Combination Treatment of Deep Sea Water and Fucoidan Attenuates High Glucose-Induced Insulin-Resistance in HepG2 Hepatocytes

Abstract
:
1. Introduction
2. Results
2.1. Analysis of Element Contents in DSW of Hardness 1000 ppm
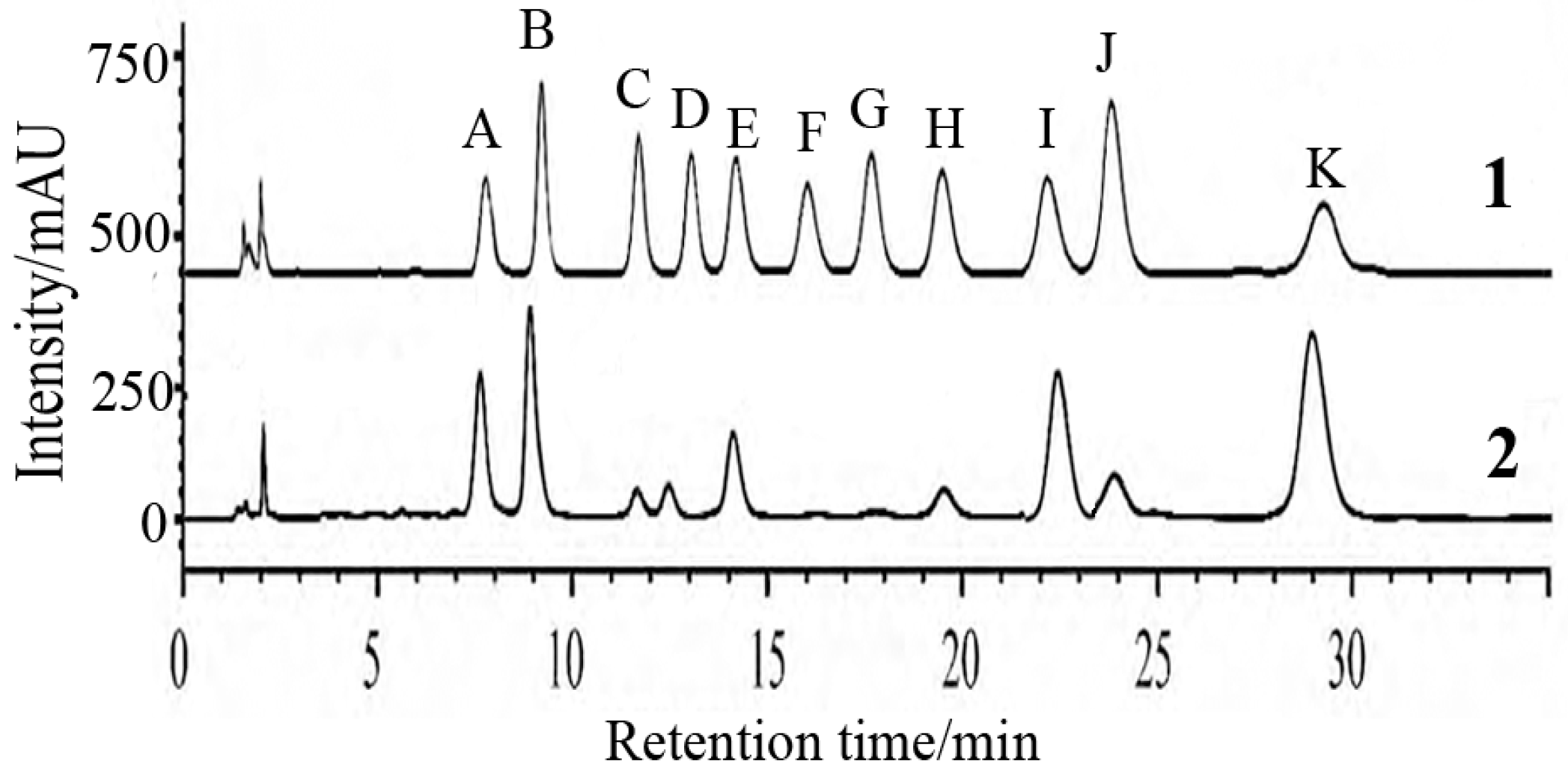
2.2. Analysis of FPS Physicochemical Property
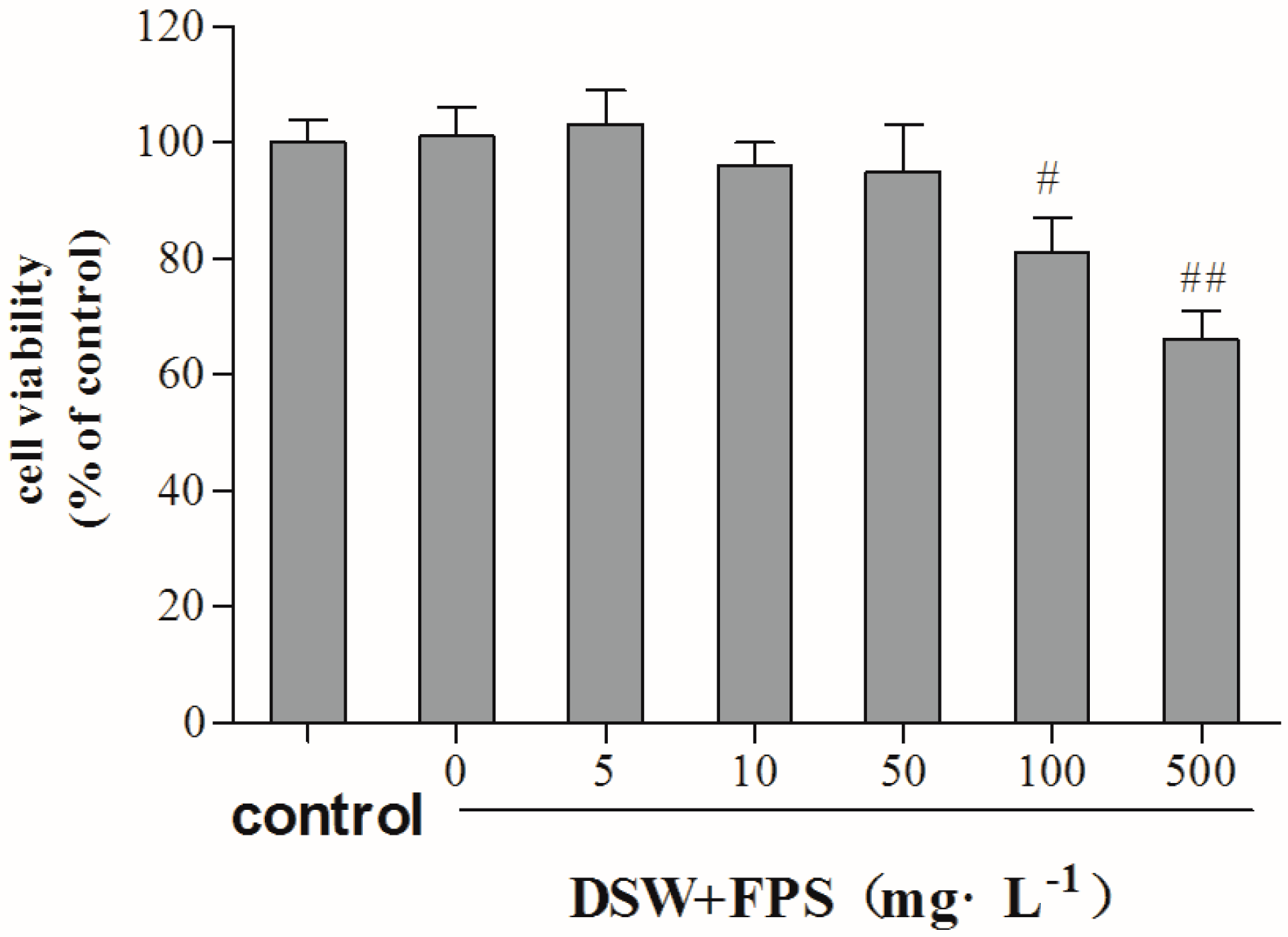
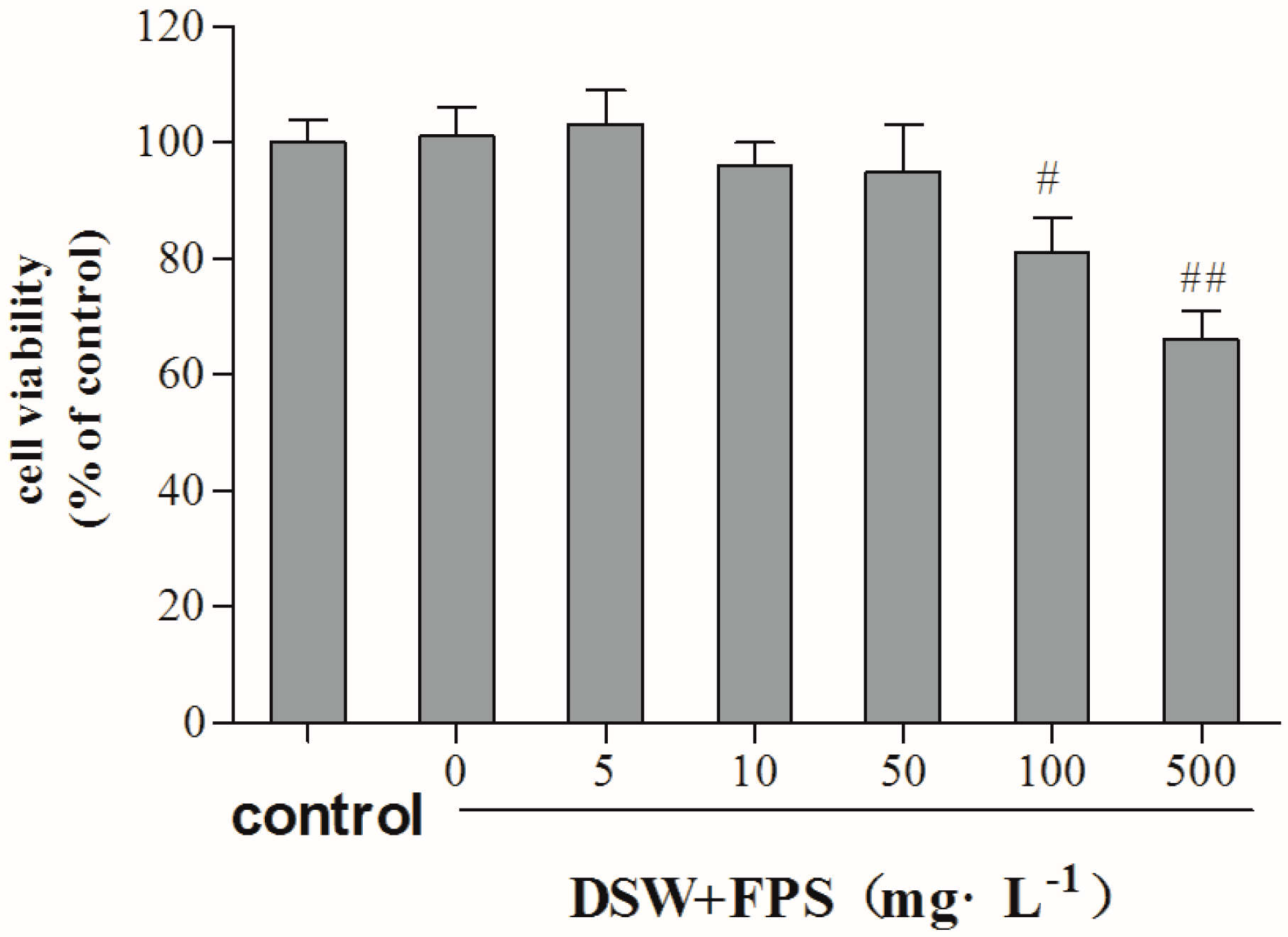
2.3. Cytotoxity of Combination Treatment of DSW and FPS on Hepg2 Cells
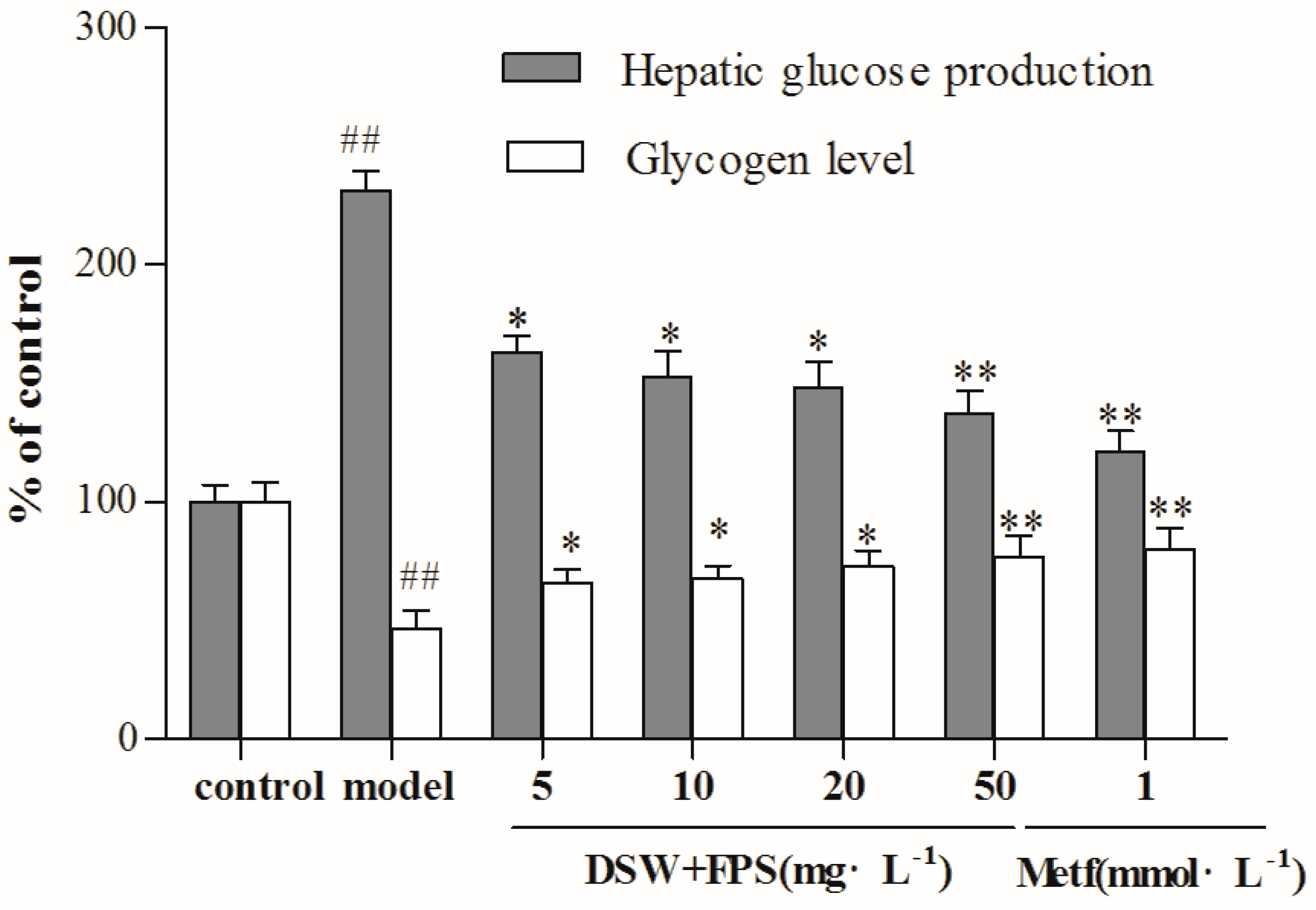
2.4. Combination Treatment of DSW and FPS Repressed Hepatic Glucose Production and Increased Glycogen Level in IR-Hepg2 Cells
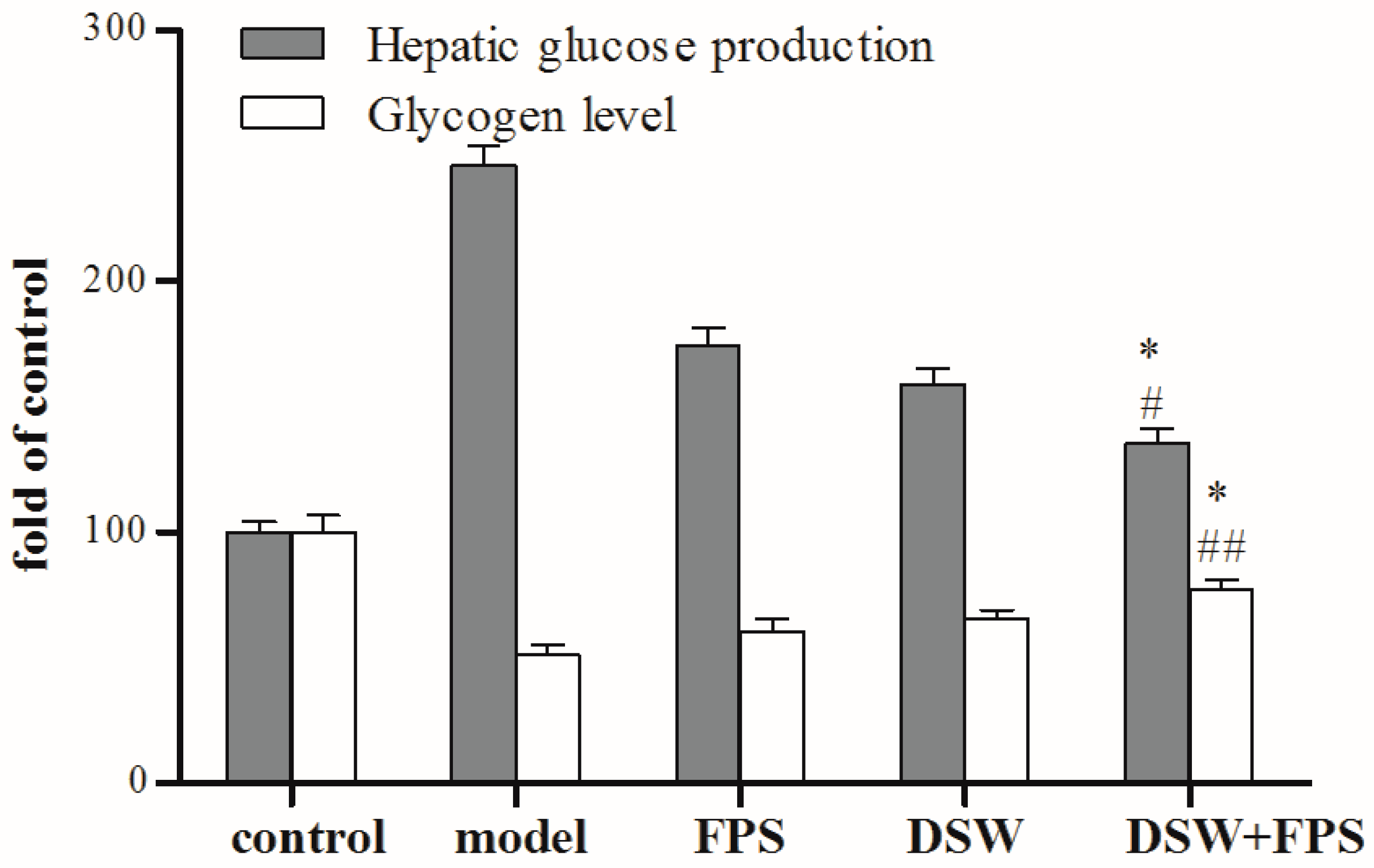
2.5. Comparison on Hepatic Glucose Production and Glycogen Level between Combination Treatment of DSW and FPS and Single Application of DSW or FPS in IR-HepG2
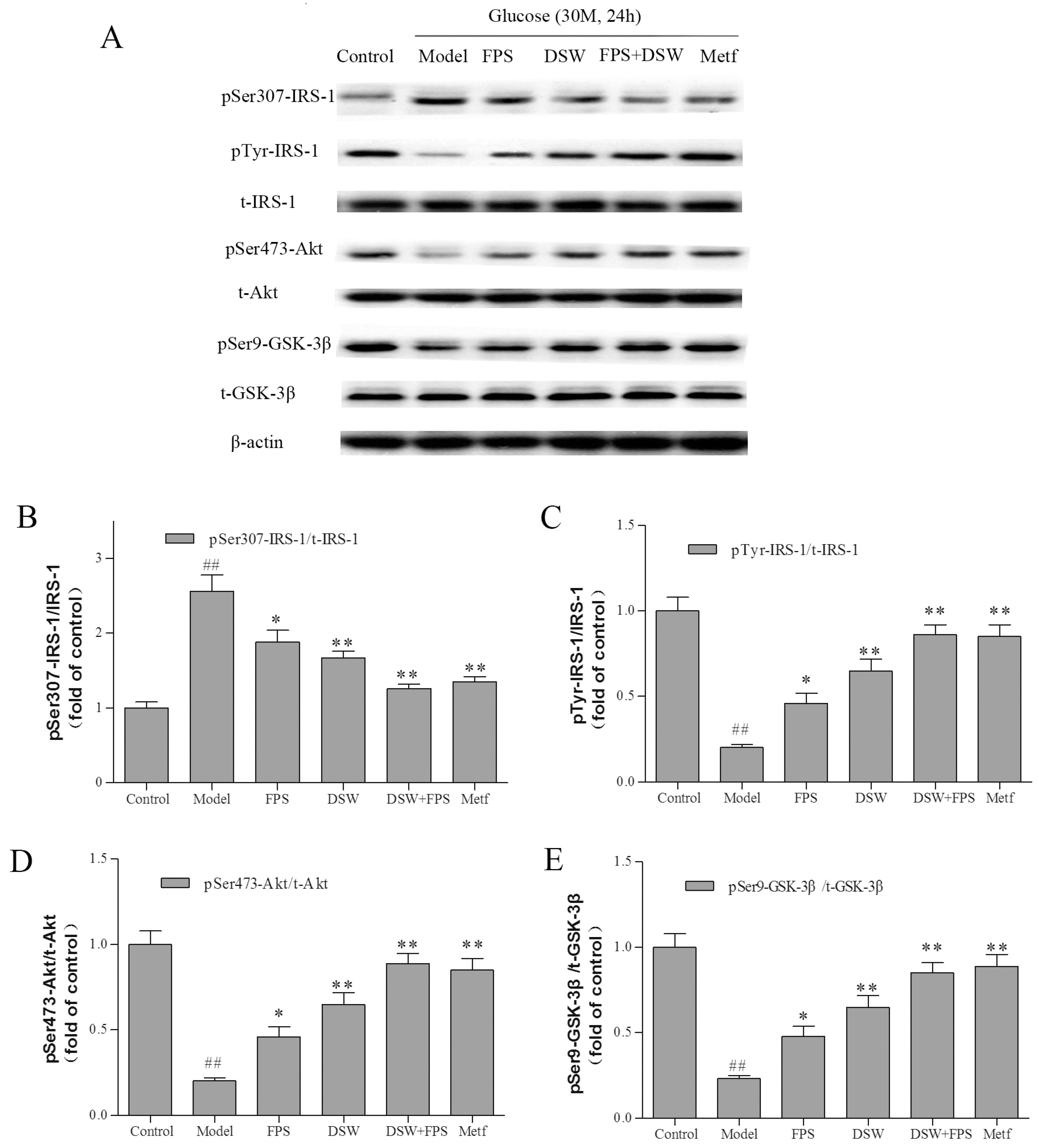
2.6. Effects of Combination Treatment of DSW and FPS on Insulin Signaling in HepG2 Cells under High Glucose Condition
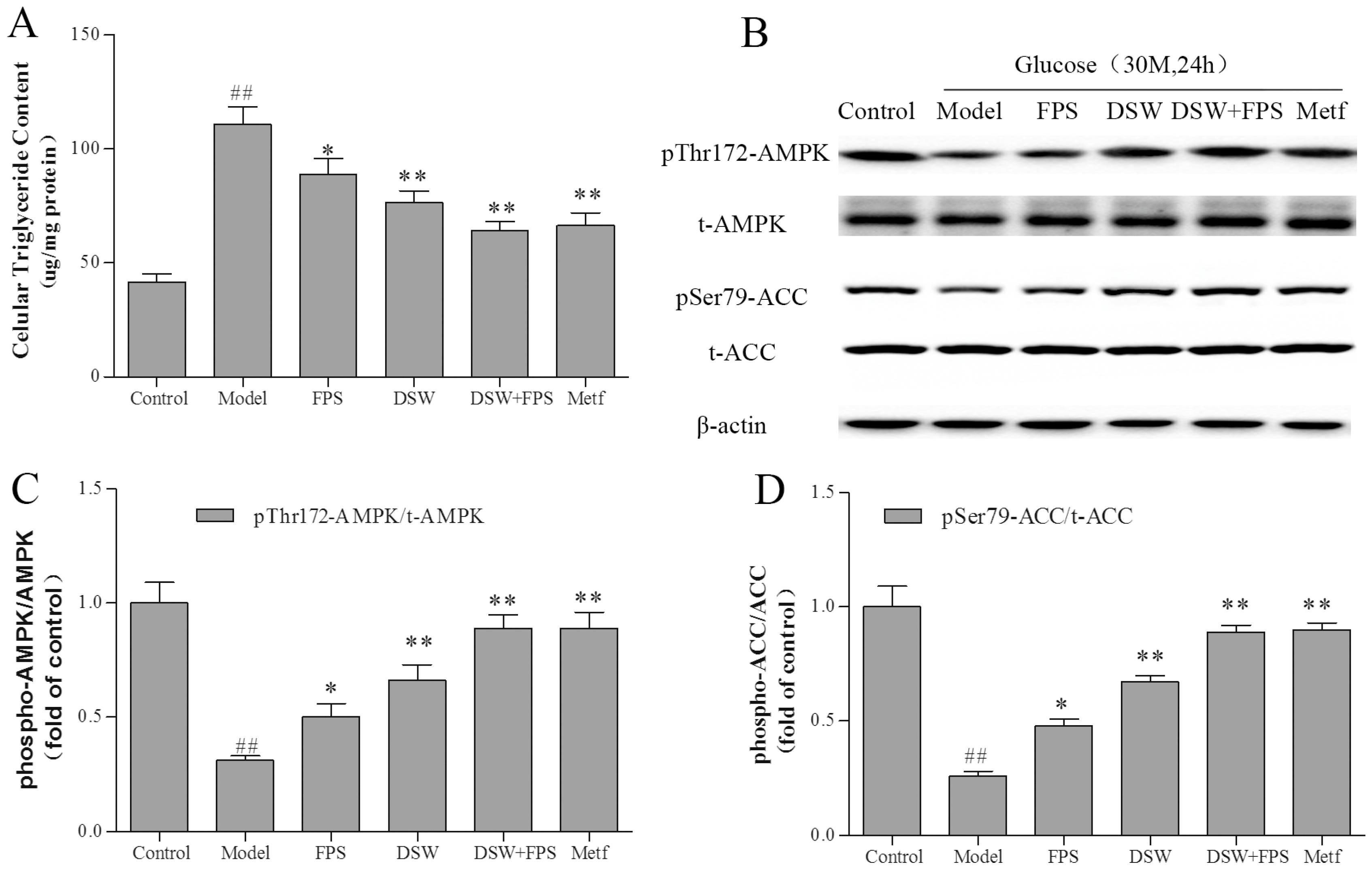
2.7. Combination Treatment of DSW and FPS Reversed the Suppression of AMPK and ACC Phosphorylation and the Accumulation of Triglyceride by High Glucose Concentration
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation and Elemental Analyses of DSW of Hardness 1000 ppm
4.3. Preparation and Physicochemical Property Analyses of FPS
4.4. Cell Culture and Drug Administration
4.5. Cytotoxity Assay
4.6. Establishment of IR-Hepg2 Cell Model by High Glucose Concentration
4.7. Hepatic Glucose Production Assay
4.8. Analysis of Glycogen Contents
4.9. Preparation of Protein Extract of HepG2 Cells
