New Bis-Alkenoic Acid Derivatives from a Marine-Derived Fungus Fusarium solani H915


Abstract
:1. Introduction
2. Results
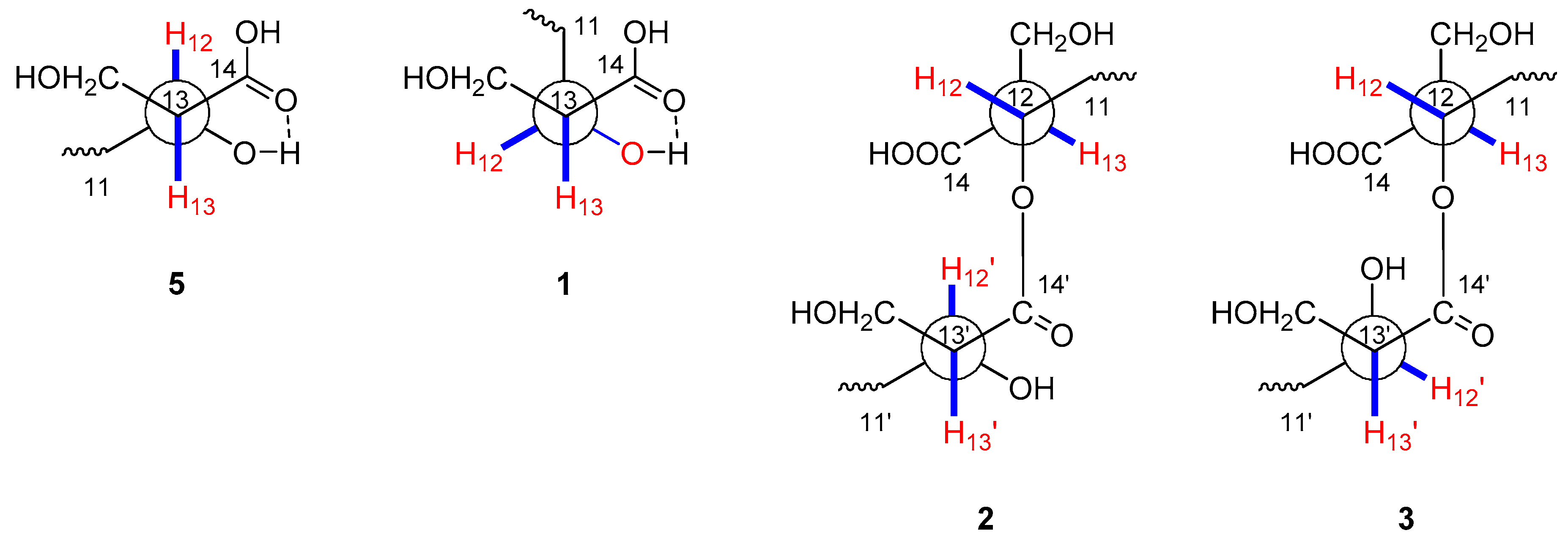
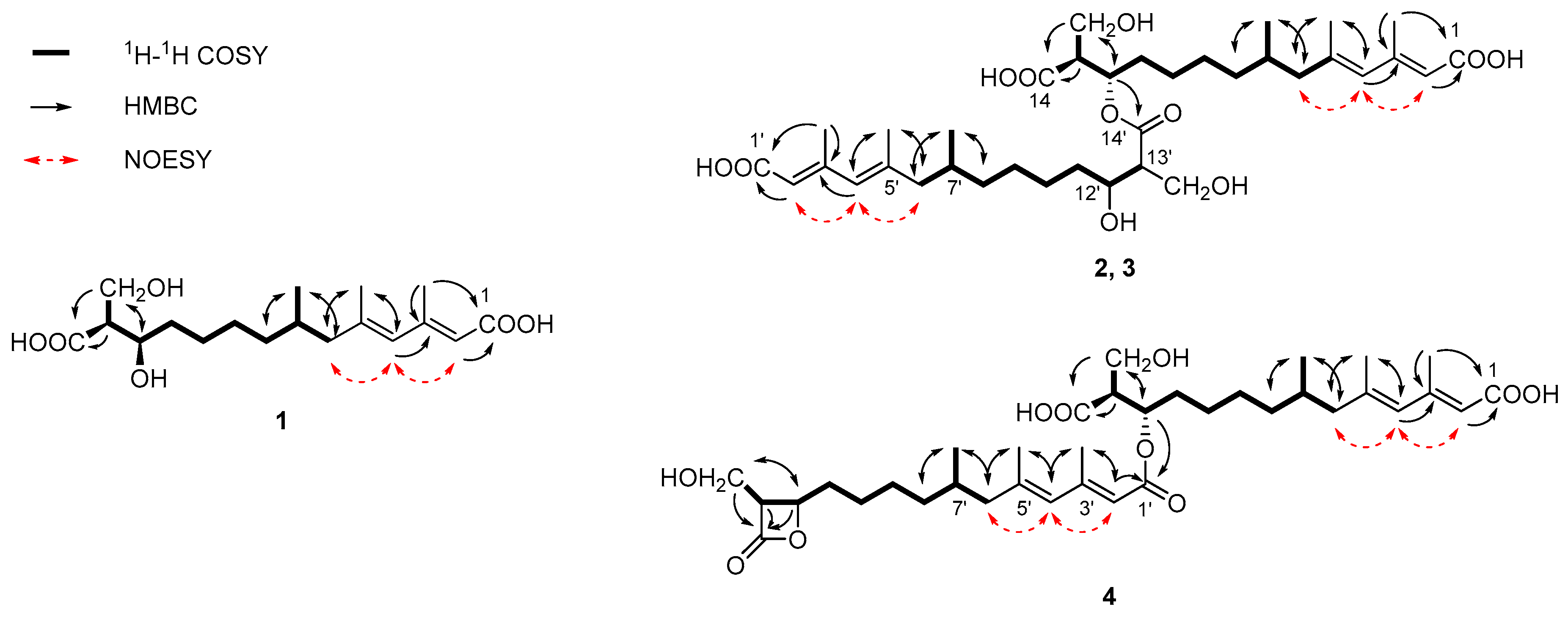
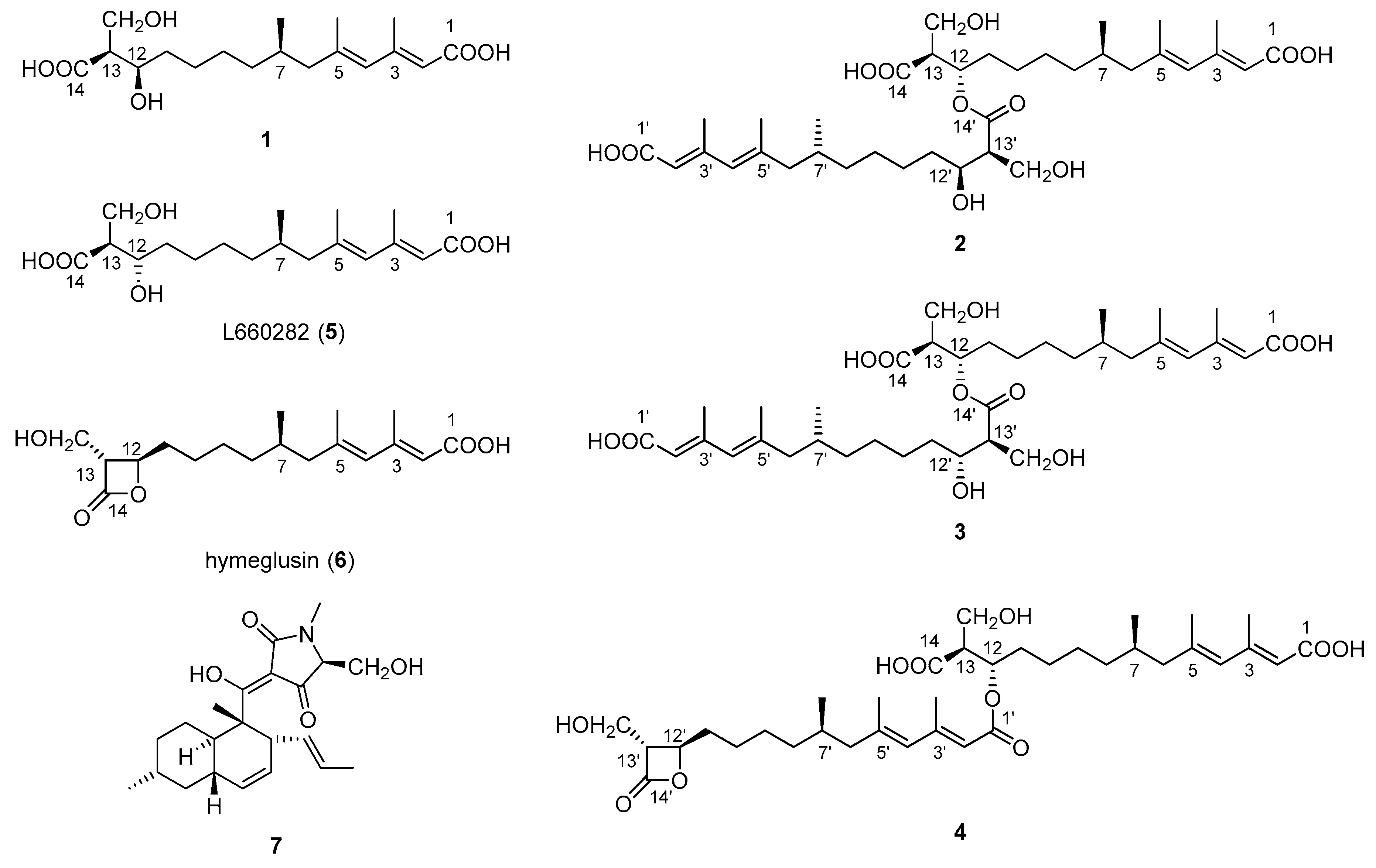
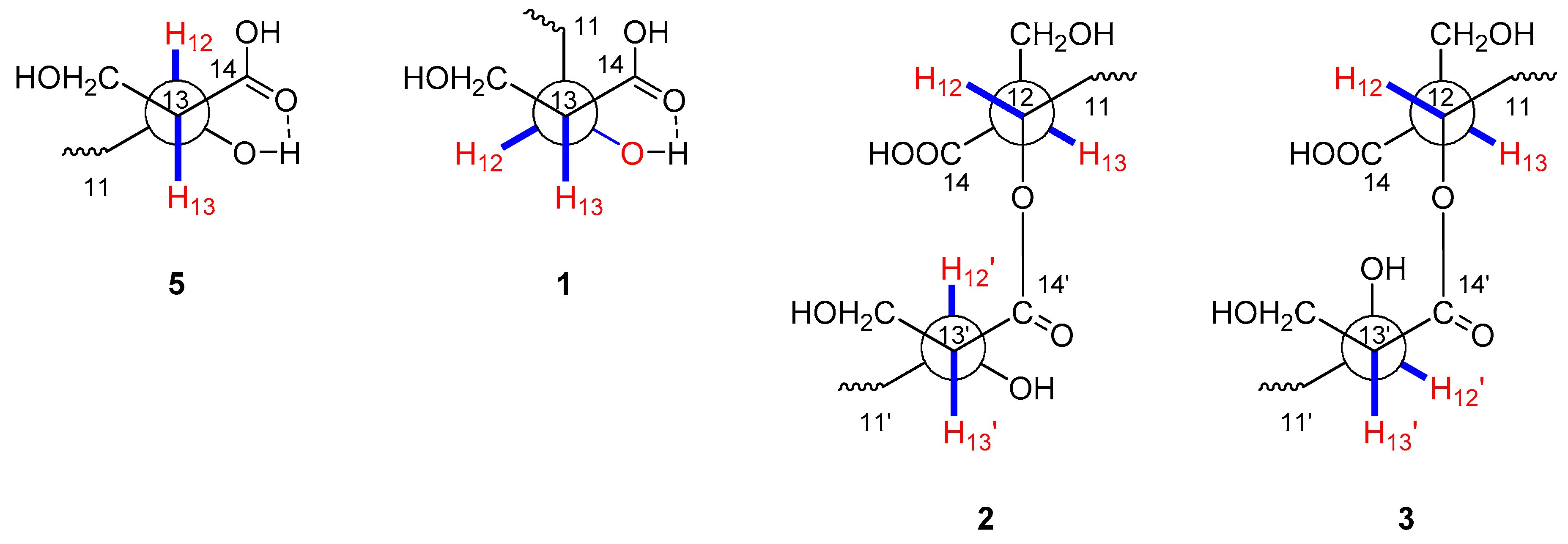
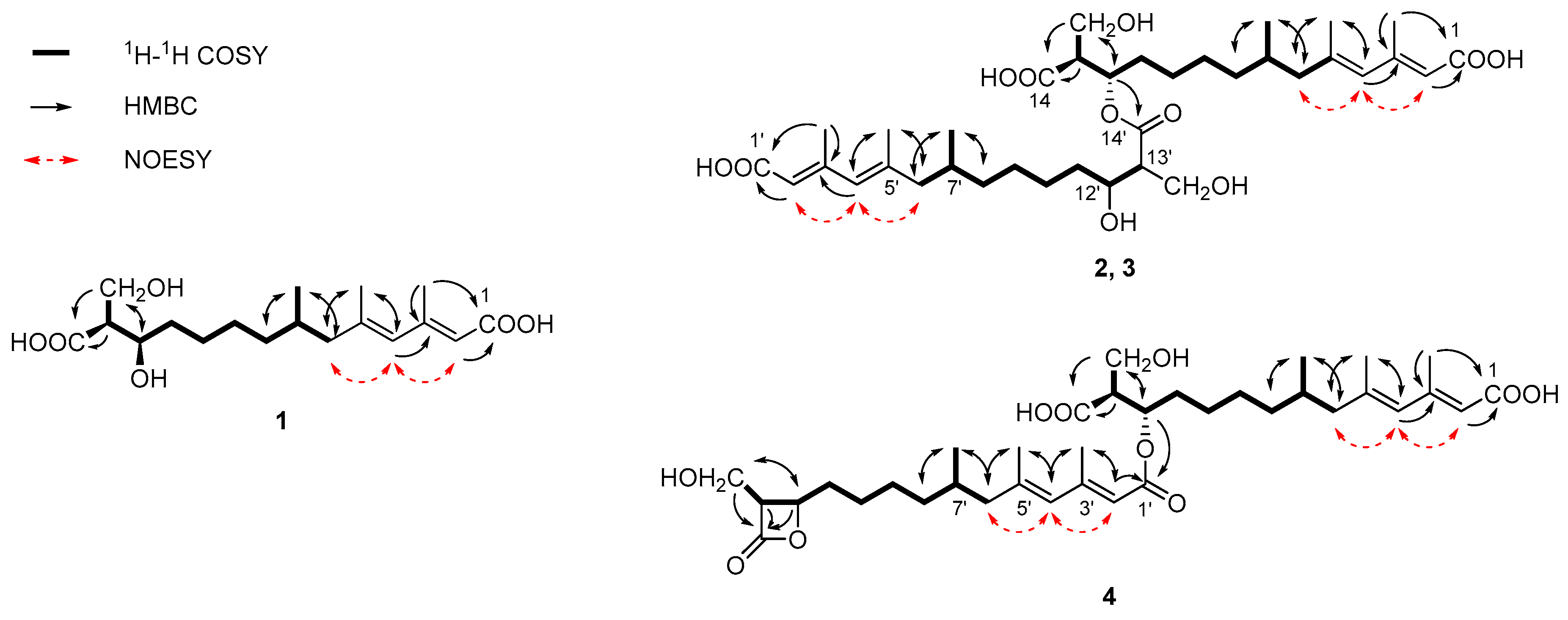
2.1. Structural Identification of New Compounds
2.2. Antifungal Activity
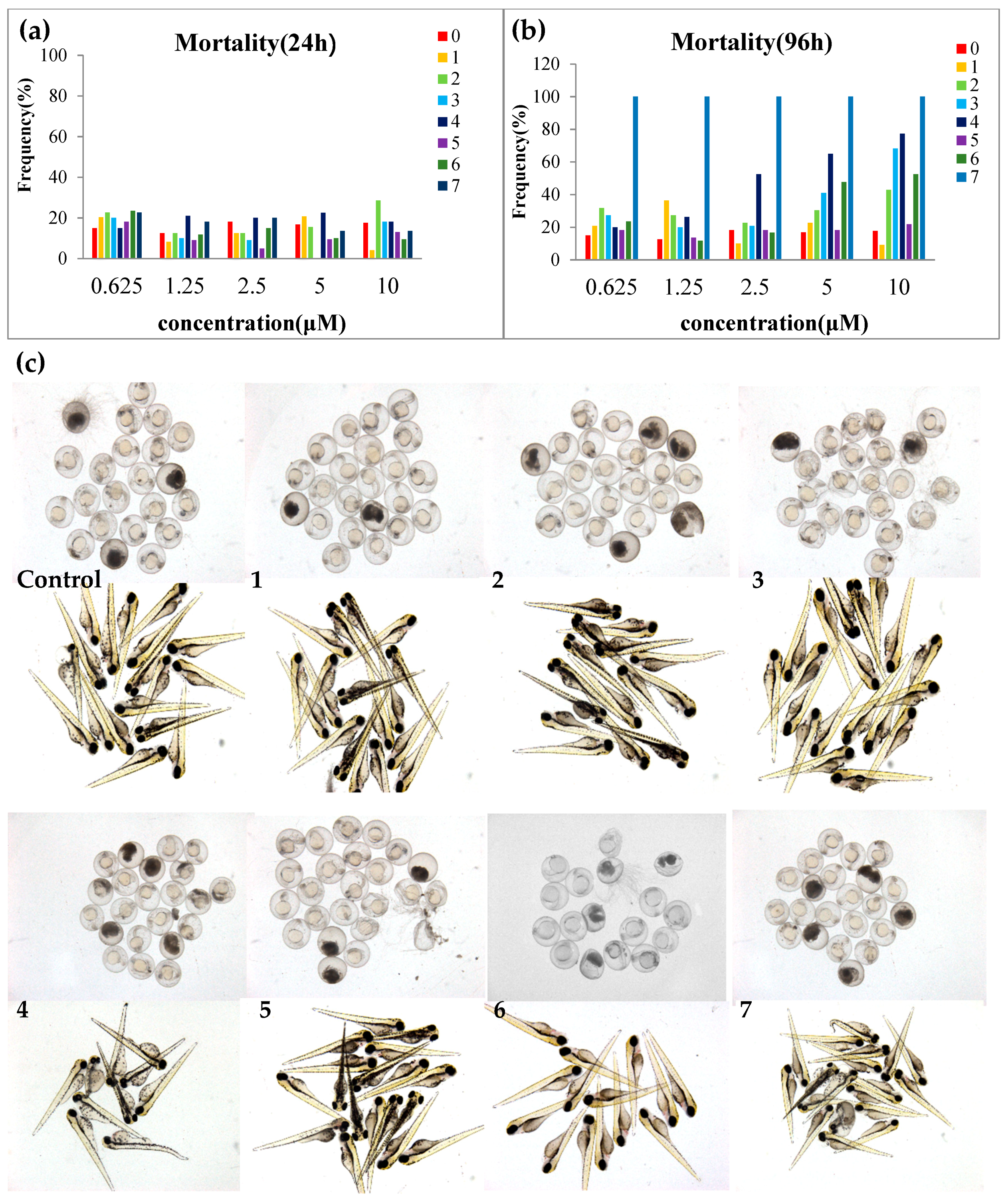
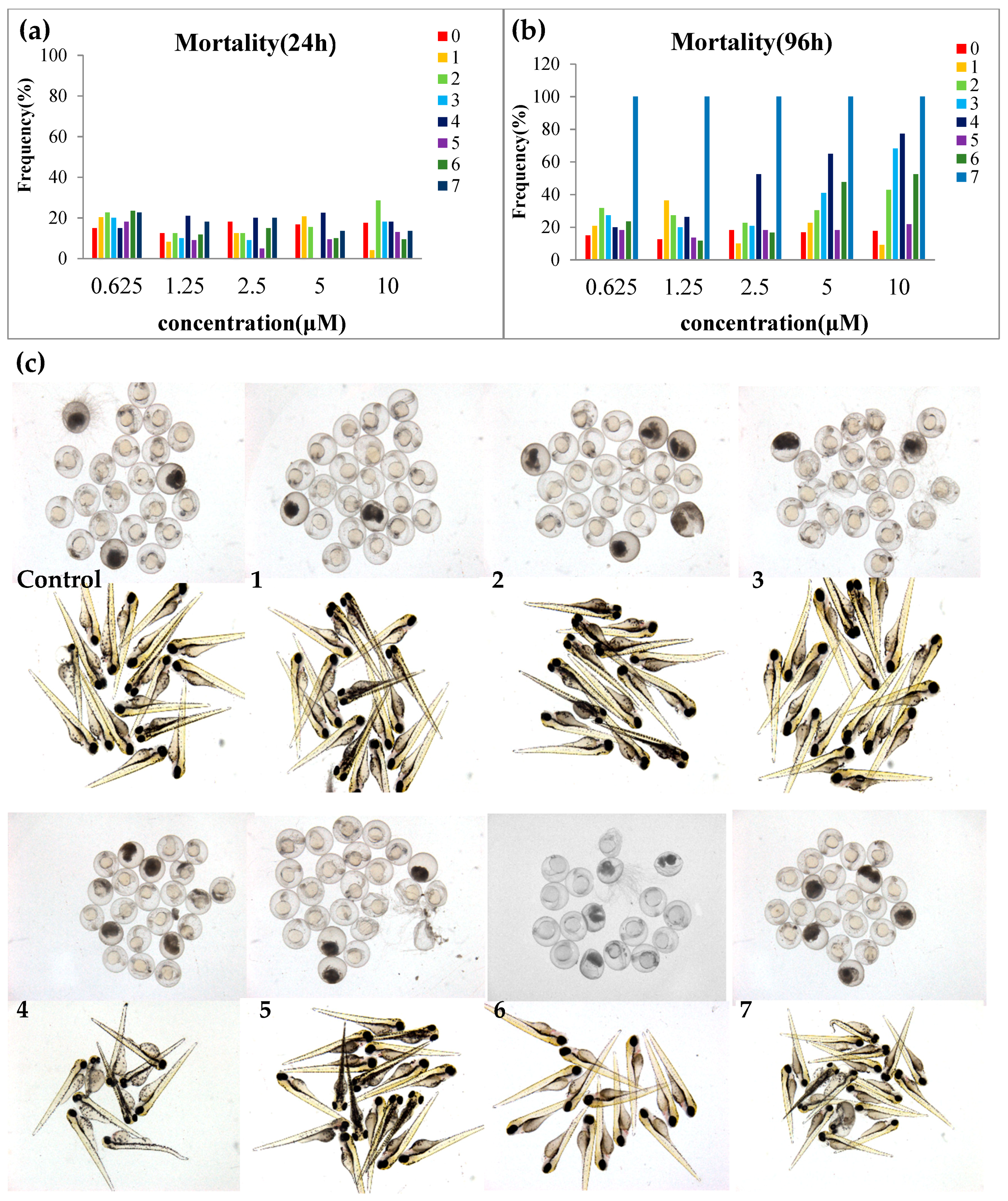
2.3. Toxicity Evaluation
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Fungal Strain and Fermentation
4.3. Extraction and Isolation
4.4. Alkaline Hydrolysis of 2–4 and 6
4.5. Antifungal Activity
4.6. Antiproliferative Effects on Zebrafish Embryo
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feller, I.C.; Lovelock, C.E.; Berger, U.; Mckee, K.L.; Joye, S.B.; Ball, M.C. Biocomplexity in Mangrove Ecosystems. Annu. Rev. Mar. Sci. 2010, 2, 395. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Bartlett, J.G.; Gilbert, D.N. The future of antibiotics and resistance. N. Engl. J. Med. 2013, 368, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, D.; Vijayakumar, C.; Kumar, N.; Samiyappan, R. PGPR-induced defense responses in the tea plant against blister blight disease. Crop Protect. 2007, 26, 556–565. [Google Scholar] [CrossRef]
