E) 1H-13C HMBC spectrum. G2/G1 refers to a cross-peak between G H-2 and G C-1, etc.
The non-reducing ends of KS chains are generally capped with NeuAc at the C-3 or C-6 of the terminal Gal [4]. In the 1H-NMR spectrum, signals at 1.74 and 2.68 ppm were consistent with H3ax and H3eq of NeuAc (Figure 1A). The signal at 39.38 ppm in 13C-NMR spectrum belonged to the unique C3 of NeuAc (Figure 1B). In addition, signals in the 1H-1H COSY and 1H-13C HSQC also demonstrated the presence of NeuAc (Figure S3).
Moreover, a signal of 1.33 ppm in the 1H-NMR spectrum corresponded to the H-6 of α (1,3)-linked fucose (Figure 1A). The 1H-1H COSY showed a connection from H-6 to H-5 at 1.33/4.27 ppm. A cross-peak at 1.33/19.16 ppm in the 1H-13C HSQC was assigned to the methyl group of fucose (Figure S3). Most signals of fucose were not identifiable since the low content of fucose resulted in signals covering by other sugars [32].
In 1H-13C HSQC, two characteristic β-anomeric 1H/13C-signals at 4.66/102.73 and 4.48/102.68 ppm were attributed to GlcNAc (denoted N1) and Gal (denoted G1) residues, respectively (Figure 1D). These signals were in approximately equimolar proportions, consistent with the disaccharide-repeating unit of KS. In the 1H-13C HMBC, two clear signals involved in glycosidic bonds were at 4.48/78.54 and 4.66/81.95 ppm (denoted N1/G3 and G1/N4, Figure 1E), which demonstrated β (1,3)- and β (1,4)-linkage types between GlcNAc and Gal, respectively. The signals of glycosidic bonds were also confirmed by 1H-13C HSQC due to the downfield shift of 13C-signals (denoted N4 and G3, Figure 1D).
Furthermore, in the 1H-13C HSQC, there were two cross-peaks at 4.29/66.21 and 4.13/67.49 ppm (denoted N6 and G6), assigned to 6-O-sulfo GlcNAc and 6-O-sulfo Gal residues, respectively (Figure 1D). Compared to the non-sulfated residues (denoted N6′ and G6′), these signals were shifted ~0.6 ppm downfield on the 1H-scale and ~5 ppm downfield on the 13C-scale. The results indicated that 6-sulfation primarily occurred at both of the repeating residues and the non-sulfated residues accounted for a very minor portion.
2.3. HILIC-FTMS Analysis of KS
Keratanase II hydrolyzes β (1,3)-glucosaminidic linked to galactose in KS [33]. On cleavage, the enzyme requires a sulfo group at 6-O-position of GlcNAc but acts independently of a sulfo at 6-O-position of the Gal linked to the GlcNAc [34]. The KSO generated by keratanase II digestion were analyzed using HILIC-FTMS. The total ion chromatogram (TIC) of KSO sample was shown in Figure 2A. The results confirmed that the major repeating unit of the isolated KS was the disulfated disaccharide, (-3Gal6Sβ1→4GlcNAc6Sβ1-). The raw data was deconvoluted using DeconTools, and then the output of DeconTools was processed by GlycResoft to generate matching structures and to provide relative quantitative information [35]. A total of 31 oligosaccharides were matched by GlycResoft, which ranged from degree of polymerization (dp) 2 to dp 10 and included chains with both an even- and odd-number of sugar residues as well as sialylated oligosaccharides. Approximately 90% of the oligosaccharides were disaccharides. Relative quantitative results on the major oligosaccharides were shown graphically in Figure 2B. The extracted ion chromatograms of these major oligosaccharides were presented in Figure S4 and structural assignments relied on a mass accuracy of <5 ppm.
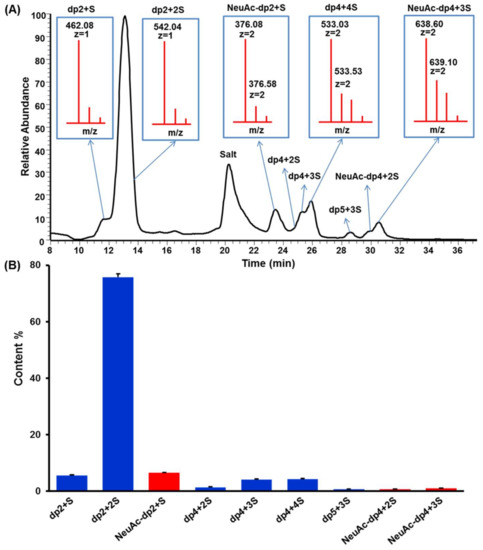
Figure 2.