4.10. Measurement of Cellular Triglyceride
4.11. Western Blot Analysis
4.12. Immumoprecipitation
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Petersen, K.F.; Shulman, G.I. New insights into the pathogenesis of insulin resistance in humans using magnetic resonance spectroscopy. Obesity 2006, 14, S34–S40. [Google Scholar] [CrossRef] [PubMed]
- Rachel, J.P.; Varman, T.S.; Kitt, F.P.; Gerald, I.S. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar]
- Nakajima, K.; Yamauchi, K.; Shigematsu, S.; Ikeo, S.; Komatsu, M.; Aizawa, T.; Hashizume, K. Selective attenuation of metabolic branch of insulin receptor down-signaling by high glucose in a hepatoma cell line, HepG2 cells. J. Biol. Chem. 2000, 275, 20880–20886. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Lin, J.K. Epigallocatechin gallate (EGCG) attenuates high glucose-induced insulin signaling blockade in human hepG2 hepatoma cells. Mol. Nutr. Food Res. 2008, 52, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, S.N.; Hardie, D.G.; Morrice, N.; Tornqvist, H.E. 5′-AMP-activated protein kinase phosphorylates IRS-1 onSer-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J. Biol. Chem. 2001, 276, 46912–46916. [Google Scholar] [CrossRef] [PubMed]
- Tahrani, A.A.; Piya, M.K.; Kennedy, A.; Barnett, A.H. Glycaemic control in type 2 diabetes: Targets and new therapies. Pharmacol. Ther. 2010, 125, 328–361. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.Y.; Qian, K.; Morris-Natschke, S.L.; Hsu, C.S.; Lee, K.H. Recent discovery of plant-derived anti-diabetic natural products. Nat. Prod. Rep. 2012, 29, 580–606. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.J.; Joo, E.J. Effect of the supply of natural water from deep sea rock on the immune response and antioxidant activity in rats. J. Anim. Sci. 2006, 48, 211–215. [Google Scholar]