- Saha, D.; Dasgupta, S.; Saha, A. Antifungal activity of some plant extracts against fungal pathogens of tea (Camellia sinensis). Pharm. Biol. 2005, 43, 87–91. [Google Scholar] [CrossRef]
- Gunasekera, T.; Paul, N.; Ayres, P. The effects of ultraviolet-B (UV-B: 290–320 nm) radiation on blister blight disease of tea (Camellia sinensis). Plant Pathol. 1997, 46, 179–185. [Google Scholar] [CrossRef]
- Ponmurugan, P.; Baby, U.; Rajkumar, R. Growth, photosynthetic and biochemical responses of tea cultivars infected with various diseases. Photosynthetica 2007, 45, 143–146. [Google Scholar] [CrossRef]
- Sanjay, R.; Ponmurugan, P.; Baby, U. Evaluation of fungicides and biocontrol agents against grey blight disease of tea in the field. Crop Protect. 2008, 27, 689–694. [Google Scholar] [CrossRef]
- Sarkar, S.; Ajay, D.; Pradeepa, N.; Balamurugan, A.; Premkumar, R. Evaluation of chemical and neem pesticides against Pestalotiopsis theae causing grey blight disease of tea. Ann. Plant Protect. Sci. 2009, 17, 252–253. [Google Scholar]
