Abstract
Four sesterterpenes, erectusolides B, C, D, and seco-manoalide-25-methyl ether, two 2-furanone derivatives, erectusfuranones A and B, together with thirteen known sesterterpenes, (6Z)-neomanoalide-24-acetate, two diastereomers of 24-O-methylmanoalide, luffariolide B, manoalide, (6E)- and (6Z)-neomanoalide, seco-manoalide, scalarafuran, 12-acetylscalarolide, 12-epi-O-deacetyl-19-deoxyscalarin, 12-epi-scalarin, and 12-O-deacetyl-12-epi-scalarin, three indole alkaloids, 5-hydroxy-1H-indole-3-carbaldehyde, hyrtiosine A, and variabine B, and one norterpene, cavernosine were isolated from the marine sponge Hyrtios erectus. Their structures were determined by means of spectroscopic methods and the absolute configurations of the asymmetric centers were determined using the modified Mosher’s method. The cytotoxic activities for the isolated compounds have been reported.
1. Introduction
Marine organisms have always been an attractive source of natural products with novel and exotic structures and useful biological activities [1,2,3,4,5,6,7,8,9,10,11]. Marine sponges of the genus Hyrtios (order Dictyoceratida family Thorectidae) have yielded scalarane [6,8,12,13,14] and manoalide [12] types sesterterpenes, which are important groups of active secondary metabolites. They have been reported to possess many biological activities, such as cytotoxic [3,6,15,16], antibacterial [9,11], antibiotic [17] activities, inhibition of the DNA-relaxing activity of mouse DNA topoisomerase I [12], and enhancement nerve growth factor synthesis in cultured astroglial cells [13]. As a part of our ongoing research program focused on the discovery of cytotoxic compounds from Thai marine sponges [18,19,20], drawing from the previous report, one sesterterpene, erectusolide A, six phenolic alkenes, erectuseneols A–F, together with nine known compounds, were isolated from the EtOAc soluble extract of Hyrtios erectus (Chulabhorn Research Institute (CRI) 588) [3]. Of these, some sesterterpenes exhibited significant cytotoxic activities against the MOLT-3 cell line with an IC50 values of 3.79–5.82 μM [3]. Accordingly, further investigation of the EtOAc soluble extract was carried out, leading to the isolation of four new sesterterpenes, erectusolides B (1), C (2), D (4), and seco-manoalide-25-methyl ether (3), two new 2-furanone derivatives, erectusfuranones A and B (5 and 6), together with seventeen known sesterterpenes, which were identified as (6Z)-neomanoalide-24-acetate (7) [9], two diastereomers of 24-O-methylmanoalide (8 and 9) [10], luffariolide B (10) [15], manoalide (11) [10,12,17], (6E)- and (6Z)-neomanoalides (12 and 13) [12,15,17], seco-manoalide (14) [12,17] scalarafuran (15) [8,16], 12-acetylscalarolide (17) [21], 12-epi-O-deacetyl-19-deoxyscalarin (18) [8,16], 12-epi-scalarin (19) [13,16], and 12-O-deacetyl-12-epi-scalarin (20) [13,14,16], three indole alkaloids, 5-hydroxy-1H-indole- 3-carbaldehyde (21) [14,22], hyrtiosine A (22) [14,22], and variabine B (23) [7], and one norterpene, cavernosine (16) [3,23]. All of the known compounds (Figure 1) were readily identified by extensive study of their spectral data, including high resolution atmospheric pressure chemical ionization mass spectrometry (HRAPCIMS) or high resolution electrospray ionization mass spectrometry (HRESIMS), 1D and 2D nuclear magnetic resonance (NMR) data, as well as by comparison with those reported in the literatures.
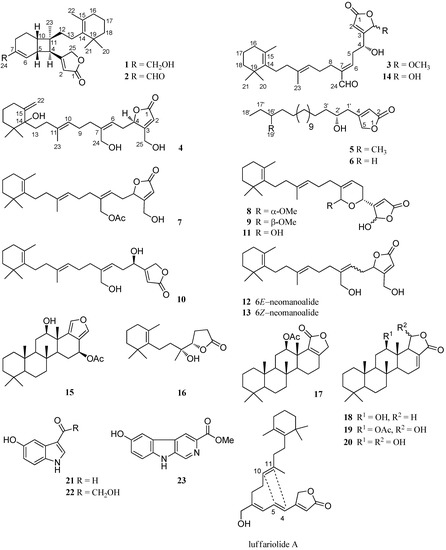
Figure 1.
Isolated compounds 1–23 from sponge Hyrtios erectus and luffariolide A.
2. Results
Two separate collections of H. erectus were studied; and they were collected in February 2011 from different locations of the Similan Island, Phangnga province, Thailand. The MeOH extract of the Thai marine sponge H. erectus from collection CRI 588 was dissolved in H2O and partitioned with EtOAc. The EtOAc soluble extract was fractionated by column chromatography over silica gel, Sephadex LH-20, and reversed-phase high performance liquid chromatography (HPLC) to afford three new sesterterpenes, erectusolides B (1), C (2), and seco-manoalide-25-methyl ether (3), two new 2-furanone derivatives, erectusfuranones A (5) and B (6) together with eight known sesterterpenes, 7–14. One additional new sesterterpene, erectusolide D (4), and ten known compounds (13, 15–23) were obtained from the H. erectus collection CRI 572. All new compounds showed considerable structural similarity with the co-occurring known sesterterpenes.