- Sheu, M.J.; Chou, P.Y.; Lin, W.H.; Pan, C.H.; Chien, Y.C.; Chung, Y.L.; Liu, F.C.; Wu, C.H. Deep sea water modulates blood pressure and exhibits hypolipidemic effects via the AMPK-ACC athway: An in vivo study. Mar. Drugs 2013, 11, 2183–2202. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Hao, J.J.; Peng, W.; Qiu, P.; Li, C.; Guan, H. Modulation of Lipid Metabolism by Deep-Sea Water in Cultured Human Liver (HepG2) Cells. Mar. Biotechnol. 2014, 16, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Y.; Yang, F.L.; Hsu, H.W.; Lu, Y.F. Drinking deep seawater decreases serum total and low-density lipoprotein-cholesterol in hypercholesterolemic subjects. J. Med. Food 2012, 15, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, M.; Yoshioka, S.; Hamada, A.; Takuma, D.; Yokota, J.; Kusunose, M.; Kyotani, S.; Kawakita, H.; Odani, K.; Tsutsui, Y.; et al. Difference between deep seawater and surface seawater in the preventive effect of atherosclerosis. Biol. Pharm. Bull. 2004, 27, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Hamada, A.; Cui, T.; Yokota, J.; Yamamoto, S.; Kusunose, M.; Miyamura, M.; Kyotani, S.; Kaneda, R.; Tsutsui, Y.; et al. Pharmacological activity of deep-sea water: Examination of hyperlipemia prevention and medical treatment effect. Biol. Pharm. Bull. 2003, 26, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Shin, E.J.; Park, J.E.; Shon, Y.H. Anti-diabetic effect of balanced deep-sea water and its mode of action in high-fat diet induced diabetic mice. Mar. Drugs 2013, 11, 4193–4212. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Park, J.E.; Shon, Y.H. Stimulatory Effect of Balanced Deep-Sea Water Containing Chitosan Oligosaccharides on Glucose Uptake in C2C12 Myotubes. Mar. Biotechnol. 2016, 18, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, S.H.; Yoo, Y.G.; Chu, Y.S.; Shon, Y.H.; Nam, K.S.; Yun, J.W. Inhibitory effect of deep-sea water on differentiation of 3T3-L1 adipocytes. Mar. Biotechnol. 2009, 11, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Park, J.E.; Shin, E.J.; Shon, Y.H. Modulation of glucose metabolism by balanced deep-sea water ameliorates hyperglycemia and pancreatic function in streptozotocin-induced diabetic mice. PLoS ONE 2014, 9, e102095. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.T.; Hwang, D.F. Effect of deep sea water on the exercise-induced fatigue of rats. J. Food Drug Anal. 2009, 17, 133–136. [Google Scholar]
- Tsuchiya, Y.; Watanabe, A.; Fujisawa, N. Effects of desalted deep seawater on hematologic and blood chemical values in mice. Tohoku J. Exp. Med. 2004, 203, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A. Fucoidan as a Potential Therapeutic for Major Blinding Diseases—A Hypothesis. Mar. Drugs 2016, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Teruya, K.; Yoshida, T.; Eto, H.; Shirahata, S. Fucoidan extract enhances the anti-cancer activity of chemotherapeutic agents in MDA-MB-231 and MCF-7 breast cancer cells. Mar. Drugs 2013, 11, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Ludmylla, C.; Ana, G. Sulfated Seaweed Polysaccharides as Multifunctional Materials in Drug Delivery Applications. Mar. Drugs 2016, 14, 42. [Google Scholar]