(A) Total ion chromatography of HILIC-FTMS profiling of fully digested KS domain structure. (B) Composition analysis of KSO calculated by GlycResoft. The analytical error for each GAG was <1%. The major composition unit of the oligomers was highly sulfated domains (approximately one sulfo group per saccharide). The minor component of NeuAc capped oligomers was also detected (degree of polymerization (dp)2 + sulfate (S), dp4 + 2S and dp4 + 3S with one NeuAc residue at the non-reducing end).
Moreover, the integration ratio between H3ax of NeuAc and H1 of Gal in 1H-NMR spectrum was 1:22, facilitating the calculation of NeuAc content as 2.69%, which closely agreed with the results calculated by GlycResoft. Based on the determined molecular weight and NeuAc content, we deduced that four NeuAc residues were present in each KS chain. This suggested that KS might have at least four branches since NeuAc was only believed to occupy the non-reducing ends of chains.
2.4. Preparation and Mapping of KSO
KS sample was partly digested by keratanase II, and then fractionated on a Bio-Gel P6 column. The elution curve illuminated five prominent oligosaccharide fractions (Figure 3). Based on ESI-MS analysis, the five fractions corresponded to oligosaccharides of dp2, dp4, dp5, dp6, and dp8 (Figure S5). Surprisingly, oligosaccharide of dp5 was mainly a sialylated KS tetrasccharide with three sulfo-substitution.
Figure 3.
Elution profile of KSO on Bio-Gel P6 gel filtration chromatography.
The sialylated KS tetrasccharide was determined by 1D and 2D NMR spectroscopy (Figure 4) to confirm the linkage type between NeuAc and Gal. In 1H-13C HMBC, C2, C4, and C5 of NeuAc were assigned by the cross-peaks with H3ax and H3eq. Then the proton in Gal (H-X) correlating with the C2 of NeuAc was identified (Figure 4A), and the 1H-1H COSY showed H-X correlating with H-2 which also coupled with H-1 of Gal. Therefore, we inferred H-X was H-3, which demonstrated that NeuAc was α (2,3)-linked to Gal (Figure 4B). No sign of any α (2,6)-linkages was observed. However, in bovine articular cartilage, NeuAc α(2,3)- and α(2,6)-linked types were both identified [34].
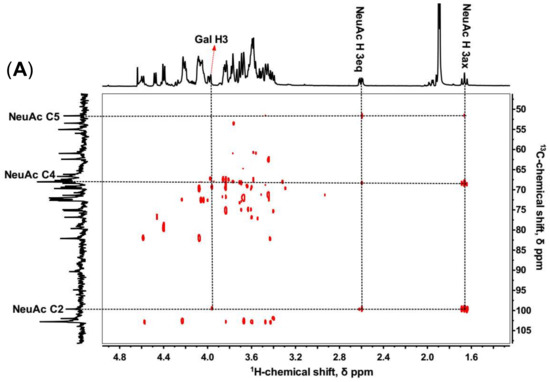
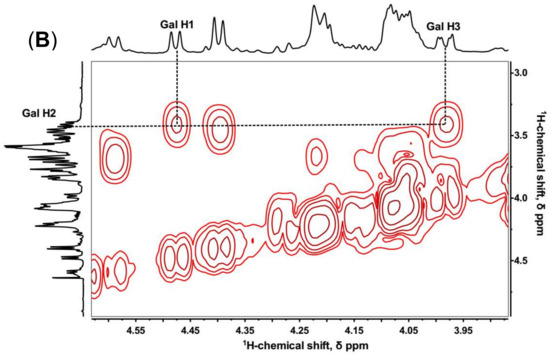
Figure 4.
1H-13C HMBC (A) and 1H-1H COSY (B) of sialylated KS tetrasaccharide isolated from Bio-Gel P6 column.
Multistage mass spectrometric sequencing of sialylated KS tetrasccharide revealed that the unsulfated hexose might be Gal or GlcNAc in the reducing end (Figure S6). Thus, a generalized chemical structure of shark cartilage KS could be described (Figure 5).
