Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers



Abstract
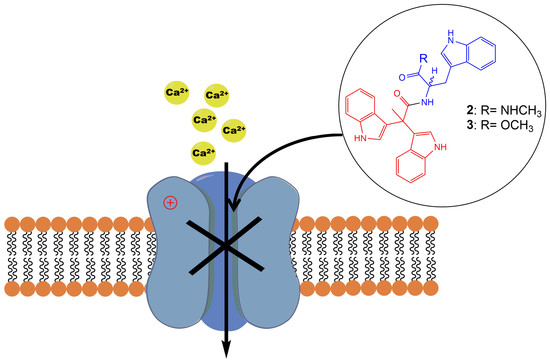
1. Introduction
2. Results
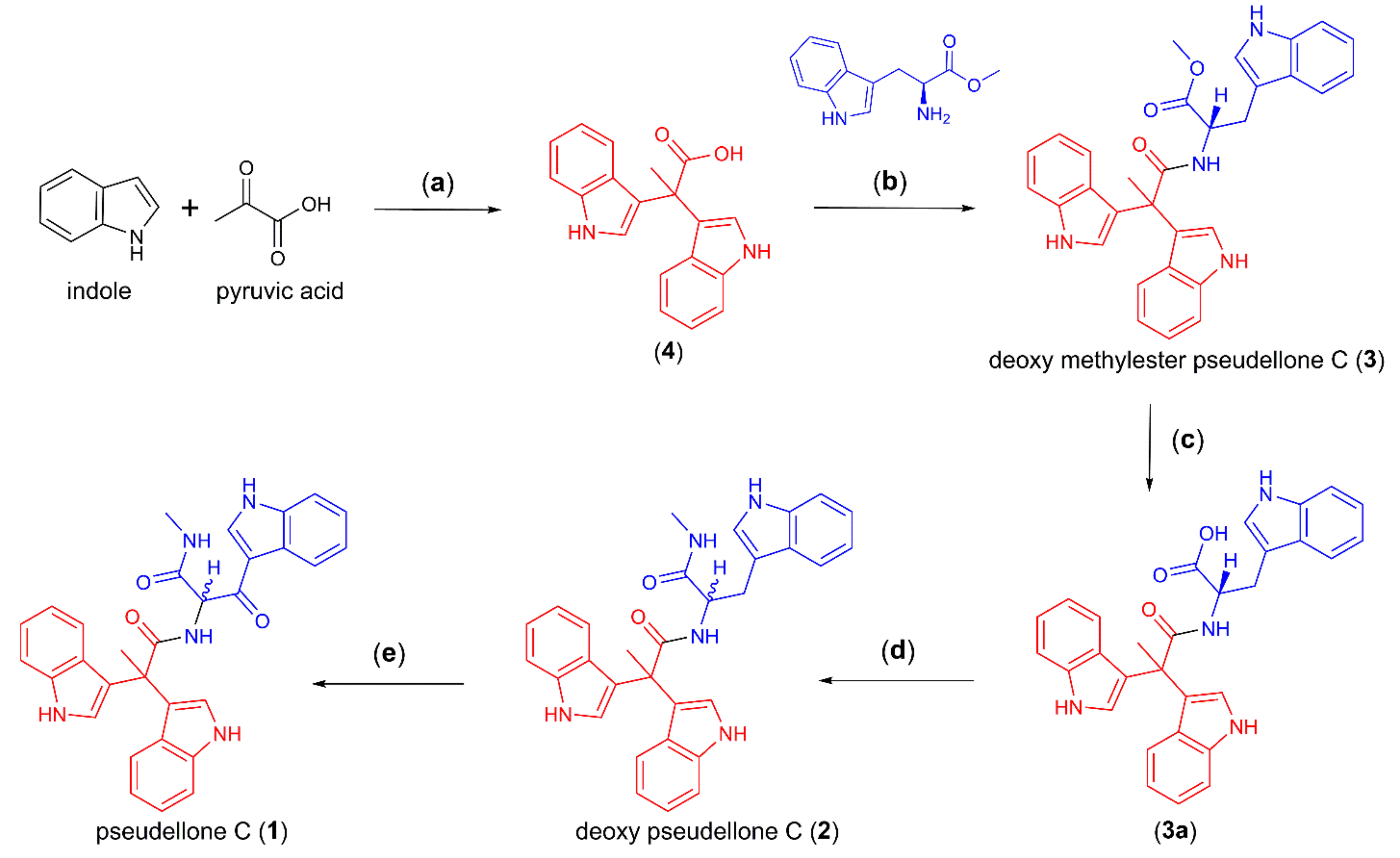
2.1. Synthesis of Pseudellone C and Its Bisindole Alkaloid Analogs
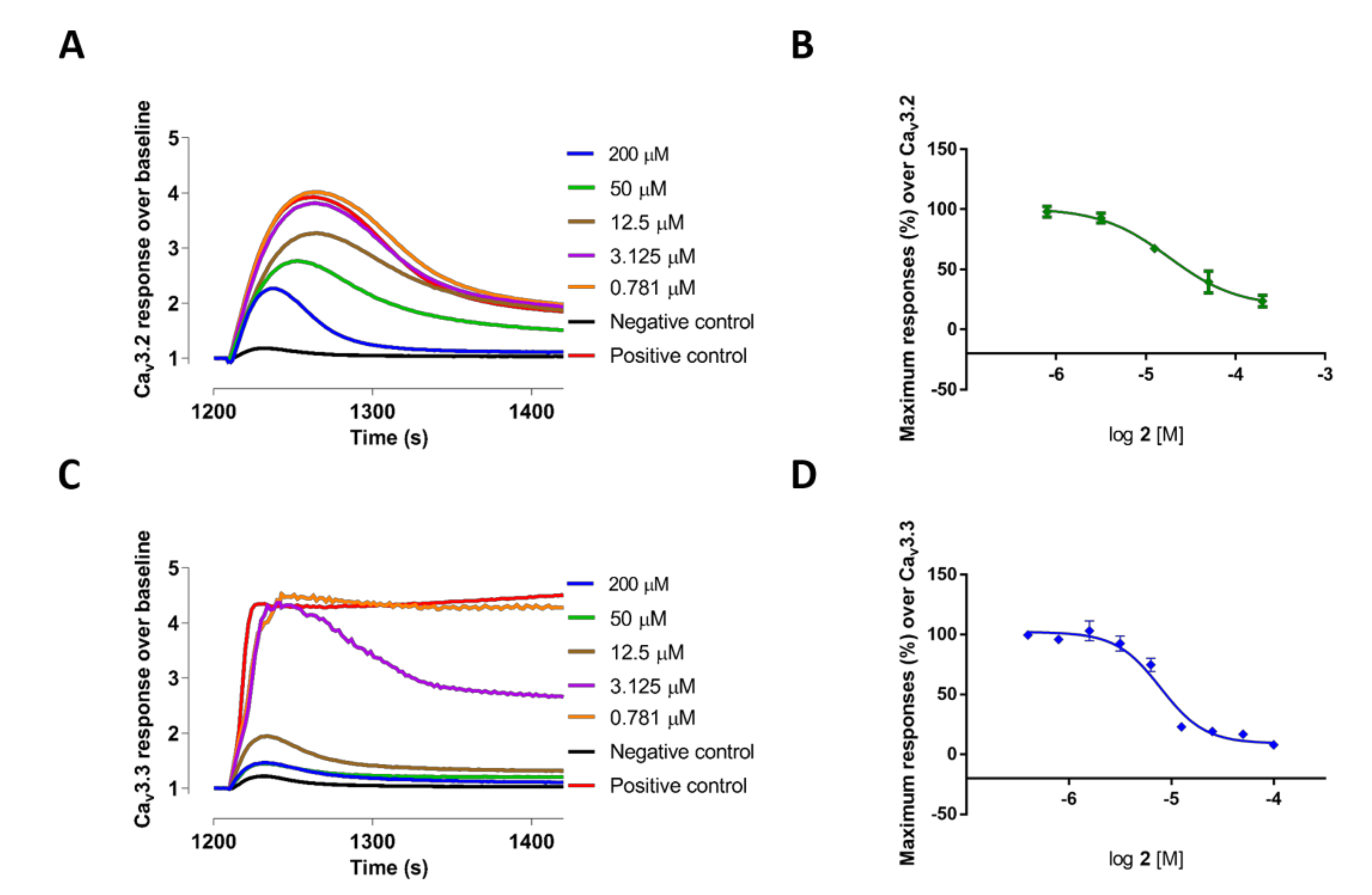
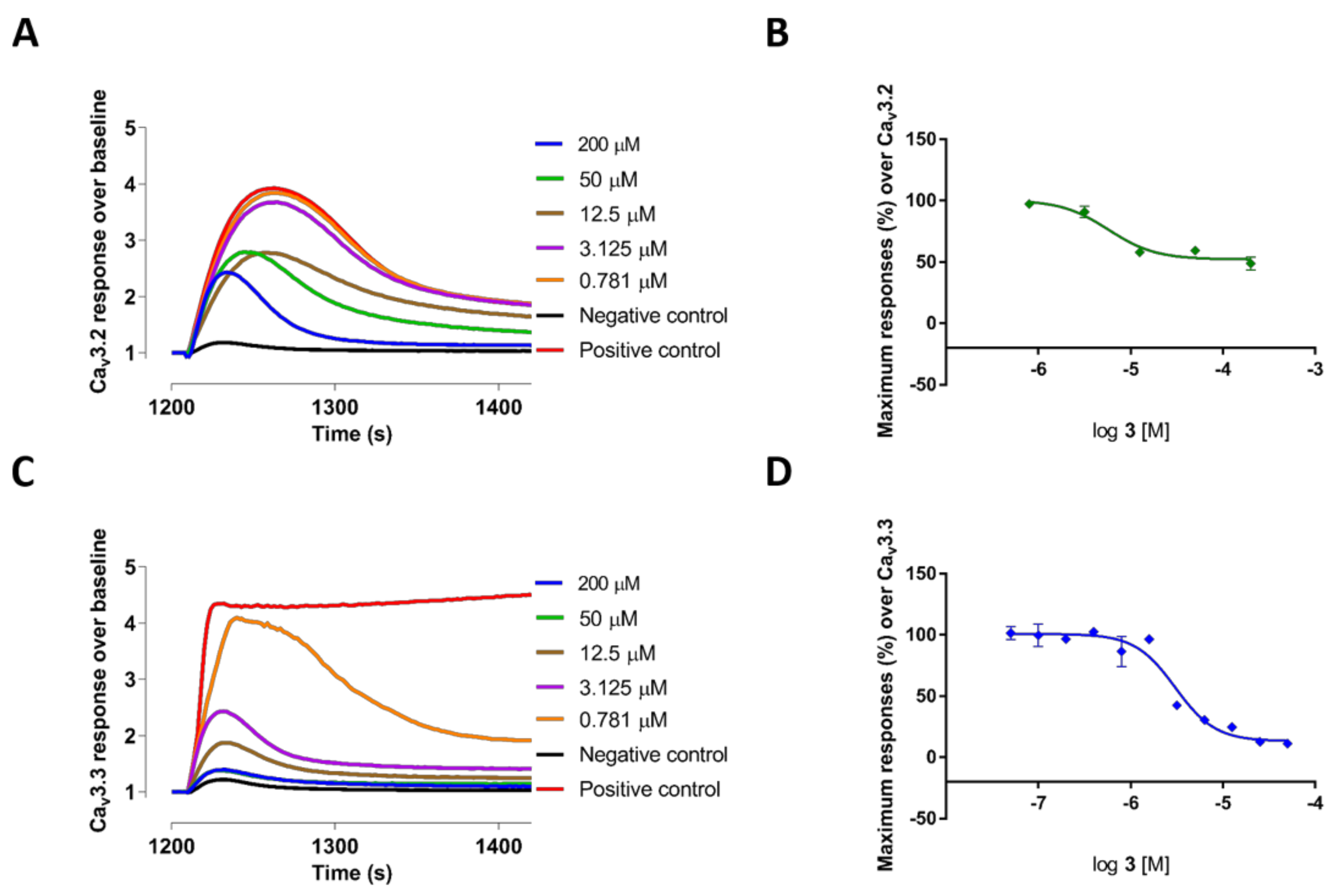
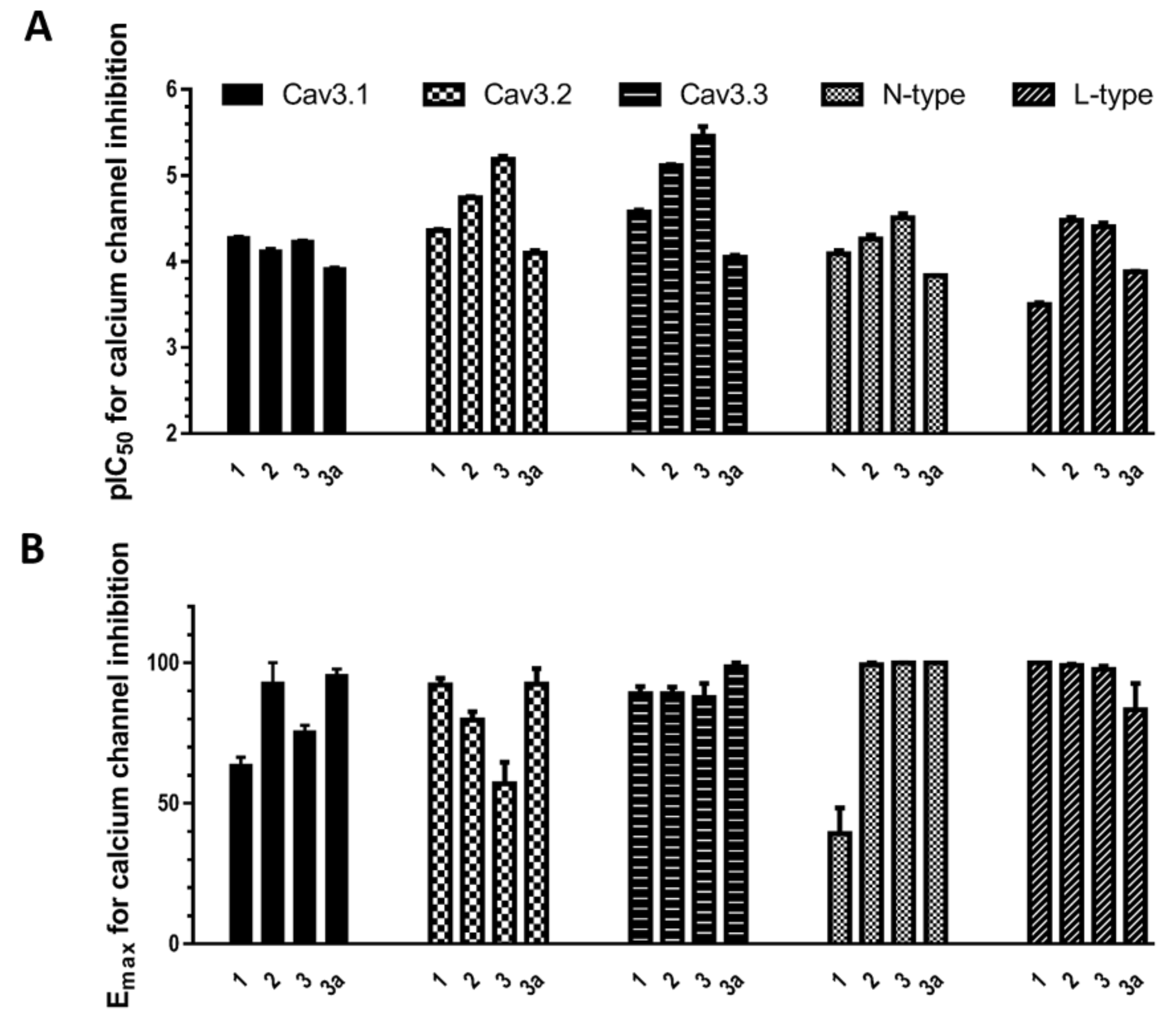
2.2. Evaluation of VGCC Activities of the Synthetic Compounds Using FLIPR Cell-Based Assays
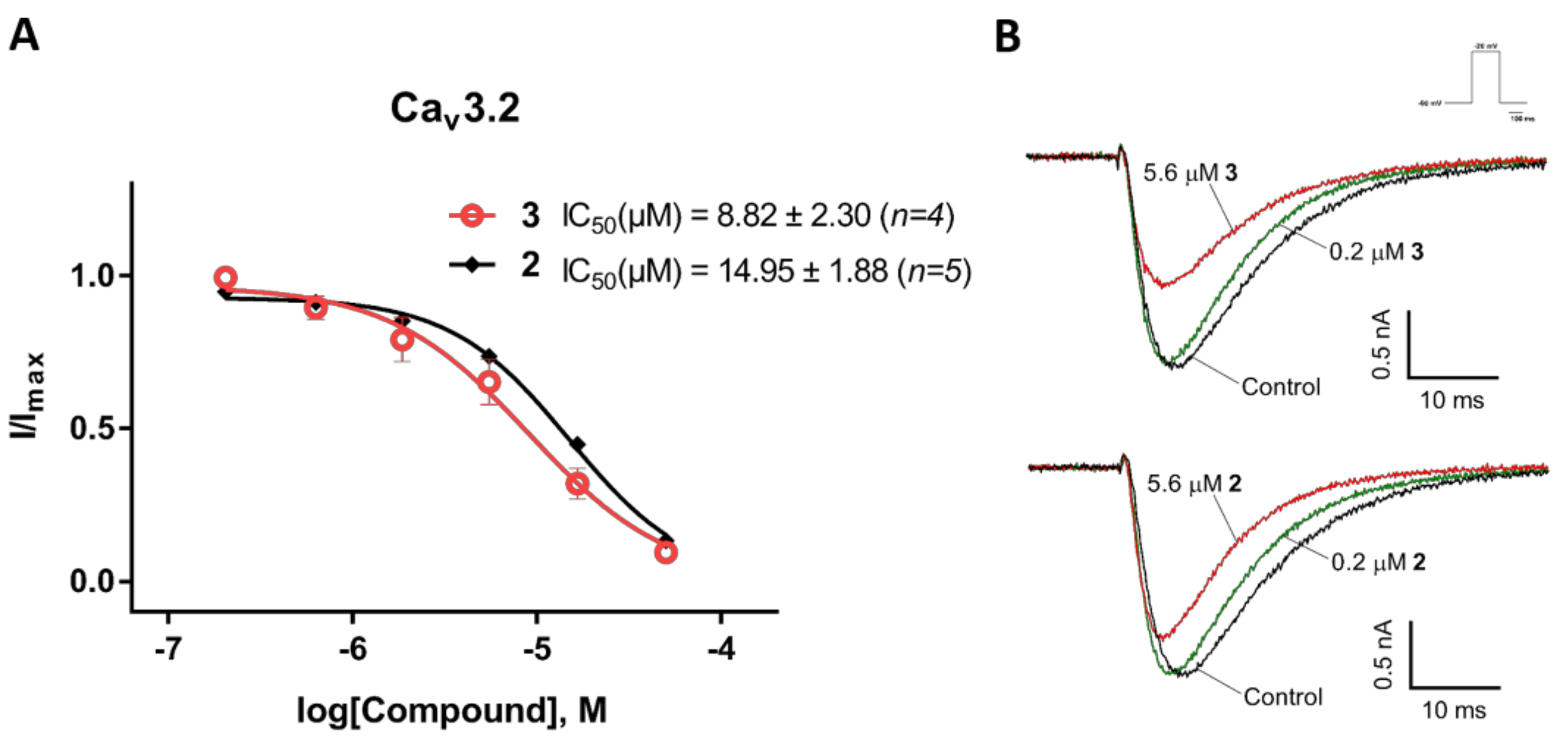
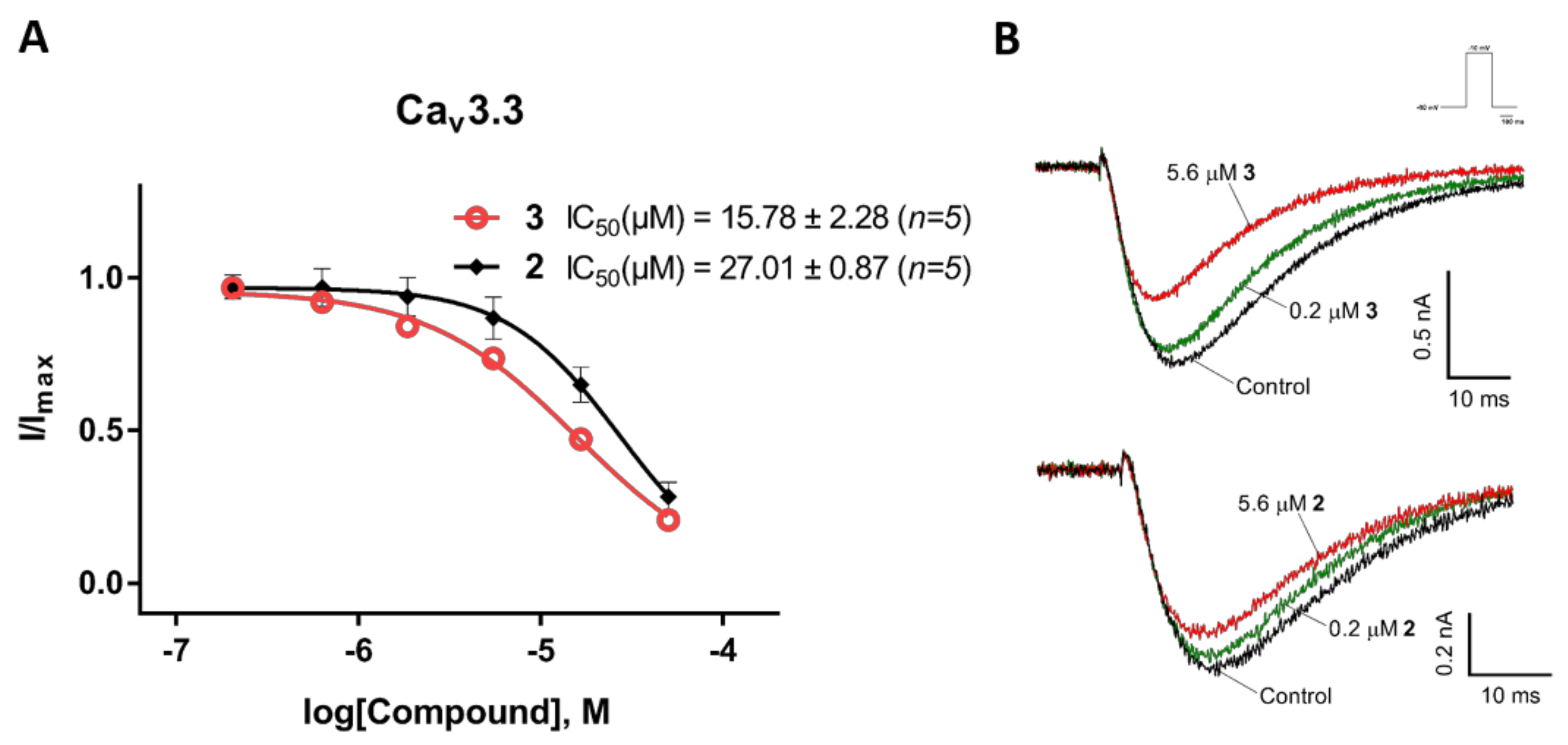
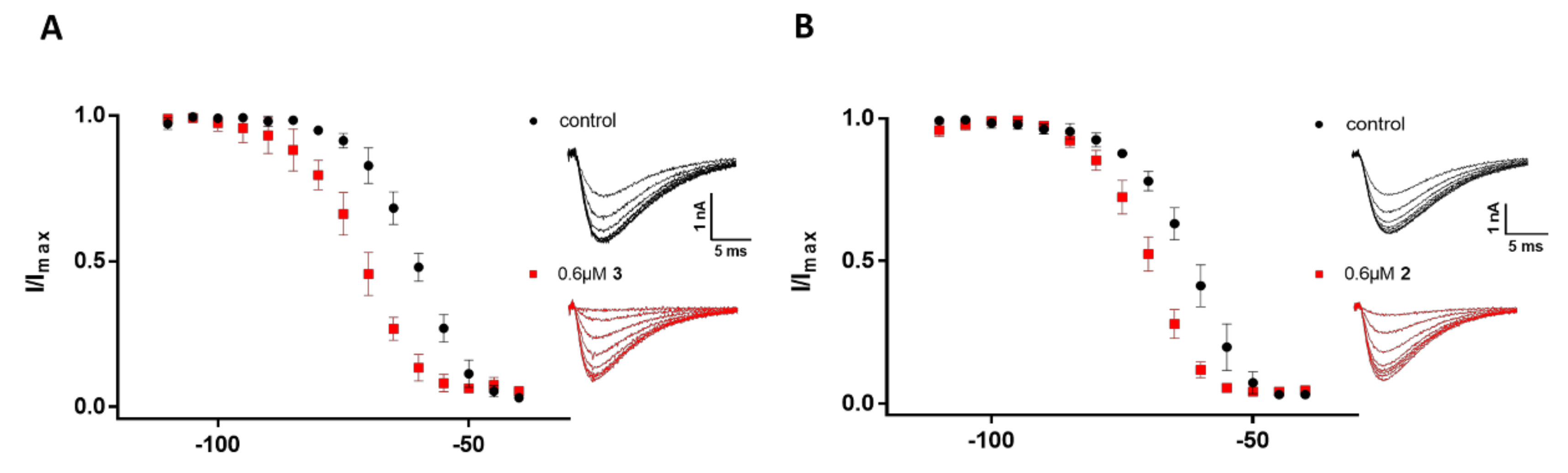
2.3. Electrophysiological Characterization of the Selective CaV3.x Blockers in QPatch Assays
3. Discussion
4. Materials and Methods
4.1. General Procedure for Chemical Synthesis
4.1.1. Synthesis of 2,2-di(1H-indol-3-yl)propanoic Acid (4)
4.1.2. Synthesis of l-Tryptophan Methyl Ester
4.1.3. Synthesis of Methyl (2,2-di(1H-indol-3-yl)propanoyl)-l-tryptophanate (3)