- Bell, D.S. The case for combination therapy as first-line treatment for the type 2 diabetic patient. Treat. Endocrinol. 2006, 5, 131–137. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Jeong, S.K.; Kim, H.R.; Kim, D.S.; Chae, S.W.; Chae, H.J. Effects of triglyceride on ER stress and insulin resistance. Biochem. Biophys. Res. Commun. 2007, 363, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Gammon, S.R.; Knippers, J.D.; Paulsen, S.R.; Rubink, D.S.; Winder, W.W. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 2002, 92, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: Molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef] [PubMed]
- De Lordes Lima, M.; Cruz, T.; Pousada, J.C.; Rodrigues, L.E.; Barbosa, K.; Cangucu, V. The effect of magnesium supplementation in increasing doses on the control of type 2 diabetes. Diabetes Care 1998, 21, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moran, M.; Guerrero-Romero, F. Oral magnesium supplementation improves insulin sensitivity and metabolic control in type 2 diabetic subjects: A randomized double-blind controlled trial. Diabetes Care 2003, 26, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Manson, J.E.; Buring, J.E.; Liu, S. Dietary magnesium intake in relation to plasma insulin levels and risk of type 2 diabetes in women. Diabetes Care 2004, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zeng, C.; Li, X.X.; Gong, Q.Y.; Lei, G.H.; Yang, T.B. Association among dietary magnesium, serum magnesium, and diabetes: A cross-sectional study in middle-aged and older adults. J. Health Popul. Nutr. 2016, 35, 33. [Google Scholar] [CrossRef] [PubMed]