- Chakraborty, B.; Basu, P.; Das, R.; Saha, A.; Chakraborty, U. Detection of cross reactive antigens between Pestalotiopsis theae and tea leaves and their cellular location. Ann. Appl. Biol. 1995, 127, 11–21. [Google Scholar] [CrossRef]
- Staeubert, C.; Krakowsky, R.; Bhuiyan, H.; Witek, B.; Lindahl, A.; Broom, O.; Nordstroem, A. Increased lanosterol turnover: A metabolic burden for daunorubicin-resistant leukemia cells. Med. Oncol. 2016, 33, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tang, X.X.; Yan, X.; Wu, Z.; Yi, Z.W.; Fang, M.J.; Su, X.; Qiu, Y.K. A new macrolactin antibiotic from deep sea-derived bacteria Bacillus subtilis B5. Nat. Prod. Res. 2016, 30, 2777–2782. [Google Scholar] [CrossRef]
- Aldridge, D.C.; Giles, D.; Turner, W.B. Antibiotic 1233A, a fungal β-lactone. J. Chem. Soc. C 1971, 3888–3891. [Google Scholar] [CrossRef]
- Kumagai, H.; Tomoda, H.; Omura, S. Biosynthesis of antibiotic 1233A (F-244) and preparation of [14C]1233A. J. Antibiot. 