Compound 1 was obtained as a weak optical rotation value ( −1.3), and its molecular formula was determined to be C25H36O3 (eight degrees of unsaturation) by HRAPCIMS. Infrared (IR) absorption bands of compound 1 suggested the presence of β-substituted α,β-unsaturated γ-lactone (β-substituted butenolide) at 1779 and 1746 cm−1 and hydroxyl group at 3443 cm−1. Four of the eight degrees of unsaturation implied by the molecular formula of 1 were taken up in one carbon–oxygen double bonds and three carbon–carbon double bonds, thus indicating the tetracyclic nature of the molecule. The 13C and (distortionless enhancement by polarization transfer (DEPT) NMR spectra of 1 showed 25 carbons including, 4 tertiary methyls, 7 methylenes, 2 oxymethylenes, 3 methines, 2 olefinic methines, and 7 quaternary carbons. The 1H and 13C NMR spectral data of 1 (Table 1, Figures S1 and S2) were comparable to those of luffariolides A (Figure 1) and B (10) and implied that all compounds possessed identical two terminal units, which included a polyalkylated-cyclohexene (C12–C22) and β-substituted butenolide moieties. The major differences were found in the C4–C11, and C-23. In the 1H–1H COSY correlation of 1 (Figure 2), H-6 (δH 5.67) showed allylic coupling to Hax-8 (1H, δH 1.90–2.00) and H2-24 (2H, δH 4.03/4.06) and H-5 was coupled to H-4, H-6, and H-10. From its 1H–1H correlation spectroscopy (COSY) spectrum of 1 (Figure 2), typical allylic coupling of the olefinic methines at δH 5.86 (H-2) and 5.67 (H-6) with the respective methine signal at δH 2.74 (H-4) and oxymethylene at δH 4.03/4.06 (H2-24) were discernable. Both the COSY and heteronuclear multiple bond correlation (HMBC) data indicated that the double bonds were not conjugated to each other. The 1H, 13C, and COSY NMR data confirmed the presence of a 3-hydroxymethylbicyclo [4.2.0] oct-2-ene (C4–C11 and C-24) in the molecule. The HMBC correlations of 1 (Figure 2 and Figure S4), the proton signal at δH 5.86 (H-2) correlated to methine carbon at δC 51.0 (C-4), H-4 at δH 2.74 correlated to quaternary carbon at δC 169.7 (C-3), methine carbon at δC 115.3 (C-2), and methylene carbon at δC 73.2 (C-25) allowed the connectivity of the butenolide with the bicyclic ring through the linkage of C-3 and C-4. The linkage of the polyalkylated-cyclohexene and CH3-23 at quaternary C-11 on the molecule were deduced from the HMBC correlations of H3-23 (δH 1.13) with C-4, C-10 (δ 37.0), C-11 (δ 43.0), and C-12 (δ 36.6) and the correlations of H2-12 (δH 1.28–1.36 and 1.58–1.66) with C-4, C-10, C-14 (δ 136.3), and C-23 (δ 21.5). The large trans-diaxial coupling constant between Hax-9 and Hax-10 (J = 12.2 Hz) on the cyclohexene ring suggested the β-configuration of Hax-10. The relative stereochemistry of 1 was established by analysis of the (nuclear overhauser effect spectroscopy (NOESY) spectrum (in CDCl3 and C6D6; Figure 2). NOESY cross-peaks resonance of H-4, Hα-9, and H3-23 and H-5, H-10, H-12, and H-2 implied a cis-junction for rings B/C and H-5, Hax-10, β-substituted butenolide moiety, and polyalkylated-cyclohexene (C12–C22) are on the same face while the methyl group (CH3-23) and H-4 on the opposite face of the molecule. Structure 1 with relative stereochemistry as shown was thus assigned to erectusolide B, which contained fused cyclohexene and cyclobutane rings. It could derive from luffariolide A (Figure 1) [11], through a biogenetic pathway involving a 2 + 2 cycloaddition of double bonds at C-4/C-5 and C-10/C-11 as suggested by Lin, H.W. and co-workers [4]. Furthermore, compounds 1 and 2 represent two important compounds which lend further support for the proposed biosynthetic pathway of Lin et. al. for the cyclobutane formation [4]. This substance has an optical rotation near zero ( −1.3); attempting to separate the substance using various chiral columns was found to be inseparable.

Table 1.
1H (600 MHz) and 13C (150 MHz) NMR data (a in CDCl3 b in C6D6) of compounds 1 and 2.
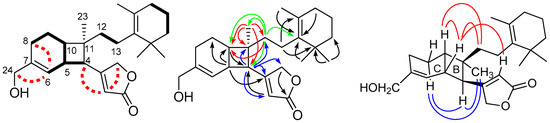
Figure 2.
Key COSY ( and
and  ), HMBC (
), HMBC ( ), and NOESY (
), and NOESY ( ) correlations of 1.
) correlations of 1.
and ), HMBC (), and NOESY () correlations of 1.
Compound 2 was obtained as optically active ( −18.5), and its molecular formula was determined to be C25H34O3 (nine degrees of unsaturation) by HRAPCIMS. Infrared (IR) absorption bands of compound 2 suggested the presence of β-substituted α,β-unsaturated γ-lactone at 1779 and 1746 cm−1 and α,β-unsaturated carbonyl group at 1682 cm−1. The 1H and 13C NMR spectroscopic data of 2 (Table 1, Figures S4–S8) indicated that it was essentially identical to compound 1, except for the presence of an aldehyde group (δH 9.45, s; δC 192.9) for 2 in place of the hydroxylmethyl group (δH 4.03/4.06, each d, J = 13.2 Hz, δC 66.8) for 1. This was further confirmed by the HMBC correlations (Figures S11 and S12) between olefinic H-6 (δH 6.74) and aldehydic carbon (δC 192.9) and between proton aldehyde (δH 9.45) and C-7 (δC 143.7) and C-8 (δC 19.4). The NOESY spectrum of 2 was similar to that observed of 1 indicating the same relative stereochemistry. Thus, compound 2 was suggested to be the formaldehyde analog of 1, and named erectusolide C. This compound has low optical rotation ( −18.5); attempting to separate the compound using various chiral columns was unsuccessful.
Compound 3 was obtained as a pale yellow solid, exhibiting similar ultraviolet (UV), infrared (IR), 1H, and 13C NMR spectra (Table 2, Figures S13 and S14) as (6E) seco-manoalide (14) [17]. Accurate mass measurement by HRAPCIMS of 3 indicated a pseudo molecular ion peak at m/z 465.2414 [M + Cl]− (calcd for C26H38ClO5, 465.2413), consistent with the molecular formula C26H38O5. This difference of 14 amu compared to the molecular formula of 14, and the appearance of a signal of methoxy group in 1H, and 13C NMR spectra of 3 at δH 3.65 and δC 57.9. It is suggested that the hydroxyl group at C-25 was replaced by a methoxyl group. This was further confirmed by the HMBC correlations (Figure S16) between the methoxyl protons (δH 3.65) and hemiacetal carbon (δC 103.3) and between H-25 (δH 5.84) and methoxyl carbon (δC 57.9). The absolute stereochemistry at C-4 of 3 was determined by the modified Mosher’s method [24]. The hydroxyl group of 3 was converted into both the S- and R-MTPA esters 3a and 3b, respectively. The 1H NMR chemical shifts were assigned by the analysis of the 1H–1H COSY NMR data for each MTPA ester (experimental section). The calculated ΔδS−R values were positive for the H2-5 (+0.03 and +0.03), H-6 (+0.15), H2-8 (+0.04), and H-24 (+0.11) and negative for the H-2 (−0.13), 25-OMe (−0.004), and H-25 (−0.02) (Figure 3), implying that the absolute configuration of C-4 was R. Thus, compound 3 was characterized as (4R,6E) seco-manoalide-25-methyl ether.