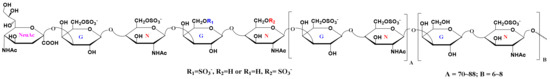
Figure 5.
Schematic diagram of the chemical structure of KS.
These results demonstrated a higher degree of sulfation for Prionace glauca cartilage KS than KS derived from brain, bovine corneal, articular cartilage, and chicken egg white, all of which possessed less sulfation on the Gal residues [7,8,29,30]. Additionally, KS was modified with ~2.69% NeuAc through α(2,3)-linked to Gal.
2.5. RCA120 Binding Activities of KS and KSO
Although the strong binding properties among RCA120 and Galβ1→4GlcNAc (LacNAc) had been described previously through SPR, the interaction between RCA120 and KS polysaccharide in vitro was scarce [36,37,38]. However, our SPR results showed that both KS from shark cartilage and chicken egg white strongly bound to RCA120 in a concentration-dependent manner with KD values of 1.22 × 10−7 M and 1.37 × 10−7 M, respectively (Figure 6 and Table S3). Wang et al. once reported that RCA120 binding to Galβ1→4 linked oligosaccharide was enhanced by 2-O- or 6-O-sulfation but abolished by 4-O-sulfation [39]. However, concerning heterogeneity and flexibility of polysaccharide, the degree of sulfation at Gal residues hardly influenced RCA120 binding ability to a certain extent.
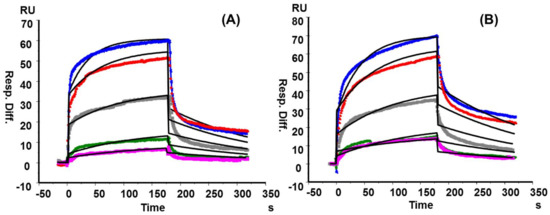
Figure 6.
SPR sensor grams for interactions of RCA120 with KS from shark cartilage (A) and chicken egg white (B). The concentrations of each protein (from top to bottom): 500, 250, 125, 63, and 31 nM, respectively. The black curves were the fitting curves using models from BIAevaluate 4.0.1.
In order to assess the influence of sugar chain length on RCA120 binding, KSO from dp2 to dp8 were eluted directly onto an SPR imaging chip at 1 mg/mL. Compared to KS, the relative binding strength was listed in Figure 7. As the results showed, along with the degree of polymerization increasing, the interaction with RCA120 was gradually enhanced. The conclusion was consistent with the MOE docking results in Table 2. The binding modes between RCA120 and KSO were shown as Figure 8. The growing numbers of hydrogen bond between the ligands and the receptor were accompanied by the extension of chain. Unexpectedly, the binding affinity of dp8 was superior to polysaccharide, which meant the potential application of oligosaccharide of dp8 in antidote exploitation.
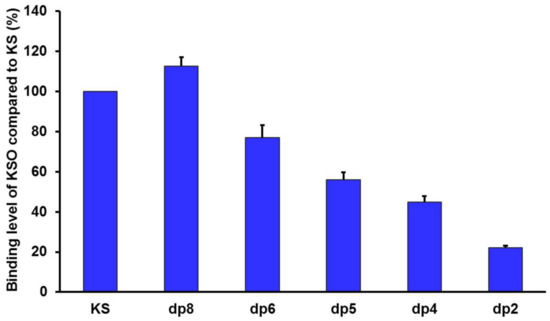
Figure 7.
Normalized binding of shark cartilage KSO (from dp2 to dp8) and polysaccharide to RCA120. The concentrations of KSO (from dp2 to dp8) and polysaccharide were 1 mg/mL.
Table 2.
The score of KSO predicted using MOEDock.
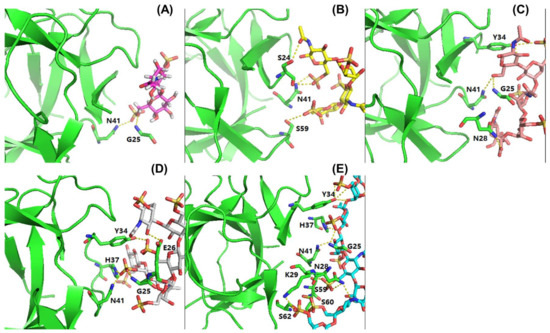
Figure 8.