1992, 45, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Ratnaweera, P.B.; de Silva, E.D.; Williams, D.E.; Andersen, R.J. Antimicrobial activities of endophytic fungi obtained from the arid zone invasive plant Opuntia dillenii and the isolation of equisetin, from endophytic Fusarium sp. BMC Complement. Altern. Med. 2015, 15, 220. [Google Scholar] [CrossRef]
- Parng, C.; Seng, W.L.; Semino, C.; McGrath, P. Zebrafish: A preclinical model for drug screening. Assay Drug Dev. Technol. 2002, 1, 41–48. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 * |
|---|---|---|---|---|---|---|
| 2 | 5.50, br.s | 5.60, br.s | 5.58, br.s | 5.56, br.s | 5.50, s | |
| 2′ | 5.56, br.s | 5.57, br.s | 5.59, br.s | 5.57, br.s | ||
| 4 | 5.67, br.s | 5.70, br.s | 5.71, br.s | 5.71, br.s | 5.66, s | |
| 4′ | 5.71, br.s | 5.72, br.s | 5.76, br.s | 5.73, br.s | ||
| 6a | 2.00, dd (13.0, 6.1) | 2.02, m | 2.05, dd (5.0, 3.3) | 2.04, dd (13.1, 6.0) | 2.00, dd (13.1, 6.0) | |
| 6b | 1.75, dd (13.3, 8.3) | 1.82, m | 1.82, m | 1.80, dd (12.1, 4.4) | 1.75, dd (13.1, 8.3) | |
| 6′a | 2.06, dd (13.6, 6.2) | 2.08, m | 2.08, dd (13.3, 6.0) | 2.07, dd (13.2, 6.1) | ||
| 6′b | 1.79, m | 1.79, m | 1.83, dd (13.2, 8.4) | 1.83, dd (12.8, 8.3) | ||
| 7 | 1.58, m | 1.60, m | 1.62, m | 1.61, m | 1.57, br.dd (12.7, 6.4) | |