Table 2.
1H (600 MHz) and 13C (150 MHz) NMR data (CDCl3) of compounds 3 and 4.
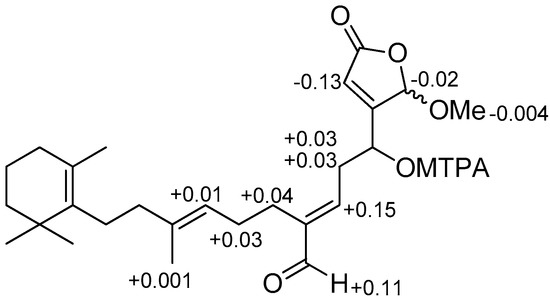
Figure 3.
ΔδS-R values in ppm for S- and R-MTPA esters of compound 3 in CDCl3.
Compound 4 was obtained as an optically active ( −20.9), and its molecular formula was determined to be C25H38O5 (seven degrees of unsaturation) by HRAPCIMS. Five of the seven degrees of unsaturation implied by the molecular formula of 4 were taken up in one carbon–oxygen double bonds and four carbon–carbon double bonds, thus indicating the bicyclic nature of the molecule. The IR spectrum exhibited absorption bands corresponding to a hydroxyl group (3424 cm−1), an ester carbonyl (1739 cm−1), and an exomethylene substituent (898 cm−1). Similarities in the NMR spectra between compounds 4 (Table 2, Figures S17 and S18) and 6Z-neomanoalide (13) suggested that compound 4 was also a neomanoalide-type sesterterpene [17].
The main differences in the 1H NMR spectra of 4 and compound 13 were the absence of one olefinic methyl group resonance in compound 4 and the appearance of a resonance attributable to an exomethylene moiety (δH 4.83 and 4.91, H2-22). Observation of a sp2 methylene carbon resonance (δC 108.3) and a corresponding quaternary carbon (δC 150.5) further supported the presence of a exocyclic methylene functionality in the structure of 4. An oxygenated quaternary carbon (δC 80.4, C-14) was observed by 13C NMR and DEPT experiments. The assignment and placement of the hydroxyl substituent at C-14 and the exocyclic methylene at C-15 were deduced by observation of long range 1H–13C HMBC correlations (Figure 4 and Figure S20) (H2-13, H2-16, H2-18, H3-21/22 to C-14, H2-17 to C-15, and H2-16 to C-22). The relative stereochemistry of the methylenecyclohexane unit was elucidated mainly on the basis of NOESY correlations (Figure 4). The correlations among axial Hα-18 (δH 1.63, td, J = 13.0, 5.7 Hz), axial methylene H2-13, and equatorial CH3-20 (δH 0.96) and correlations among axial H3-21 (δH 0.88), exocyclic methylene H2-22, and equatorial Hβ-18 (δH 1.37, brd, J = 13.7 Hz) that were observed in 4 indicated that the equatorial hydroxyl group at C-14 occupied the β-face. The geometry of the olifinic bonds was assigned as 6Z,10E on the basis of NOESY correlations between H2-5 and H2-24 and between H2-9 and H3-23, respectively. Compound 4 exhibited a negative optical rotation { −20.9 (c 1.64, CHCl3)}, similar to that of 6Z-neomanoalide (13) { −28 (c 0.8, CH2Cl2)} [12] the relative stereochemistry at C-4 of 4 was then assigned to be R. Thus, the structure 4 was concluded as shown in Figure 1 and was named erectusolide D.
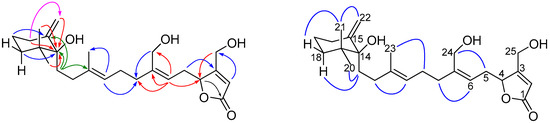
Figure 4.
Key HMBC ( ), and NOESY (
), and NOESY ( ) correlations of 4.
) correlations of 4.
), and NOESY () correlations of 4.
Compound 5 was obtained as optically active ( +1.3), and its molecular formula was determined to be C23H42O3 (three degrees of unsaturation) by HRESIMS. The IR absorption bands data of compound 5 suggested the presence of β-substituted α, β-unsaturated γ-lactone (β-substituted butenolide for three degrees of unsaturation in the structure at 1777 and 1739 cm−1 in addition to the hydroxyl group at 3442 cm−1. The 1H and 13C NMR spectra of 5 as shown in Table 3 (Figures S11 and S22) suggests that compound 5 possess β-substituted butenolide moiety to which a saturated long chain hydrocarbon group is attached. The 1H NMR signals at δH 5.93 (1H) and 4.85 (2H) assignable to an olefinic proton and methylene protons at the α- and γ-positions of the α,β-unsaturated γ-lactone, respectively, reveals that the long chain hydrocarbon group is attached at the β-position (C-4). The 2-hydroxy-16-methyloctadecane group (long chain hydrocarbon) was assigned by a combination of HRESIMS, 1H and 13C NMR, DEPT, COSY, HSQC (Figure S23), and HMBC analyses. The HMBC spectrum of 5 (Figure 5 and Figure S24) showed the correlations from two of the methyl protons at δH 0.85 (H3-18′, t, J = 7.2 Hz) and δH 0.84 (H3-19′, d, J = 6.0 Hz) to the methine carbon at δC 34.4 (C-16′) and methylene carbon at δC 29.42 (C-17′) suggesting that the position of the second methyl group was attached at C-16′. In the HMBC spectrum of 5 also showed the correlations between methylene H2-1′ (δH 2.52/2.64) and C-3 (δC 117.3), C-2′ (δC 70.2), C-3′ (δC 37.7), and C-5 (δC 73.8) and between H-2′ (δH 3.84–3.90) and C-4 (δC 167.3), C-4′ (δC 25.5) indicating clearly that the methylene carbon (C-1′) of 2-hydroxy-16-methyloctadecyl attached to the C-4 of the α,β-unsaturated γ-lactone. The absolute stereochemistry at C-2′ of 5 was determined by the modified Mosher’s method [24]. The hydroxyl group of 5 was converted into both the S- and R-MTPA esters 5a and 5b, respectively, each of which was a single diastereoisomer by 1H and COSY NMR experiments. The calculated ΔδS-R values were positive for the H2-3′ (+0.04) and H2-4′ (+0.06), while ΔδS-R values were negative for H2-1′ (−0.07 and −0.06), H-3 (−0.09), and H2-5 (−0.13 and −0.27) (Figure 6), which implied the absolute configuration of C-2′ was R. Thus, compound 5 was characterized as 4-((2R)-2-hydroxy-16-methyloctadecyl)furan-2(5H)-one and was named erectusfuranone A.