4.1.4. Synthesis of (2,2-di(1H-indol-3-yl)propanoyl)-l-tryptophan (3a)
4.1.5. Synthesis of (S)-N-(3-(1H-indol-3-yl)-1-(methylamino)-1-oxopropan-2-yl)-2,2-di(1H-indol-3-yl)propanamide (2)
4.1.6. Synthesis of Pseudellone C (1)
4.2. Cell Culture and Transient Expression
4.3. T-Type Calcium Channel Window Current FLIPR Assays
4.4. HVA Calcium Channel FLIPR Assays
4.5. Whole-Cell Patch-Clamp Electrophysiology
4.6. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fattorusso, E.; Taglialatela-Scafati, O. Modern Alkaloids: Structure, Isolation, Synthesis, and Biology; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [PubMed]
- Sheftell, F.D.; Bigal, M.E.; Tepper, S.J.; Rapoport, A.M. Sumatriptan: A decade of use and experience in the treatment of migraine. Expert Rev. Neurother. 2004, 4, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed]
- Knaus, H.-G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garcia-Calvo, M.; Helms, L.M.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Tyagarajan, S.; Chakravarty, P.K.; Park, M.; Zhou, B.; Herrington, J.B.; Ratliff, K.; Bugianesi, R.M.; Williams, B.; Haedo, R.J.; Swensen, A.M. A potent and selective indole N-type calcium channel (Ca v 2.2) blocker for the treatment of pain. Bioorg. Med. Chem. Lett. 2011, 21, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H.-J.; Xu, M.-Y.; Ju, Y.-C.; Wang, L.-Y.; Xu, J.; Yang, D.-P.; Lan, W.-J. Pseudellones A–C, three alkaloids from the marine-derived fungus Pseudallescheria ellipsoidea F42–3. Org. Lett. 2015, 17, 5156–5159. [Google Scholar] [CrossRef] [PubMed]
- Sathieshkumar, P.P.; Latha, P.; Nagarajan, R. Total Synthesis of Pseudellone C. Eur. J. Org. Chem. 2017, 2017, 3161–3164. [Google Scholar] [CrossRef]
- Garbe, T.R.; Kobayashi, M.; Shimizu, N.; Takesue, N.; Ozawa, M.; Yukawa, H. Indolyl carboxylic acids by condensation of indoles with α-keto acids. J. Nat. Products 2000, 63, 596–598. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Talapatra, B. Quinine. Cinchona Alkaloids (Tryptophan Derived Quinoline Alkaloids). In Chemistry of Plant Natural Products; Springer: Berlin, Germany, 2015; pp. 855–874. [Google Scholar]
- Ramani, S.; Patil, N.; Nimbalkar, S.; Jayabaskaran, C. Alkaloids derived from tryptophan: Terpenoid indole alkaloids. In Natural Products; Springer: Berlin, Germany, 2013; pp. 575–604. [Google Scholar]
- Crunelli, V.; Lightowler, S.; Pollard, C. AT-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J. Physiol. 1989, 413, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Tringham, E.; Powell, K.L.; Cain, S.M.; Kuplast, K.; Mezeyova, J.; Weerapura, M.; Eduljee, C.; Jiang, X.; Smith, P.; Morrison, J.-L. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci. Transl. Med. 2012, 4, 121ra119. [Google Scholar] [CrossRef] [PubMed]
- Uebele, V.N.; Nuss, C.E.; Fox, S.V.; Garson, S.L.; Cristescu, R.; Doran, S.M.; Kraus, R.L.; Santarelli, V.P.; Li, Y.; Barrow, J.C. Positive allosteric interaction of structurally diverse T-type calcium channel antagonists. Cell Biochem. Biophys. 