| 7′ | 1.60, m | 1.62, m | 1.64, m | 1.64, m | ||
| 8a | 1.18, m | 1.24, m | 1.24, m | 1.25, m | 1.19, m | |
| 8b | 1.01, m | 1.06, m | 1.05, m | 1.05, m | 1.00, m | |
| 8′a | 1.24, m | 1.24, m | 1.28, m | 1.28, m | ||
| 8′b | 1.02, m | 1.05, m | 1.10, m | 1.10, m | ||
| 9a | 1.20, m | 1.19, m | 1.25, m | 1.27, m | 1.26, m | |
| 9b | 1.20, m | 1.19, m | 1.25, m | 1.20, m | 1.12, m | |
| 9′a | 1.30, m | 1.25, m | 1.27, m | 1.27, m | ||
| 9′b | 1.30, m | 1.25, m | 1.20, m | 1.20, m | ||
| 10a | 1.29, m | 1.32, m | 1.35, m | 1.36, m | 1.36, m | |
| 10b | 1.20, m | 1.20, m | 1.19, m | 1.20, m | 1.15, m | |
| 10′a | 1.41, m | 1.36, m | 1.36, m | 1.36, m | ||
| 10′b | 1.26, m | 1.24, m | ||||
| 11a | 1.34, m | 1.54, m | 1.51, m | 1.57, m | 1.30, m | |
| 11b | 1.24, m | 1.55, m | 1.23, m | |||
| 11′a | 1.31, m | 1.37, m | 1.80, m | 1.80, m | ||
| 11′b | 1.31, m | 1.74, m | 1.73, m | |||
| 12 | 3.55, m | 5.00, m | 5.00, td (7.8, 4.7) | 5.05, q (6.5) | 3.44, td (8.3, 2.9) | |
| 12′ | 3.50, m | 3.62, m | 4.53, td (6.6, 4.2) | 4.53, td (6.7, 4.2) | ||
| 13 | 2.34, dt (8.3, 5.7) | 2.57, td (8.3, 4.6) | 2.60, td (8.5, 4.9) | 2.65, td (8.0, 4.9) | 2.29, td (8.5, 4.6) | |