Table 3.
1H (600 MHz) and 13C (150 MHz) NMR data (CDCl3) of compounds 5 and 6.
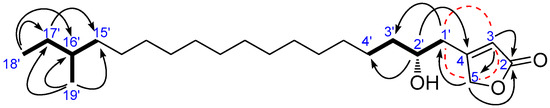
Figure 5.
Key COSY ( ), long rang COSY (
), long rang COSY ( ), and HMBC (
), and HMBC ( ) correlations of 5.
) correlations of 5.
), long rang COSY (), and HMBC () correlations of 5.
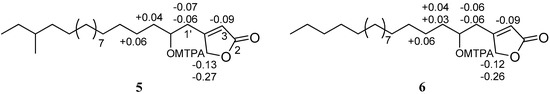
Figure 6.
ΔδS-R values in ppm for S- and R-MTPA esters of compounds 5 and 6 in CDCl3.
Compound 6 was obtained as optically active ( −4.9), and its molecular formula was determined to be C22H40O3 (three degrees of unsaturation) by HRESIMS. The UV, IR, 1H and 13C NMR (Figures S25 and S26) spectroscopic data of 6 indicated that it was essentially identical to compound 5. The signals of a methine moiety (C-16′; δC 34.3, δH 1.23–1.37) and a methyl group (C-19′; δC 19.2, δH 0.84) were not evident in 6, instead a methylene group (C-16′; δC 31.9, δH 1.22–1.31) was observed. The absolute stereochemistry at C-2′ of 6 was determined by the modified Mosher’s method [24]. The hydroxyl group of 6 was converted into both the S- and R-MTPA esters 6a and 6b, respectively, each of which was a single diastereoisomer judged by 1H and COSY NMR experiments. The ΔδS-R (Figure 6) values observed in the 1H NMR spectra were calculated and the resulting positive Δδ values for H2-6 (+0.04 and +0.04) and H2-7 (+0.06), and negative Δδ values for H2-1′ (−0.06 and −0.06), H-3 (−0.09), and H2-5 (−0.12 and −0.26) were consistent with the 2′R configuration. Thus, compound 6 was characterized as (R)-4-(2-hydroxyoctadecyl) furan-2(5H)-one and was named erectusfuranone B.
Cytotoxicity of the isolated compounds 3, 5, 7–20 and 23 were evaluated against several cancer cell lines such as (Table 4), MOLT-3 (acute lymphoblastic leukemia), HepG2 (hepatocarcinoma), HeLa (human cervical carcinoma), HuCCA-1 (human chlolangiocarcinoma), A549 (non-small-cell lung cancer), H69AR (multidrug resistance small-cell lung cancer), KB (human epidermoid carcinoma in the mouth), T47D (hormone dependent breast cancer), MDA-MB-231 (hormone independent breast cancer), and MRC-5 (normal embryonic lung cell). Compounds 8, 9, 14 and 20 showed good cytotoxic activity against MOLT-3 cell line with IC50 values of 1.77, 1.30, 6.68, and 7.64 μM, respectively, compounds 8 and 9 also showed cytotoxic activity against HeLa, HuCCA-1, and A549 cell lines with IC50 values of 1.51–16.77 μM. In addition, compounds 8 and 20 selectively exhibited cytotoxic activity toward the MOLT-3 (IC50 1.77 and 7.64 µM, respectively) cancer cell line with the selectivity index (SI) value of 7 (IC50 13.00 µM for normal cell line, MRC-5) and 9 (IC50 70.97 µM for MRC-5), respectively. The SI value is the ratio of IC50 of normal cell (MRC-5) and IC50 of cancer cell line. Compound 9 selectively exhibited cytotoxic activity against MOLT-3 (IC50 1.30 µM) and HeLa (IC50 1.51 µM) cell lines with respective SI values of 8 and 7 (IC50 10.84 µM for MRC-5). Manoalides 8 and 9, the acetal derivatives of the hemiacetal 11, showed higher activity than 11, suggesting that the presence of 24-O-methyl of manoalides 8 and 9 were important for cytotoxic activity (Table 4). Manoalide 11 was previously reported to possess good cytotoxicity against L1210 (mouse lymphocytic leukemia) and KB (mouth epidermal carcinoma) cell lines with the IC50 value of 0.053 and 0.63 µM, respectively [12]. Compound 20 (12-O-deacetyl-12-epi-scalarin) exhibited cytotoxic activity against A549 cells with an IC50 of 73.38 µM (Table 4), while its IC50 have been reported in the literature to be 36.82 μM [16]. Luffariolide B (10), (6E)- and (6Z)-neomanoalides (12 and 13) showed weak cytotoxicity against the MOLT-3 cell line with IC50 of 35.45, 37.81, and 34.10 μM, respectively, but they exhibited cytotoxicity against the L1210 cell line with IC50 of 3.23, 24.38, and 13.93 μM, respectively [15].
Table 4.
Cytotoxicity of compounds 3, 5, 7–20 and 23 (IC50, μM).
