Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study

 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
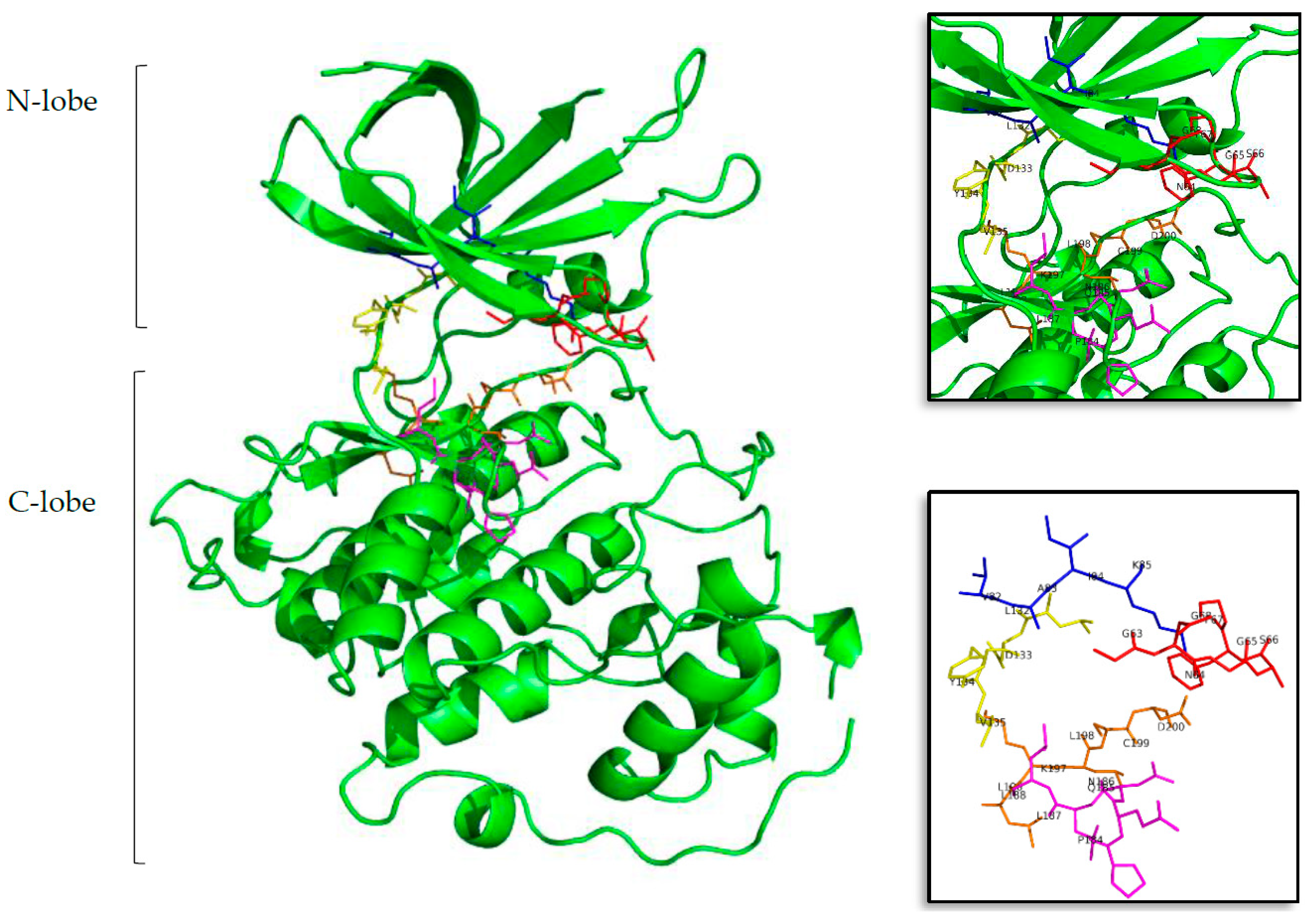
2.1. New Possible GSK3β, CK1δ, DYRK1A, and CLK1 ATP-Competitive Inhibitors