Binding modes of KSO to RCA120. (A–E) showed the binding modes of dp2, dp4, dp5, dp6, and dp8 to RCA120, respectively. The receptor was shown in green. The dashed lines showed hydrogen bonds between the ligands and the receptor.
3. Materials and Methods
3.1. Materials and Chemicals
Prionace glauca cartilage was obtained from Rushan Wantongming Biotech Company (Weihai, Shandong Province, China). The chondroitin ABC lyase and heparin lyases I, II, III were performed in our laboratory following the reported method [40]. XB-SAX chromatography column was from Welch, Shanghai. High performance gel permeation chromatography column (Shodex OHpak SB-804 HQ and SB-802.5 HQ) was from Showa Denko K.K., Tokyo, Japan. Packing materials for Q-Sepharose fast flow anion-exchange (QFF) column were from GE Healthcare Biosciences AB, Boston, MA, USA. Disaccharide standards of chondroitin sulfate were from Iduron, Cheshire, UK. Luna HILIC chromatography column was from Phenomenex, Torrance, CA, USA. Recombinant keratanase II expressed in Escherichia coli was prepared as previously described [33]. Acetonitrile and ammonium acetate were of HPLC grade (Sigma Aldrich, St. Louis, MO, USA). All other chemicals were of analytical grade.
3.2. Isolation and Purification of KS
KS from Prionace glauca cartilage was prepared by methods reported previously with minor modification [28]. The crude polysaccharide, generated by proteolysis, was separated on a QFF column, and eluted with a linear gradient of 0.7–2 mol/L NaCl at a flow rate of 4 mL/min. The eluate was tested by the phenol-sulfuric acid method at 490 nm [41]. The fraction containing polysaccharide was collected, concentrated, and desalted. The isolated fractions were then digested using chondroitin ABC lyase and heparinases (I, II, and III) and the products were detected by thin layer chromatography.
3.3. Molecular Weight and Chemical Composition Analysis
Uronic acid content was determined by modified carbazole method [42]. Sulfate content was determined by BaCl2-Gelatin method [43]. Purity and relative molecular weight (Mw) were determined by high performance gel filtration chromatography system and detected by refractive index detector (RID) and multi-angle laser scattering system (MALLS) (Wyatt Technology, Santa Barbara, USA) [44]. Monosaccharide composition was determined by a pre-column 1-phenyl-3-methyl-5-pyrazolone (PMP) derivatization–HPLC method [45].
3.4. Profiling of KSO Generated by Keratanase II Digestion Through HILIC-FTMS
The KS sample was completely degraded by enzymatic hydrolysis. KS (100 µg) was dissolved in 100 μL of digestion buffer containing 50 mM ammonium acetate (pH 7.0). Excess keratanase II (50 mU) was added to KS sample and incubated at 37 °C overnight with gentle agitation. Enzymatic digestion was terminated by heating in a 100 °C water bath, and then spun down at 12,000 rpm for 5 min; supernatant was used directly for HILIC-FTMS analysis.
HILIC-FTMS analysis was performed on an Agilent 1290 LC ultra-performance liquid chromatography (UPLC) system (Agilent Technologies, Wilmington, DE, USA) equipped with a LTQ ORBITRAP XL mass spectrometer (Thermo, Scientific, Waltham, MA, USA). The KSO were separated by a Luna HILIC column (150 × 2.00 mm, 3 μm, Phenomenex) at 25 °C. The mobile phase was a mixture of 5 mM NH4OAc/98% acetonitrile (solvent A) and 5 mM NH4OAc/H2O (solvent B) at a flow rate of 150 μL/min. The gradient was programmed as 92% A initially and then linearly changed to 60% A over 58 min. The analysis was performed in the negative ion mode using a capillary temperature of 275 °C. The spray voltage was 4.2 kV and Nitrogen dry gas flowed at 40 L/min. Data acquisition and analysis were performed using Xcalibur 2.0 software (Thermo, Scientific, Waltham, MA, USA) and GlycReSoft 1.0 software (Publicly archived, http://code.google.com/p/glycresoft/downloads/list).