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were recorded on a JASCO DIP 1020 polarimeter using cylindrical glass cell (10 mm inner diameter (I.D.) × 10 mm). UV spectra were measured with a UV-1700 Pharma Spec (Shimadzu, Kyoto, Japan) spectrophotometer. Fourier transform infrared (FTIR) spectra were obtained using a universal attenuated total reflectance attached on Perkin Elmer Spectrum One spectrometer (PerkinElmer, Waltham, MA, USA). Nuclear magnetic resonance (NMR) spectra were recorded in a CDCl3 or C6D6 solution containing Me4Si as internal standard on Bruker AM400 or AVANCE600 spectrometer (Bruker Corporation, Billerica, MA, USA). HR–MS was performed on a Bruker (Micro ToF, Bruker Corporation, Billerica, MA, USA) spectrometer. HPLC was carried out on a Waters 600 system (Warers Corporation, Milford, MA, USA) equipped with a Waters Delta 600 pump, a Waters 600 Controller, a Waters 2998 photodiode array detector, and Waters Empower 2 software. Sephadex™ LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was used for a column gel filtration. All commercial grade solvents were distilled prior to use and spectral grade solvents were used for spectroscopic measurements.
3.2. Sponge Material
Sponges (Hyrtios erectus) CRI 572 and CRI 588 were collected by hand using scuba at a depth of 30–40 feet in the Similan Island at the Andaman Sea (Phangnga province, Thailand) on 22 and 23 February 2011, respectively. The sponges were identified by Dr. Sumaitt Putchakarn, Head of Marine Biodiversity Research, Unit Curator of Porifera and Echinodermata, Institute of Marine Science, Burapha University, Bangsaen, Chonburi, Thailand. The voucher specimens (CRI 572 and CRI 588) were presently deposited at the Laboratory of Natural Products, Chulabhorn Research Institute, Bangkok, Thailand.
3.3. Extraction and Isolation
A frozen sample (2.6 kg) of H. erectus collection CRI 572 was cut into small pieces and extracted exhaustively with MeOH. The extract was filtered through cotton, and then evaporated under reduced pressure to give an aqueous residue, which was partitioned with EtOAc. The organic layer was concentrated to give a dark brown solid (7.52 g). The EtOAc–soluble fraction was subjected to vacuum liquid chromatography on silica gel and eluted with an EtOAc–hexane gradient (0→100% of EtOAc). Nine fractions (F1–F9) were obtained. F2 (340.5 mg) was subjected to column chromatography on Sephadex LH-20, using CH2Cl2–MeOH (1:1) to give compound 15 (16 mg). F4 (293.3 mg) was repeatedly fractionated by Sephadex LH-20, using CH2Cl2–MeOH (1:1) to give a mixture compounds (96.8 mg) that was separated using on a HPLC column (Hichrome C18, 5 µm, 21.2 mm × 250 mm with MeOH–H2O (gradient, 77→97% MeOH for 40 min), flowrate 12 mL/min, λ 220, 265 nm) to afford compounds 16 (28 mg, at 12.7 min), 17 (4.9 mg, at 32.5 min). F5 (574.4 mg) was subjected to column chromatography on Sephadex LH-20, using 100% MeOH to obtain a mixtures compound (9.3 mg) that was separated using on a HPLC column (Sunfire C18, 5 µm, 10 mm × 250 mm, with 80% CH3CN–H2O, flowrate 3 mL/min, λ 220 nm) to afford 18 (4.0 mg, at 25.7 min). F6 (976.4 mg) was subjected to column chromatography on Sephadex LH-20, using CH2Cl2–MeOH (1:1) to give three fractions (f1–f3). Fraction f1 was subjected to repeated chromatography on Sephadex LH-20, using 100% MeOH to give a mixtures compound (73.5 mg) that was separated using a HPLC column (Hichrome C18, 5 µm, 21.2 mm × 250 mm, with MeOH–H2O (gradient, 75→100% MeOH for 50 min), flowrate 12 mL/min, λ 220 nm) to afford compound 19 (2.3 mg, at 35.2 min). Fraction f2 was subjected to column chromatography on Sephadex LH-20, using 100% MeOH to give a mixtures compound (45.6 mg) that was separated using on a HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm, with 88% MeOH–H2O, flowrate 12 mL/min, λ 220 nm) to afford 20 (20 mg, at 16.7 min). Fraction f3 was subjected to repeated chromatography on Sephadex LH-20, using 100% MeOH to give a mixtures compound (6.9 mg) that was separated using on a preparative thin layer chromatography (PTLC) [hexane–CH2Cl2–acetone (2:2:1) as eluent] to afford 21 (1.4 mg). F7 (508.1 mg) was subjected to column chromatography on Sephadex LH-20, using 100% MeOH to give a mixtures compound (12.1 mg) that was separated using on a HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm, with 20% MeOH–H2O, flowrate 12 mL/min, λ 254 nm) to afford compound 22 (7.5 mg, at 12.8 min). F8 (298.1 mg) was repeatedly fractionated by Sephadex LH-20, using CH2Cl2–MeOH (1:1) to give a mixture compound (127.6 mg) that was separated using a HPLC column (Hichrome C18, 5 µm, 21.2 mm × 250 mm with 75% MeOH–H2O, flowrate 12 mL/min, λ 222 nm) to afford compounds 4 (16.4 mg, at 11.3 min), and 13 (16.1 mg, at 33.5 min). F9 (249.2 mg) was subjected to repeated chromatography on Sephadex LH-20, using CH2Cl2–MeOH (1:1) to give a mixtures compound (20.3 mg) that was separated using a HPLC column (Hichrome C18, 5 µm, 21.2 mm × 250 mm, with MeOH–H2O (45–55% MeOH for 20 