- Konishi, K.; Wada, K.; Tamura, T.; Tsuji, M.; Kawachi, T.; Nagata, C. Dietary magnesium intake and the risk of diabetes in the Japanese community: Results from the Takayama study. Eur. J. Nutr. 2017, 56, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Drouillet, P.; Balkau, B.; Charles, M.A.; Vol, S.; Bedouet, M.; Ducimetière, P.; Desir Study Group. Calcium consumption and insulin resistance syndrome parameters. Data from the Epidemiological Study on the Insulin Resistance Syndrome (DESIR). Nutr. Metab. Cardiovasc. Dis. 2007, 17, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Ferrat, A.; Lavandero, S.; Jaimovich, E.; Klip, A. Calcium signaling in insulin action on striated muscle. Cell Calcium. 2014, 56, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.W.; Kouta, S.; Robinson, R.; MacNeil, S.; Heller, S. Chromium supplementation improves insulin resistance in patients with type 2 diabetes mellitus. Diabet. Med. 2000, 17, 684–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Zhang, X.H.; Russell, J.C.; Hulver, M.; Cefalu, W.T. Chromium picolinate enhances skeletal muscle cellular insulin signaling in vivo in obese, insulin-resistant JCR:LA-cp rats. J. Nutr. 2006, 136, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Refaie, F.M.; Esmat, A.Y.; Mohamed, A.F. Effect of chromium supplementation on the diabetes induced-oxidative stress in liver and brain of adult rats. Biometals 2009, 22, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Van