3.5. Preparation and Sequence Analysis of KSO
KS sample (200 mg) was partly digested by keratanase II (5 IU) at 37 °C for 5 h with gentle agitation. The reaction was terminated in 100 °C water bath, and the precipitant was removed by centrifugation at 12,000 r/min. The supernatant was loaded on Bio-Gel P6 column connected to an ÄKTA-fast protein liquid chromatography (FPLC) system (General Electric Company, Boston, MA, USA). The column was then eluted by 0.2 mol/L NH4HCO3 at a flow rate of 0.2 mL/min. Next, each fraction was analyzed by ESI-MS at the negative-ion mode. For collision induced dissociation (CID)-MSn scanning, helium was used as collision gas with collision energy of 20–25 eV [46].
3.6. NMR Spectroscopy Analysis
Samples of KS and KSO were dissolved in 500 μL 99.9% deuterium oxide (D2O) respectively and freeze-dried three times to replace all exchangeable protons with deuterium, then redissolved in 500 μL D2O. One-dimensional (1D) 1H-NMR, DEPTQ NMR, and two-dimensional (2D) 1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC were performed at 298K on Bruker BioSpin GmbH 600 MHz (Billerica, MA, USA) with Topspin 2.1.6 software (Bruker, Billerica, MA, USA). Chemical shifts were displayed relative to internal deuterated acetone for 1H and 13C.
3.7. Surface Plasmon Resonance (SPR) Binding Kinetics of RCA120-KS/KSO Interactions
SPR measurements were performed using a Biacore 3000 SPR instrument (General Electric Company, Boston, MA, USA). For the polysaccharides binding, biotinylated shark cartilage KS and chicken egg white KS (From lab of Robert J. Linhardt) [30] sensor chip were prepared by reaction of sulfo-N-hydroxysuccinimide long-chain biotin (Pierce, Rockford, IL, USA) with the free amino groups and the residue with the reducing end in the polysaccharide chain following a published procedure [29]. Two-fold serial dilutions of RCA120 were injected over the sensor chip at a flow-rate of 30 µL/min for a period of 3 min followed by 3 min dissociation period. For the oligosaccharides binding, RCA120 was immobilized to CM5 chip using amine coupling based on the manufacturer’s protocol. The successful immobilization of RCA120 was confirmed by the observation of a ~2000 RU increase in the sensor chip. KSO from dp2 to dp8 and polysaccharide (1 mg/mL) in HBS-EP buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, and 0.005% surfactant P20, (pH 7.4)) were injected at a flow rate of 30 µL/min for 3 min. At the end of the sample injection, the same buffer was flowed over the sensor surface to facilitate dissociation. The sensor chip was regenerated by injecting with 30 µL of 2 M NaCl and 30 µL of running buffer to get a fully regenerated surface. SPR experiments were performed in triplicate at each concentration, confirming reproducibility. The binding sensor grams (RU versus time) were pooled, trimmed, double referenced, and experimentally fit to different kinetic models using BIAevaluation software v4.0.1 (General Electric Company, Boston, MA, USA).
3.8. MOE Binding Affinity Calculation
Molecular docking was performed using MOE with the AMBER12: EHT force field. Ricin B-like lectin crystal structures used for the ADT calculations above were utilized. The induced fit docking approach was applied with consideration of the side chain flexibility of KSO at the binding site. The ligand binding site was defined using the bound ligands in the crystal structures. Ten docking conformations of the ligands were produced, and the best scored conformation with minimum binding energy was selected for analysis.
4. Conclusions
Herein, we reported a purified KS from Prionace glauca cartilage. A series of KSO were also obtained by KS digestion with keratanase II. The structure of KS and KSO were confirmed by multi-dimensional NMR spectra and HILIC-FTMS. The results showed that shark cartilage KS was highly sulfated and the major disaccharide repeating unit was -3Gal6Sβ1→4GlcNAc6Sβ1-. Moreover, this KS was modified by NeuAc capped non-reducing ends of chains, and the NeuAc was α (2,3)-linked to galactose.
SPR showed that KS bound to RCA120 in a concentration-dependent manner with the KD value of 1.22 × 10−7 M. Furthermore, KS oligosaccharides from dp2 to dp8 bound to RCA120 in the increasing trend, while the binding affinity of dp8 was superior to polysaccharide. MOE docking assays also verified the results. In conclusion, these results define novel structural features for KS from Prionace glauca cartilage and demonstrate the potential application in ricin-antidote exploitation.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/4/128/s1, Table S1: Physicochemical properties analysis of KS and CS, Table S2: Disaccharides composition of CS, Table S3: Summary of kinetic data of shark KS and egg KS-RCA120 interactions, Figure S1: Elution profile of GAGs from Prionace glauca cartilage on a QFF ion-exchange column, Figure S2: Separation chromatography of CS disaccharides analysis on SAX-HPLC, Figure S3: NMR signals of NeuAc and fucose in KS, Figure S4: The extracted ion chromatograms (EICs) of KSO based on HILIC–MS analysis, Figure S5: Negative-ion mass spectra of KS oligosaccharides isolated by Bio-Gel P6, Figure S6: Negative-ion ESI-MSn product-ion spectra of sialylated KS tetrasaccharide (Sia-dp4+3S) isolated by Bio-Gel P6.