min), flowrate 12 mL/min, λ 234 nm) to afford compound 23 (5.4 mg, at 14.7 min). The flesh sponge Hyrtios erectus collection CRI 588 (14.0 kg) was cut into small pieces and extracted repeatedly with MeOH (20 L) three times (3 × 20 L). After the evaporation of the solvent, the concentrated MeOH extracts was partitioned between EtOAc and water and the EtOAc fraction was chromatographed on a silica gel column with CH2Cl2–hexane (1:1) containing increasing proportions of acetone as eluent, to give 7 fractions (A–G). Fraction C (4.0 g) was fractionated by Sephadex LH-20 with MeOH–CH2Cl2 (1:1) as eluent, to give 2 fractions (C1–C2). C2 (230.3 mg) was repeatedly chromatography on Sephadex LH-20, using CH2Cl2–MeOH (4:1) to give a mixtures compound (200 mg) that was separated using a HPLC column (Sunfire Prep C18, 5 µm, 10 mm × 250 mm) with MeOH–H2O (93:7), flowrate 2.5 mL/min, λ 224 nm, to afford compounds 5 (4.1 mg at 45.5 min), and 6 (7.2 mg at 48.2 min). Fraction D (9.0 g) was further fractionated on a Sephadex LH-20 column chromatography with MeOH–CH2Cl2 (1:1) to yield 4 fractions (D1–D4). D4 (400 mg) was further purified by HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm) with CH3CN–H2O (80:20), flowrate 8 mL/min, λ 224, 236 nm, to afford compounds 1 (5.9 mg, at 23.0 min), 2 (1.6 mg, at 29.6 min), and 3 (7.2 mg, at 35.6 min). Fraction E (25.0 g) was further fractionated by vacuum liquid chromatography eluted with CH2Cl2–hexane (1:1) containing increasing proportions of acetone as eluent, to give 4 fractions (E1–E4). Fraction E1 (2 g) was further purified by a HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm) with CH3CN–H2O (85:15), flowrate 8 mL/min, λ 224, 236 nm, to afford compounds 7 (74 mg, at 32.3 min), 8 (52 mg, at 47.9 min), and 9 (229 mg, 52.8 min). Fraction E3 (3 g) was further purified by a HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm) with CH3CN–H2O (80:20), flowrate 8 mL/min, λ 224, 236 nm, to afford compounds 10 (27 mg, at 19.9 min), and 11 (56 mg, at 32.5 min). Fraction E4 (1.4 g) was further purified by HPLC column (Sunfire C18, 5 µm, 19 mm × 250 mm) with CH3CN–H2O (82:18), flowrate 8 mL/min, λ 224, 236 nm, to afford compounds 13 (72 mg, at 27.35 min) and 14 (33 mg, at 29.3 min). Fraction G (4.0 g) was further filtrated on a Sephadex LH-20 column chromatography with MeOH to give a residue (655 mg), which was further purified by HPLC column (Cosmosil, 5 μ C18-MS-II, 20 mm × 250 mm) with CH3CN–H2O (60:40), flowrate 8 mL/min, λ 224, 236 nm, to afford compound 12 (70.0 mg, at 64.2 min).
Preparation of (S)- and (R)-MTPA Esters of compound 3: To a solution of 3 (3.1 mg) in pyridine (0.9 mL) was added (R)-MTPA-chloride (17 µL). The mixture was stirred at room temperature for 3 h, checked with thin layer chromatography (TLC) to make sure that the reaction was complete, quenched by the addition of 2 mL of H2O, and the mixture was subsequently extracted with CH2Cl2 (2 mL) three times (3 × 2 mL). The CH2Cl2 soluble layers were combined, dried over anhydrous MgSO4, and evaporated. The residue was subjected to short silica gel column chromatography using hexane–EtOAc (4:1) to give the (S)-MTPA ester 3a (1.6 mg).
Selected 1H NMR (CDCl3 600 MHz) of 3a: δH 9.32, s, H-24; 6.28, t (J = 7.1 Hz), H-6; 5.86, brs, H-2; 5.79, s, H-25; 5.74, dd (J = 7.2, 4.2 Hz), H-4; 5.09, t (J = 7.1 Hz), H-10; 3.03, ddd (J = 16.1, 6.6, 4.2 Hz), Ha-5; 2.90, dt (J = 16.1, 8.0 Hz), Hb-5; 2.24, t (J = 7.6 Hz), H2-8; 2.02, m, H2-9.
The same procedure was used to prepare the (R)-MTPA ester 3b (2.5 mg from 3.9 mg of 3) with (S)-MTPA chloride.
Selected 1H NMR (CDCl3 600 MHz) of 3b: δH 9.21, s, H-24; 6.13, t (J = 7.1 Hz), H-6; 6.00, brs, H-2; 5.81, s, H-25; 5.74, dd (J = 7.0, 4.2 Hz), H-4; 5.07, t (J = 6.9 Hz), H-10; 3.00, ddd (J = 16.2, 6.8, 4.3 Hz), Ha-5; 2.87, dt (J = 16.2, 7.5 Hz), Hb-5; 2.20, t (J = 7.7 Hz), H2-8; 1.99, m, H2-9
(S)-MTPA esters 5a (1.3 mg from 1.4 mg of 5) and 6a (1.8 mg from 1.4 mg of 6) and (R)-MTPA esters 5b (1.5 mg from 1.4 mg of 5), 6b (1.7 mg from 1.2 mg of 6) were prepared by the method mentioned above.
Selected 1H NMR (CDCl3, 400 MHz) of 5a: δH 5.77, brs, H-3; 2.72, dd (J = 15.7, 6.9 Hz), Ha-1′; 2.62, dd (J = 15.7, 3.1 Hz), Hb-1′; 5.28, m, H-2′; 1.67, m, H2-3′; 4.56, dd (J = 17.8, 1.8 Hz), Ha-5; 4.25, brd (J = 17.6 Hz), Hb-5.
Selected 1H NMR (CDCl3, 400 MHz) of 5b: δH 5.86, s, H-3; 2.78, dd (J = 15.7, 7.3 Hz), Ha-1′; 2.69, dd (J = 15.7, 3.1 Hz), Hb-1′; 5.29, m, H-2′; 1.63, m, H2-3′; 4.69, brd (J = 17.4 Hz), Ha-5; 4.52, brd (J = 17.4 Hz), Hb-5.
Selected 1H NMR (CDCl3, 600 MHz) of 6a: δH 5.77, brs, H-3; 2.72, dd (J = 15.6, 7.1 Hz), Ha-1′; 2.63, dd (J = 15.6, 3.4 Hz), Hb-1′; 5.28, m, H-2′; 1.71, m, Ha-3′; 1.59, m, Hb-3′; 1.29, m, H2-4′; 4.57, dd (J = 17.8, 1.6 Hz), Ha-5; 4.26, brd (J = 16.7 Hz), Hb-5.
Selected 1H NMR (CDCl3, 600 MHz) of 6b: δH 5.86, brs, H-2; 2.78, dd (J = 15.4, 6.58 Hz), Ha-1′; 2.69, dd (J = 15.4, 3.6 Hz), Hb-1′; 5.29, m, H-2′; 1.67, m, Ha-3′; 1.55, m, Hb-3′; 1.23, m, H2-4′; 4.69, d (J = 17.5 Hz), Ha-5; 4.52, d (J = 17.5 Hz), Hb-5.