Oort, M.M.; Yao, M.; Van der Horst, D.J.; Rodenburg, K.W. Insulin and Chromium Picolinate Induce Translocation of CD36 to the Plasma Membrane through Different Signaling Pathways in 3T3-L1 Adipocytes, and with a Differential Functionality of the CD36. Biol. Trace Elem. Res. 2011, 142, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Rosanoff, A.; Seelig, M.S. Comparison of Mechanism and Functional Effects of Magnesium and Statin Pharmaceuticals. J. Am. Coll. Nutr. 2004, 23, 501S–505S. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.T.; Kim, Y.D.; Jung, Y.M.; Park, D.C.; Lee, D.S.; Ku, S.K.; Li, X.; Lu, Y.; Chao, G.H.; Kim, K.J.; et al. Low molecular weight fucoidan (LMWF) improves ER stress-reduced insulin sensitivity through AMPK activation in L6 myotubes and restores lipid homeostasis in a mouse model of type 2 diabetes. Mol. Pharmacol. 2013, 84, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Heeba, G.H.; Morsy, M.A. Fucoidan ameliorates steatohepatitis and insulin resistance by suppressing oxidative stress and inflammatory cytokines in experimental non-alcoholic fatty liver disease. Environ. Toxicol. Pharmacol. 2015, 40, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yu, J.; Ma, Z.; Zhang, H.; Xie, F. Effects of fucoidan on insulin stimulation and pancreatic protection via the cAMP signaling pathway in vivo and in vitro. Mol. Med. Rep. 2015, 12, 4501–4507. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Yu, G.; Wang, W.; Zhao, X.; Zhang, J.; Ewart, S.H. Properties of polysaccharides in several seaweeds from Atlantic Canada and their potential anti-influenza viral activities. J. Ocean Univ. China 2012, 11, 205–212. [Google Scholar] [CrossRef]