Acknowledgments
This work was supported by Grants from NSFC-Shandong Joint Fund for Marine Science Research Centers (U1606403), National Natural Science Foundation of China (31670811, 31600646), Natural Science Foundation of Shandong Province (ZR2016HB42), the Scientific and Technological Innovation Project Financially Supported by Qingdao National Laboratory for Marine Science and Technology (2016ASKJ08), China Postdoctoral Science Foundation funded project (2015M580610, 2016T90654), the Fundamental Research Funds for the Central Universities (201762002), and Taishan scholar project special funds.
Author Contributions
Q.L., G.L., and G.Y. conceived and designed the experiments; Q.L. performed the extraction, structural analysis and MOE docking experiments and drafted the manuscript; J.Z. and F.Z. performed the SPR experiments; Q.L., G.L., X.Z., and X.S. analyzed the data; Q.L., C.C., X.Z., and X.S. interpreted the results; C.C. and R.L. reviewed and provided useful suggestions to improve the manuscript. G.L. and G.Y. critically revised the manuscript and gave final approval of the version published.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, L.; Ly, M.; Linhardt, R.J. Proteoglycan sequence. Mol. Biosyst. 2012, 8, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan storage disorders: A review. Biochem. Res. Int. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, J.L. MINI REVIEW Keratan sulfate: Structure, biosynthesis, and function. Glycobiology 2000, 10, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, J.L. Keratan Sulfate Biosynthesis. IUBMB. Life 2002, 54, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Linker, A.; Davidson, E.A.; Weissmann, B. The mucopolysaccharides of bovine cornea. J. Biol. Chem. 1953, 205, 611–616. [Google Scholar] [PubMed]
- Funderburgh, J.L.; Caterson, B.; Conrad, G.W. Distribution of proteoglycans antigenically related to corneal keratan sulfate proteoglycan. J. Biol. Chem. 1987, 262, 11634–11640. [Google Scholar] [PubMed]
- Lauder, R.M.; Huckerby, T.N.; Nieduszynski, I.A.; Plaas, A.H. Age-related changes in the structure of the keratan sulphate chains attached to fibromodulin isolated from articular cartilage. Biochem. J. 1998, 330, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Krusius, T.; Finne, J.; Margolis, R.K.; Margolis, R.U. Identification of an O-glycosidic mannose-linked sialylated tetrasaccharide and keratan sulfate oligosaccharides in the chondroitin sulfate proteoglycan of brain. J. Biol. Chem. 1986, 261, 8237–8242. [Google Scholar] [PubMed]
- Gardell, S. Separation of mucopolysaccharides on a cellulose column. Acta Chem. Scand. 1957, 11, 668. [Google Scholar] [CrossRef]
- Seno, N.; Meyer, K.; Anderson, B.; Hoffman, P. Variations in keratosulfates. J. Biol. Chem. 1965, 240, 1005–1010. [Google Scholar] [PubMed]
- Tai, G.H.; Huckerby, T.N.; Nieduszynski, I.A. 600 MHz 1H NMR study of a fucose-containing heptasaccharide derived from a keratanase digestion of bovine articular cartilage keratan sulphate. Carbohydr. Res. 1994, 255, 303–309. [Google Scholar] [CrossRef]
- Ashwell, G.; Morell, A.G. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv. Enzymol. Relat. Areas Mol. Biol. 1974, 41, 99–128. [Google Scholar] [PubMed]
- Brown, G.M.; Huckerby, T.N.; Morris, H.G.; Nieduszynski, I.A. Degradation of articular cartilage keratan sulphates using hydrazinolysis and nitrous acid. Environment of fucose residues. Biochem. J. 1992, 286, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Quantock, A.J.; Young, R.D.; Akama, T.O. Structural and biochemical aspects of keratan sulphate in the cornea. Cell. Mol. Life Sci. 2010, 67, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Sommarin, Y.; Wendel, M.; Shen, Z.; Hellman, U.; Heinegard, D. Osteoadherin, a cell-binding keratan sulfate proteoglycan in bone, belongs to the family of leucine-rich repeat proteins of the extracellular matrix. J. Biol. Chem. 1998, 273, 16723–16729. [Google Scholar] [CrossRef] [PubMed]