2009, 55, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.S.; Macdonald, T.L. The pharmacology and regulation of T type calcium channels: New opportunities for unique therapeutics for cancer. Cell Calcium 2006, 40, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F. T-type calcium channel: A privileged gate for calcium entry and control of adrenal steroidogenesis. Front. Endocrinol. 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Kerckhove, N.; Meleine, M.; Alloui, A.; Barrere, C.; Gelot, A.; Uebele, V.N.; Renger, J.J.; Eschalier, A.; Ardid, D. State-dependent properties of a new T-type calcium channel blocker enhance CaV3. 2 selectivity and support analgesic effects. Pain 2013, 154, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.L. The discovery of the vinca alkaloids—Chemotherapeutic agents against cancer. Biochem. Cell Biol. 1990, 68, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Rahmani, R. Preclinical and clinical pharmacology of vinca alkaloids. Drugs 1992, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Finnbladder, N.B.C.S.G.; de Tratamiento Oncologico, C.U.; EORTC Genito-Urinary Group; Australian Bladder Cancer Study Group; National Cancer Institute of Canada Clinical Trials Group. Neoadjuvant cisplatin, methotrexate, and vinblastine chemotherapy for muscle-invasive bladder cancer: A randomised controlled trial. Lancet 1999, 354, 533–540. [Google Scholar]
- Shore, P. Reserpine: A survey of its pharmacology. In Alterations of Chemical Equilibrium in the Nervous System; Springer: Berlin, Germany, 1971; pp. 349–356. [Google Scholar]
- Jerie, P. Milestones of cardiovascular therapy. IV. Reserpine. Casopis Lekaru Ceskych 2007, 146, 573–577. [Google Scholar] [PubMed]
- Preskorn, S.H. The evolution of antipsychotic drug therapy: Reserpine, chlorpromazine, and haloperidol. J. Psychiatr. Pract. 