- Zang, M.; Zuccollo, A.; Hou, X.; Nagata, D.; Walsh, K.; Herscovitz, H.; Brecher, P.; Ruderman, N.B.; Cohen, R.A. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004, 279, 47898–47905. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
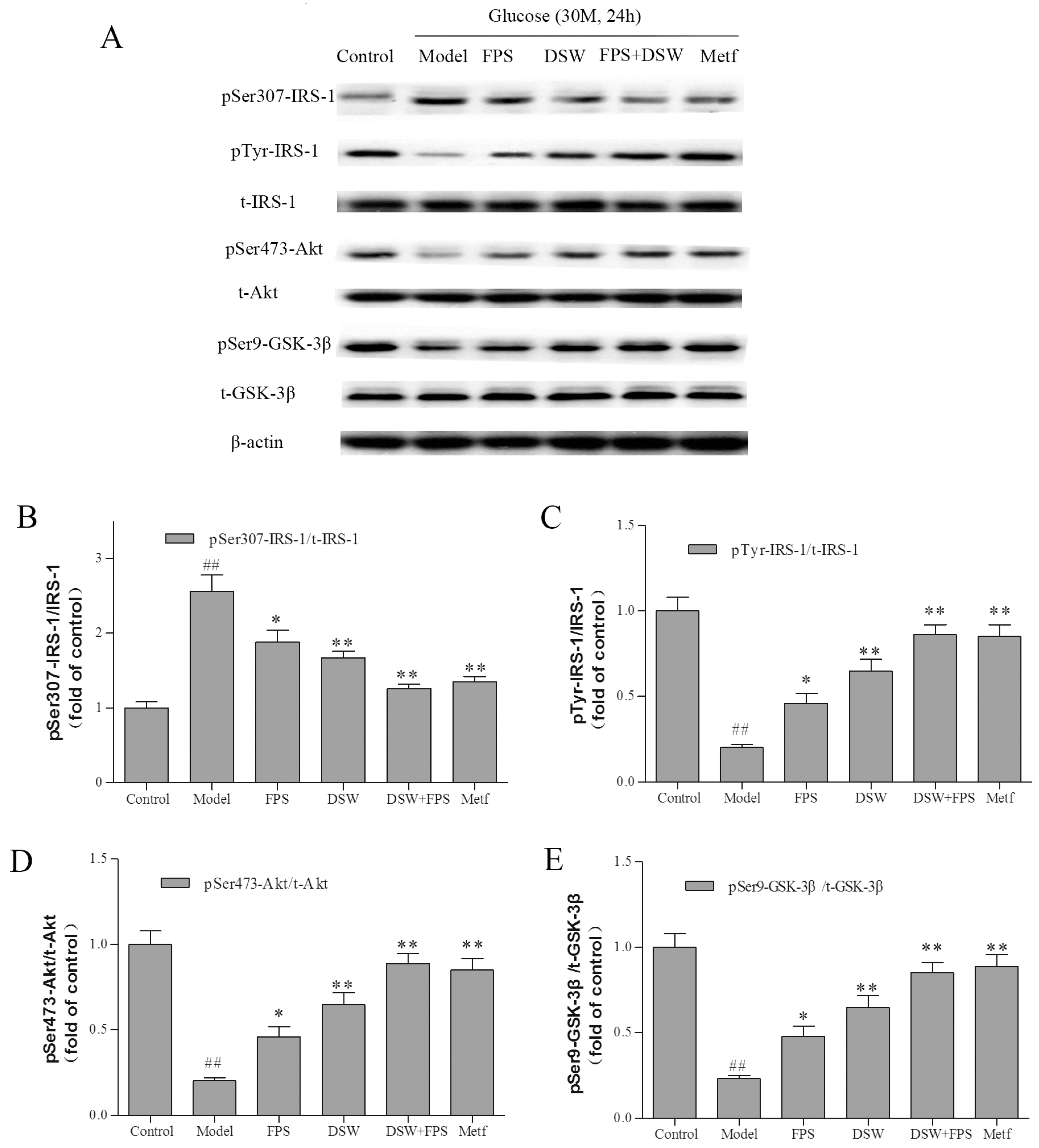

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Macro Element/mg·L−1 | Trace Element/µg·L−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Na | Ca | Mg | V | Cr | Mn | Zn | Se | Hg | Pb | |
| Content | 68.3 | 70.6 | 201.8 | 0.62 | 1.2 | 1.38 | 5.7 | 0.48 | ND | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, S.; Peng, W.-B.; Zhou, H.-L. Combination Treatment of Deep Sea Water and Fucoidan Attenuates High Glucose-Induced Insulin-Resistance in HepG2 Hepatocytes. Mar. Drugs 2018, 16, 48. https://doi.org/10.3390/md16020048
He S, Peng W-B, Zhou H-L. Combination Treatment of Deep Sea Water and Fucoidan Attenuates High Glucose-Induced Insulin-Resistance in HepG2 Hepatocytes. Marine Drugs. 2018; 16(2):48. https://doi.org/10.3390/md16020048
Chicago/Turabian StyleHe, Shan, Wei-Bing Peng, and Hong-Lei Zhou. 2018. "Combination Treatment of Deep Sea Water and Fucoidan Attenuates High Glucose-Induced Insulin-Resistance in HepG2 Hepatocytes" Marine Drugs 16, no. 2: 48. https://doi.org/10.3390/md16020048
APA StyleHe, S., Peng, W.-B., & Zhou, H.-L. (2018). Combination Treatment of Deep Sea Water and Fucoidan Attenuates High Glucose-Induced Insulin-Resistance in HepG2 Hepatocytes. Marine Drugs, 16(2), 48. https://doi.org/10.3390/md16020048