| 13′ | 2.44, td, (8.8, 4.6) | 2.50, m | 3.50, br.dd (7.8, 3.9) | 3.50, br.dd (7.7, 3.9) | ||
| 3-CH3 | 2.09, d (1.1) | 2.15, br.s | 2.15, br.s | 2.14, d (0.9) | 2.09, d (1.1) | |
| 3′-CH3 | 2.14, br.s | 2.16, br.s | 2.18, br.s | 2.16, d (1.1) | ||
| 5-CH3 | 1.70, d (1.1) | 1.74, br.s | 1.75, br.s | 1.73, br.s | 1.70, d (1.1) | |
| 5′-CH3 | 1.75, br.s | 1.76, br.s | 1.77, br.s | 1.76, d (1.1) | ||
| 7-CH3 | 0.73, d (6.6) | 0.78, d (6.6) | 0.78, d (6.6) | 0.77, d (6.6) | 0.73, d (6.6) | |
| 7′-CH3 | 0.79, d (6.4) | 0.79, d (6.6) | 0.80, d (6.6) | 0.80, d (6.6) | ||
| 13-CH2OH | 3.51, dd (10.5, 8.4) | 3.60, dd (10.4, 4.5) | 3.58, dd (10.5, 9.0) | 3.60, dd (10.1, 1.5) | 3.66, dd (10.4, 4.5) | |
| 3.46, dd (10.4, 5.4) | 3.53, dd (10.4, 9.0) | 3.54, dd (10.5, 4.3) | 3.52, dd (10.5, 4.8) | 3.55, dd (10.2, 9.1) | ||
| 13′-CH2OH | 3.76, dd (10.4, 4.5) | 3.61, dd (10.5, 5.5) | 3.72, dd (11.7, 4.2) | 3.72, dd (11.7, 4.2) | ||
| 3.61, dd (10.4, 9.0) | 3.56, dd (10.4, 5.3) | 3.63, dd (11.7, 3.3) | 3.62, dd (11.7, 3.3) |
| No. | 1 | 2 | 3 | 4 | 5 | No. | 2 | 3 | 4 | 6 * |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 168.1 | 168.3 | 168.1 | 170.8 | 168.1 | 1′ | 168.2 | 168.1 | 166.1 | 170.9 |