- Takeda-Uchimura, Y.; Uchimura, K.; Sugimura, T.; Yanagawa, Y.; Kawasaki, T.; Komatsu, Y.; Kadomatsu, K. Requirement of keratan sulfate proteoglycan phosphacan with a specific sulfation pattern for critical period plasticity in the visual cortex. Exp. Neurol. 2015, 274, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ohgomori, T.; Natori, T.; Miyamoto, K.; Kusunoki, S.; Sakamoto, K.; Ishiguro, N.; Imagama, S.; Kadomatsu, K. Keratan sulfate expression in microglia is diminished in the spinal cord in experimental autoimmune neuritis. Cell Death Dis. 2013, 4, e946. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Ishino, Y.; Jiang, W.; Yoshimura, T.; Takeda-Uchimura, Y.; Uchimura, K.; Kadomatsu, K.; Ikenaka, K. Keratan sulfate regulates the switch from motor neuron to oligodendrocyte generation during Development of the mouse spinal cord. Neurochem. Res. 2016, 41, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Ishii, K.; Asaka, R.; Suzuki, A.; Takatsu, A.; Kashima, H.; Shiozawa, T. Immunohistochemical expression of keratan sulfate: A possible diagnostic marker for carcinomas of the female genital tract. J. Clin. Pathol. 2011, 64, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Kurihara, H.; Ito, T.; Kikuchi, H.; Yoshida, K.; Yamanokuchi, H.; Asari, A. The keratan sulfate disaccharide Gal(6S03) 1,4-GlcNAc(6S03) modulates interleukin 12 production by macrophages in murine Thy-1 type autoimmune disease. J. Biol. Chem. 2005, 280, 20879–20886. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Gao, C.; Ota, F.; Angata, T.; Shogomori, H.; Ohtsubo, K.; Yoshida, K.; Lepenies, B.; Taniguchi, N. Flagellin/Toll-like receptor 5 response was specifically attenuated by keratan sulfate disaccharide via decreased EGFR phosphorylation in normal human bronchial epithelial cells. Biochem. Biophys. Res. Commun. 2013, 435, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Fujinawa, R.; Yoshida, T.; Ueno, M.; Ota, F.; Kizuka, Y.; Hirayama, T.; Korekane, H.; Kitazume, S.; Maeno, T.; et al. A keratan sulfate disaccharide prevents inflammation and the progression of emphysema in murine models. Am. J. Physiol.-Lung. Cell. Mol. Physiol. 2016, 312, L268–L276. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Itakura, Y.; Nakamuratsuruta, S.; Kominami, J.; Sharon, N.; Kasai, K.; Hirabayashi, J. Systematic comparison of oligosaccharide specificity of Ricinus communis agglutinin I and Erythrina lectins: A search by frontal affinity chromatography. J. Biochem. 2007, 142, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Huckerby, T.N.; Lauder, R.M. Keratan sulfates from bovine tracheal cartilage. Eur. J. Biochem. 2000, 267, 3360–3369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kariya, Y.; Conrad, A.H.; Tasheva, E.S.; Conrad, G.W. Analysis of keratan sulfate oligosaccharides by electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, T.; Suzuki, K.; Hirabayashi, J. Multistage mass spectrometric sequencing of keratan sulfate-related oligosaccharides. Anal. Chem. 2006, 78, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Dickenson, J.M.; Huckerby, T.N.; Nieduszynski, I.A. Two linkage-region fragments isolated from skeletal keratan sulphate contain a sulphated N-acetylglucosamine residue. Biochem. J. 1990, 269, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Weyers, A.; Yang, B.; Solakyildirim, K.; Yee, V.; Li, L.; Zhang, F.; Linhardt, R.J. Isolation of bovine corneal keratan sulfate and its growth factor and morphogen binding. FEBS J. 2013, 280, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Sun, X.; He, W.; Cai, C.; Onishi, A.; Zhang, F.; Linhardt, R.J.; Liu, Z. Keratan sulfate glycosaminoglycan from chicken egg white. Glycobiology 2016, 26, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Piquet, A.A.; Pereira, M.S.; Mourão, P.A.S. Residual keratan sulfate in chondroitin sulfate formulations for oral administration. Carbohydr. Polym. 