Erectusolide B (1): Colorless powder; −1.3 (c 0.56, CHCl3); UV λmax (MeOH) nm (log ε) 202 (3.9), 224 (3.4), IR (ATR) νmax 3443 (br), 2927, 2861, 1779, 1746, 1629, 1456, 1379, 1266, 1143, 1041, 888, 851, 735, 702 cm−1; 1H and 13C NMR data see Table 1; HRAPCIMS m/z 385.2727 [M + H]+ (calcd for C25H37O3, 385.2737).
Erectusolide C (2): Colorless powder; −18.5 (c 0.16, CHCl3); UV λmax (MeOH) nm (log ε) 234 (4.3); IR (ATR) νmax 2924, 2855, 1779, 1748, 1682, 1632, 1458, 1378, 1261, 1142, 1040, 887 cm−1; 1H and 13C NMR data see Table 1; HRESIMS m/z 405.2390 [M + Na]+ (calcd for C25H34NaO3, 405.2400).
seco-Manoalide-25-methyl ether (3): Pale yellow solid; −22.2 (c 1.86, CHCl3); UV λmax (MeOH) nm (log ε) 203.5 (4.3), 232 (sh); IR (ATR) νmax 3446 (br), 2930, 2863, 1762, 1687, 1457, 1371, 1203, 1120, 1070, 960, 898, 870, 735, 702 cm−1; 1H and 13C NMR data see Table 2; HRAPCIMS m/z 465.2414 [M + Cl]− (calcd for C26H38ClO5, 465.2413).
Erectusolide D (4): Pale yellow gum; −20.9 (c 1.64, CHCl3); UV λmax (MeOH) nm (log ε) 202 (4.3), IR (ATR) νmax 3424 (br), 2928, 2857, 1739, 1642, 1456, 1382, 1258, 1172, 1063, 898 cm−1; 1H and 13C NMR data see Table 2; HRAPCIMS m/z 453.2418 [M + Cl]− (calcd for C25H38ClO5, 453.2413).
erectusfuranone A (5): Colorless powder; +1.3 (c 0.32, CHCl3); UV λmax (MeOH) nm (log ε) 206 (3.9); IR (ATR) νmax 3442 (br), 2923, 2853, 1777, 1739, 1637, 1457, 1378, 1175, 1027, 888, 720 cm−1; 1H and 13C NMR data see Table 3; HRAPCIMS m/z 367.3213 [M + H]+ (calcd for C23H43O3, 367.3207).
erectusfuranone B (6): Colorless powder; −4.9 (c 0.61, CHCl3); UV λmax (MeOH) nm (log ε) 206 (3.9); IR (ATR) νmax 3402 (br), 2916, 2851, 1779, 1756, 1736, 1634, 1469, 1025, 892, 720 cm−1; 1H and 13C NMR data see Table 3; HRESIMS m/z 353.3053 [M + H]+ (calcd for C22H41O3, 353.3050).
3.4. Cytotoxicity Assay
Cytotoxic activity for adhesive cell lines including HepG2, HeLa, HuCCA-1, A549, H69AR, KB, T47D, MDA-MB-231, and MRC-5 cell lines were evaluated with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay [25,26]. For the non-adhesive MOLT-3 cell line, the cytotoxicity was assessed using the 2,3-bis-(2-methoxy-4-nitro-5-sulphenyl-(2H)-tetrazolium-5-carboxanilide (XTT) assay [27]. Etoposide and doxorubicin were used as positive controls (Table 4)
4. Conclusions
Two separate collections of H. erectus were studied. Three new sesterterpenes, erectusolides B (1), C (2), and seco-manoalide-25-methyl ether (3), two new 2-furanone derivatives, erectusfuranones A (5) and B (6) together with eight known sesterterpenes, 7–14 were isolated from H. erectus collection CRI 588. One additional new sesterterpene, erectusolide D (4), and ten known compounds (13, 15–23) were also obtained from the H. erectus collection CRI 572. Three sesterterpenes 8, 9, and 20 showed good cytotoxic activity against the MOLT-3 cell line with IC50 values of 1.30–7.64 μM and with SI (selectivity index) values 7–9.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/12/474/s1, Figure S1. 1H NMR spectrum (600 MHz) of compound 1 in C6D6, Figure S2. 13C NMR spectrum (150 MHz) of compound 1 in C6D6, Figure S3. HSQC spectrum of compound 1 in C6D6, Figure S4. HMBC spectrum of compound 1 in C6D6, Figure S5. 1H NMR spectrum (600 MHz) of compound 2 in CDCl3, Figure S6. 1H NMR spectrum (600 MHz) of compound 2 in C6D6, Figure S7. 13C NMR spectrum (150 MHz) of compound 2 in CDCl3, Figure S8. 13C NMR spectrum (150 MHz) of compound 2 in C6D6, Figure S9. HSQC spectrum of compound 2 in CDCl3, Figure S10. HSQC spectrum of compound 2 in C6D6, Figure S11. HMBC spectrum of compound 2 in CDCl3, Figure S12. HMBC spectrum of compound 2 in C6D6, Figure S13. 1H NMR spectrum (600 MHz) of compound 3 in CDCl3, Figure S14. 13C NMR spectrum (150 MHz) of compound 3 in CDCl3, Figure S15. HSQC spectrum of compound 3 in CDCl3, Figure S16. HMBC spectrum of compound 3 in CDCl3, Figure S17. 1H NMR spectrum (600 MHz) of compound 4 in CDCl3, Figure S18. 13C NMR spectrum (150 MHz) of compound 4 in CDCl3, Figure S19. HMQC spectrum of compound 4 in CDCl3, Figure S20. HMBC spectrum of compound 4 in CDCl3, Figure S21. 1H NMR spectrum (600 MHz) of compound 5 in CDCl3, Figure S22. 13C NMR spectrum (150 MHz) of compound 5 in CDCl3, Figure S23. HSQC spectrum of compound 5 in CDCl3, Figure S24. HMBC spectrum of compound 5 in CDCl3, Figure S25. 1H NMR spectrum (600 MHz) of compound 6 in CDCl3, Figure S26. 13C NMR spectrum (150 MHz) of compound 6 in CDCl3, Figure S27. HSQC spectrum of compound 6 in CDCl3, Figure S28. HMBC spectrum of compound 6 in CDCl3.