| 2 | 118.5 | 119.1 | 118.7 | 118.6 | 118.5 | 2′ | 118.8 | 118.6 | 117.3 | 118.6 |
| 3 | 153.2 | 152.4 | 153.0 | 153.1 | 153.1 | 3′ | 152.8 | 153.1 | 154.6 | 153.1 |
| 4 | 129.6 | 129.7 | 129.6 | 129.6 | 129.6 | 4′ | 129.6 | 129.6 | 129.5 | 129.6 |
| 5 | 141.3 | 140.8 | 141.2 | 141.2 | 141.3 | 5′ | 141.1 | 141.2 | 142.3 | 141.3 |
| 6 | 48.9 | 48.8 | 48.9 | 48.6 | 48.8 | 6′ | 48.8 | 48.8 | 48.9 | 48.8 |
| 7 | 30.8 | 30.7 | 30.7 | 30.6 | 30.8 | 7′ | 30.9 | 30.8 | 30.8 | 30.7 |
| 8 | 36.8 | 36.9 | 36.9 | 36.6 | 36.8 | 8′ | 36.4 | 36.7 | 36.5 | 36.6 |
| 9 | 26.8 | 26.9 | 26.9 | 26.3 | 26.8 | 9′ | 26.6 | 26.7 | 26.5 | 26.5 |
| 10 | 25.9 | 25.0 | 25.0 | 25.4 | 25.8 | 10′ | 25.7 | 26.0 | 25.2 | 25.2 |
| 11 | 35.0 | 32.6 | 32.6 | 32.2 | 35.7 | 11′ | 35.8 | 34.9 | 33.6 | 33.6 |
| 12 | 69.3 | 71.6 | 71.3 | 70.9 | 69.5 | 12′ | 69.3 | 69.3 | 74.7 | 74.8 |
| 13 | 55.4 | 53.3 | 53.3 | 52.9 | 56.0 | 13′ | 56.2 | 55.7 | 58.8 | 58.8 |
| 14 | 174.9 | 174.4 | 174.1 | 173.9 | 175.2 | 14′ | 173.3 | 172.7 | 168.1 | 168.1 |
| 3-CH3 | 19.5 | 19.5 | 19.5 | 19.5 | 19.5 | 3′-CH3 | 19.5 | 19.5 | 19.6 | 19.5 |