2012, 90, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Huckerby, T.N.; Nieduszynski, I.A.; Brown, G.M.; Cockin, G.H. A full assignment of proton resonances for an α (1-3)-linked fucose residue in keratan sulphate from bovine articular cartilage. Glycoconj. J. 1991, 8, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, W.; Jiang, P.; Yu, Y.; Lin, L.; Sun, X.; Koffas, M.; Zhang, F.; Linhardt, R.J. Construction and functional characterization of truncated versions of recombinant keratinase II from Bacillus circulans. Glycoconj. J. 2017, 34, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.M.; Huckerby, T.N.; Morris, H.G.; Abram, B.L.; Nieduszynski, I.A. Oligosaccharides derived from bovine articular cartilage keratan sulfates after keratanase II digestion: Implications for keratan sulfate structural fingerprinting. Biochemistry 1994, 33, 4836–4846. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, E.; Tan, Y.; Tan, Y.; Hu, H.; Benson, G.; Aizikov, K.; Conley, S.; Staples, G.O.; Slysz, G.W.; Smith, R.D.; et al. GlycReSoft: A software package for automated recognition of glycans from LC/MS data. PLoS ONE 2012, 7, e45474. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Singh, T.; Herp, A.; Wu, A.M. Carbohydrate recognition factors of the lectin domains present in the Ricinus communis toxic protein (ricin). Biochimie 2006, 88, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Fais, M.; Karamanska, R.; Allman, S.; Fairhurst, S.A.; Innocenti, P.; Fairbanks, A.J.; Donohoe, T.J.; Davis, B.G.; Russell, D.A.; Field, R.A. Surface plasmon resonance imaging of glycoarrays identifies novel and unnatural carbohydrate-based ligands for potential ricin sensor development. Chem. Sci. 2011, 2, 1952–1959. [Google Scholar] [CrossRef][Green Version]
- Baenziger, J.U.; Fiete, D. Structural determinants of Ricinus communis agglutinin and toxin specificity for oligosaccharides. J. Biol. Chem. 1979, 254, 9795–9799. [Google Scholar] [PubMed]
- Wang, Y.; Yu, G.; Han, Z.; Yang, B.; Hu, Y.; Zhao, X.; Wu, J.; Lv, Y.; Chai, W. Specificities of Ricinus communis agglutinin 120 interaction with sulfated galactose. FEBS Lett. 2011, 585, 3927–3934. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Blain, F.; Musil, R.A.; Zimmermann, J.J.; Gu, K.; Bennett, D.C. Isolation and expression in Escherichia coli of hepB and hepC, genes coding for the glycosaminoglycan-degrading enzymes heparinase II and heparinase III, respectively, from Flavobacterium heparinum. Appl. Environ. Microb. 1996, 62, 2723–2734. [Google Scholar]
- DuBois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Selvendran, R.R.; March, J.F.; Ring, S.G. Determination of aldoses and uronic acid content of vegetable fiber. Anal. Biochem. 1979, 96, 282–292. [Google Scholar] [CrossRef]
- Dodgson, K.S.; Price, R.G. A note on the determination of the ester sulphate content of sulphated polysaccharides. Biochem. J. 1962, 84, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Viebke, C.; Borgström, J.; Piculell, L. Characterisation of kappa-and iota-carrageenan coils and helices by MALLS/GPC. Carbohydr. Polym. 1995, 27, 145–154. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Xue, C.; Dong, P.; Sheng, W.; Yu, G.; Chai, W. Sequence determination of a non-sulfated glycosaminoglycan-like polysaccharide from melanin-free ink of the squid Ommastrephes bartrami by negative-ion electrospray tandem mass spectrometry and NMR spectroscopy. Glycoconj. J. 2008, 25, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Zhao, X.; Yang, B.; Ren, S.; Guan, H.; Zhang, Y.; Lawson, A.M.; Chai, W. Sequence determination of sulfated carrageenan-derived oligosaccharides by high-sensitivity negative-ion electrospray tandem mass spectrometry. Anal. Chem. 2006, 78, 8499–8505. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).