Author Contributions
Conceptualization, C.M. and S.R.; Funding acquisition, C.M.; Investigation, W.K. and H.P.; Project administration, S.R.; Supervision, S.R.; Validation, H.P.; Writing—original draft, H.P.; Writing—review & editing, P.T. and S.R.
Funding
This research received no external funding.
Acknowledgments
The authors thank P. Intachote, S. Sengsai and B. Saimanee for testing cytotoxic activity.
Conflicts of Interest
The authors declare no conflict of interest
References
- Bugni, S.T.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Kaweetripob, W.; Mahidol, C.; Wongbundit, S.; Tuntiwachwuttikul, P.; Ruchirawat, S.; Prawat, H. Sesterterpenes and phenolic alkenes from the Thai sponge Hyrtios erectus. Tetrahedron 2018, 74, 316–323. [Google Scholar] [CrossRef]
- Jiao, W.H.; Hong, L.L.; Sun, J.B.; Piao, S.J.; Chen, G.D.; Deng, H.; Wang, S.P.; Yang, F.; Lin, H.W. (±)-Hippolide J—A Pair of Unusual Antifungal Enantiomeric Sesterterpenoids from the Marine Sponge Hippospongia lachne. Eur. J. Org. Chem. 2017, 24, 3421–3426. [Google Scholar] [CrossRef]
- Hawas, U.W.; Abou El-Kassem, L.T.; Abdelfattah, M.S.; Elmallah, M.I.Y.; Eid, M.A.G.; Monier, M.; Nithiyanandam, M. Cytotoxic activity of alkyl benzoate and fatty acids from the red sea sponge Hyrtios erectus. Nat. Prod. Res. 2018, 32, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Elhady, S.S.; Al-Abd, A.M.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. Antiproliferative Scalarane-Based Metabolites from the Red Sea Sponge Hyrtios erectus. Mar. Drugs 2016, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Kato, H.; Rotinsulu, H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Yokosawa, H.; Tsukamoto, S. Variabines A and B: New β-carboline alkaloids from the marine sponge Luffariella variabilis. J. Nat. Med. 2014, 68, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Mahidol, C.; Prawat, H.; Sangpetsiripan, S.; Ruchirawat, S. Bioactive Scalaranes from the Thai Sponge Hyrtios gumminae. J. Nat. Prod. 2009, 72, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Konig, G.; Wrignt, A.D.; Sticher, O. Four new antibacterial sesterterpenes from a marine sponge of the genus Luffariella. J. Nat. Prod. 1992, 55, 174–178. [Google Scholar] [CrossRef]
- Namikoshi, M.; Suzuki, S.; Meguro, S.; Nagai, H.; Koike, Y.; Kitazawa, A.; Kobayashi, H.; Oda, T.; Yamada, J. Manoalide derivatives from a marine sponge Luffariella sp. collected in Palau. Fish. Sci. 2004, 70, 152–158. [Google Scholar] [CrossRef]
- Pailee, P.; Mahidol, C.; Ruchirawat, S.; Prachyawarakorn, V. Sterols from Thai Marine Sponge Petrosia (Strongylophora) sp. and Their Cytotoxicity. Mar. Drugs 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Okamoto, T.; Hayashi, K.; Yokoyama, N.; Sasaki, T.; Kitagawa, I. Marine Natural Products. XXXII. Absolute Configurations of C-4 of the Manoalide Family, Biologically Active Sesterterpenes from the Marine Sponge Hyrtions erecta. Chem. Pharm. Bull. 1994, 42, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Shigemori, H.; Ishibashi, M.; Mizobe, F.; Kawashima, A.; Nakaike, S.; Kobayashi, J. New Sesterterpenes with Nerve Growth Factor Synthesis-Stimulating Activity from the Okinawan Marine Sponge Hyrtios sp. Chem. Pharm. Bull. 1993, 41, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.A.; Elkhayat, E.S.; Ebel, R.; Edrada, R.; Prokschc, P. Indole alkaloid from the Red Sea sponge Hyrtios erectus. Arkivoc 2007, XV, 225–231. [Google Scholar]
- Tsuda, M.; Shigemori, H.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Luffariolides AE, new cytotoxic sesterterpenes from the Okinawan marine sponge Luffariella sp. J. Org. Chem. 1992, 57, 3503–3507. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Miura, S.; van Soest, R.W.M.; Ohta, T. Three New Cytotoxic Sesterterpenes from a Marine Sponge Spongia sp. J. Nat. Prod. 2003, 66, 438–440. [Google Scholar] [CrossRef] [PubMed]
- De Silva, E.D.; Scheuer, P.J. Three new sesterterpenoid antibiotics from the marine sponge Luffariella variabilis (Polejaff). Tetrahedron Lett. 1981, 22, 3147–3150. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Prachyawarakorn, V.; Tuntiwachwuttikul, P.; Ruchirawat, S. Sesquiterpene isocyanides, isothiocyanates, thiocyanates, and formamides from the Thai sponge Halichondria sp. Tetrahedron 2016, 72, 4222–4229. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Kaweetripob, W.; Wittayalai, S.; Ruchirawat, S. Iodo-sesquiterpene hydroquinone and brominated indole alkaloids from the Thai sponge Smenospongia sp. Tetrahedron 2012, 68, 6881–6886. [Google Scholar] [CrossRef]
- Prawat, H.; Mahidol, C.; Wittayalai, S.; Intachote, P.; Kanchanapoom, T.; Ruchirawat, S. Nitrogenous sesquiterpenes from the Thai marine sponge Halichondria sp. Tetrahedron 2011, 67, 5651–5655. [Google Scholar] [CrossRef]
- Bergquist, P.R.; Cambie, R.C.; Kernan, M.R. Scalarane sesterterpenes from Collospongia auris, a new thorectid sponge. Biochem. Syst. Ecol. 1990, 18, 349–357. [Google Scholar] [CrossRef]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Jefford, C.W.; Jaggi, D.; Bernardinelli, G.; Boukouvalas, J. The synthesis of (±)-cavernosine. Tetrahedron Lett. 1987, 28, 4041–4044. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Absolute configurations of marine diterpenes possessing a xenicane skeleton. An application of an advanced Mosher’s method. Tetrahedron Lett. 1989, 30, 3147–3150. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semi-automated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Doyle, A.; Griffiths, J.B. Mammalian Cell Culture: Essential Techniques; John Wiley & Sons: Chichester, UK, 1997. [Google Scholar]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).