2007, 13, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Van Deusen, A.L.; Vitko, I.; Babu, D.A.; Davies, L.A.; Huynh, N.; Cheng, H.; Yang, N.; Barrett, P.Q.; Perez-Reyes, E. Validation of high throughput screening assays against three subtypes of Cav3 T-type channels using molecular and pharmacologic approaches. Assay Drug Dev. Technol. 2007, 5, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.R.; Vetter, I.; Ragnarsson, L.; Lewis, R.J. Expression and pharmacology of endogenous Cav channels in SH-SY5Y human neuroblastoma cells. PLoS ONE 2013, 8, e59293. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | LVA CaVs (IC50 (µM) N = 3) | HVA CaVs (IC50 (µM) N = 3) | |||
|---|---|---|---|---|---|
| CaV3.1 | CaV3.2 | CaV3.3 | CaV2.2 | CaV1 | |
| 1 | 53.49 ± 2.72 | 43.96 ± 1.60 | 26.59 ± 1.23 | 82.55 ± 7.60 | 322.00 ± 21.63 |
| 2 | 76.36 ± 5.39 | 18.24 ± 0.49 | 7.71 ± 0.23 | 55.29 ± 5.58 | 33.66 ± 2.62 |
| 3 | 58.57 ± 2.62 | 6.59 ± 0.66 | 3.81 ± 1.08 | 31.32 ± 3.40 | 39.76 ± 3.98 |
| 3a | 122.23 ± 5.96 | 80.65 ± 6.21 | 90.05 ± 4.99 | 146.70 ± 1.72 | 132.03 ± 1.63 |
| 4 | 72.28 ± 0.41 | 132.17 ± 6.13 | 119.17 ± 8.66 | 179.30 ± 19.68 | 264.30 ± 11.89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Neupane, P.; Ragnarsson, L.; Capon, R.J.; Lewis, R.J. Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers. Mar. Drugs 2018, 16, 475. https://doi.org/10.3390/md16120475
Wang D, Neupane P, Ragnarsson L, Capon RJ, Lewis RJ. Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers. Marine Drugs. 2018; 16(12):475. https://doi.org/10.3390/md16120475
Chicago/Turabian StyleWang, Dan, Pratik Neupane, Lotten Ragnarsson, Robert J. Capon, and Richard J. Lewis. 2018. "Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers" Marine Drugs 16, no. 12: 475. https://doi.org/10.3390/md16120475
APA StyleWang, D., Neupane, P., Ragnarsson, L., Capon, R. J., & Lewis, R. J. (2018). Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers. Marine Drugs, 16(12), 475. https://doi.org/10.3390/md16120475