| 5-CH3 | 18.6 | 18.5 | 18.5 | 18.5 | 18.6 | 5′-CH3 | 18.5 | 18.6 | 18.6 | 18.6 |
| 7-CH3 | 19.7 | 19.8 | 19.7 | 19.7 | 19.7 | 7′-CH3 | 19.7 | 19.7 | 19.7 | 19.7 |
| 13-CH2OH | 60.1 | 60.6 | 60.6 | 60.1 | 61.3 | 13′-CH2OH | 61.5 | 59.9 | 56.7 | 56.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-Z.; Yan, X.; Tang, X.-X.; Lin, J.-G.; Qiu, Y.-K. New Bis-Alkenoic Acid Derivatives from a Marine-Derived Fungus Fusarium solani H915. Mar. Drugs 2018, 16, 483. https://doi.org/10.3390/md16120483
Liu S-Z, Yan X, Tang X-X, Lin J-G, Qiu Y-K. New Bis-Alkenoic Acid Derivatives from a Marine-Derived Fungus Fusarium solani H915. Marine Drugs. 2018; 16(12):483. https://doi.org/10.3390/md16120483
Chicago/Turabian StyleLiu, Shun-Zhi, Xia Yan, Xi-Xiang Tang, Jin-Guo Lin, and Ying-Kun Qiu. 2018. "New Bis-Alkenoic Acid Derivatives from a Marine-Derived Fungus Fusarium solani H915" Marine Drugs 16, no. 12: 483. https://doi.org/10.3390/md16120483
APA StyleLiu, S.-Z., Yan, X., Tang, X.-X., Lin, J.-G., & Qiu, Y.-K. (2018). New Bis-Alkenoic Acid Derivatives from a Marine-Derived Fungus Fusarium solani H915. Marine Drugs, 16(12), 483. https://doi.org/10.3390/md16120483