2.2. Kororamide A–B and Convolutamine I–J as Possible Kinase Inhibitors
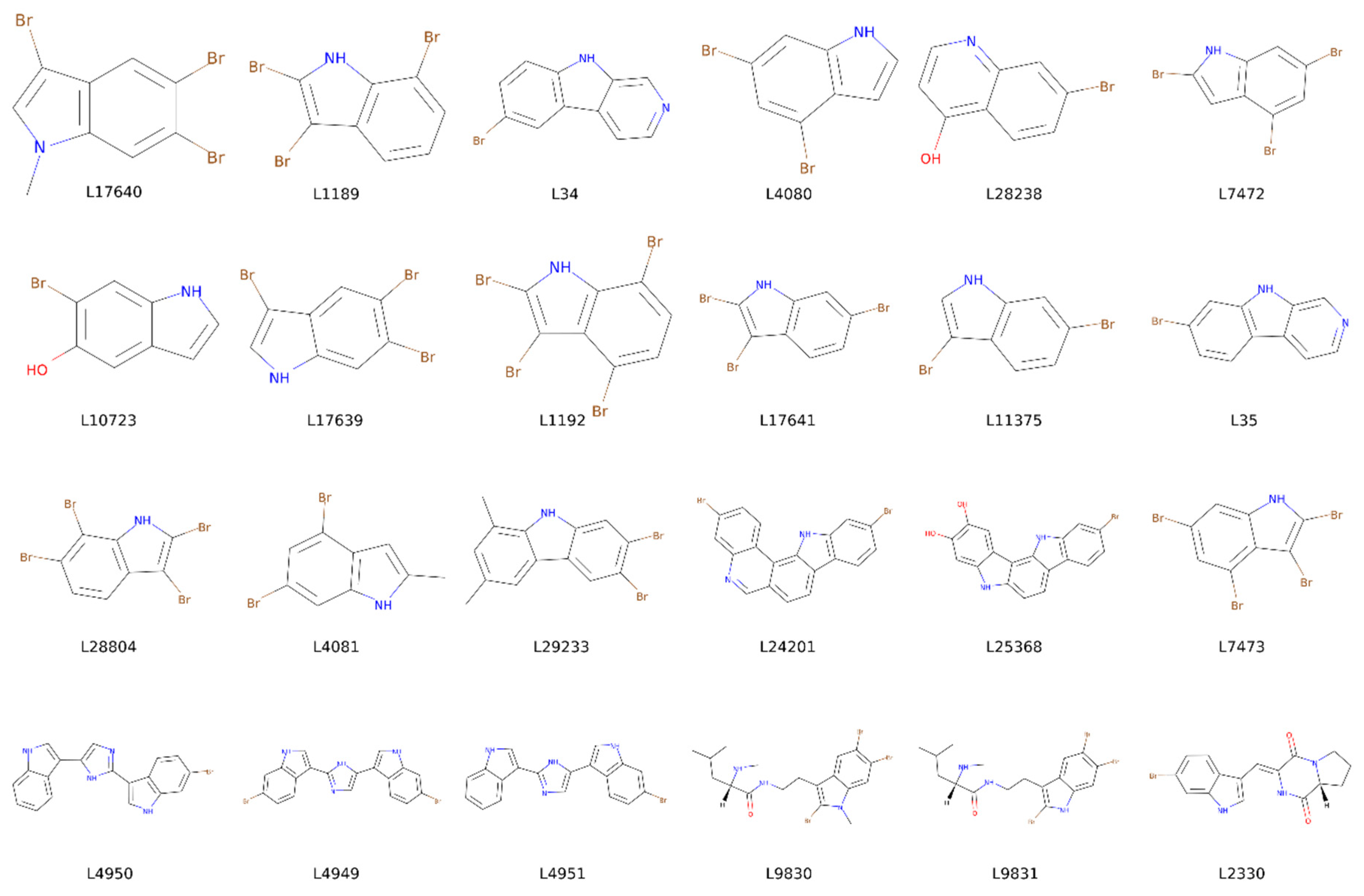
2.3. Marine Natural Products and Indole Scaffold Validation
2.4. Indole Derivatives
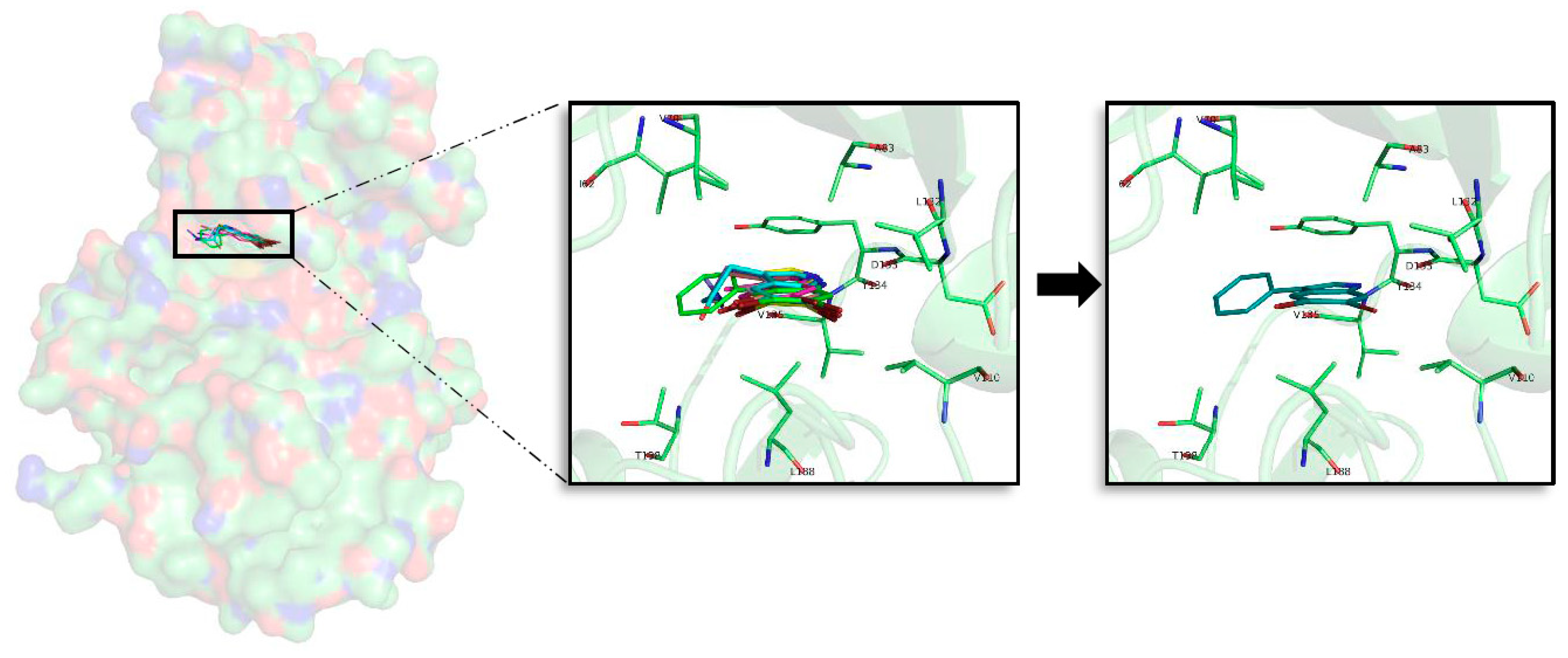
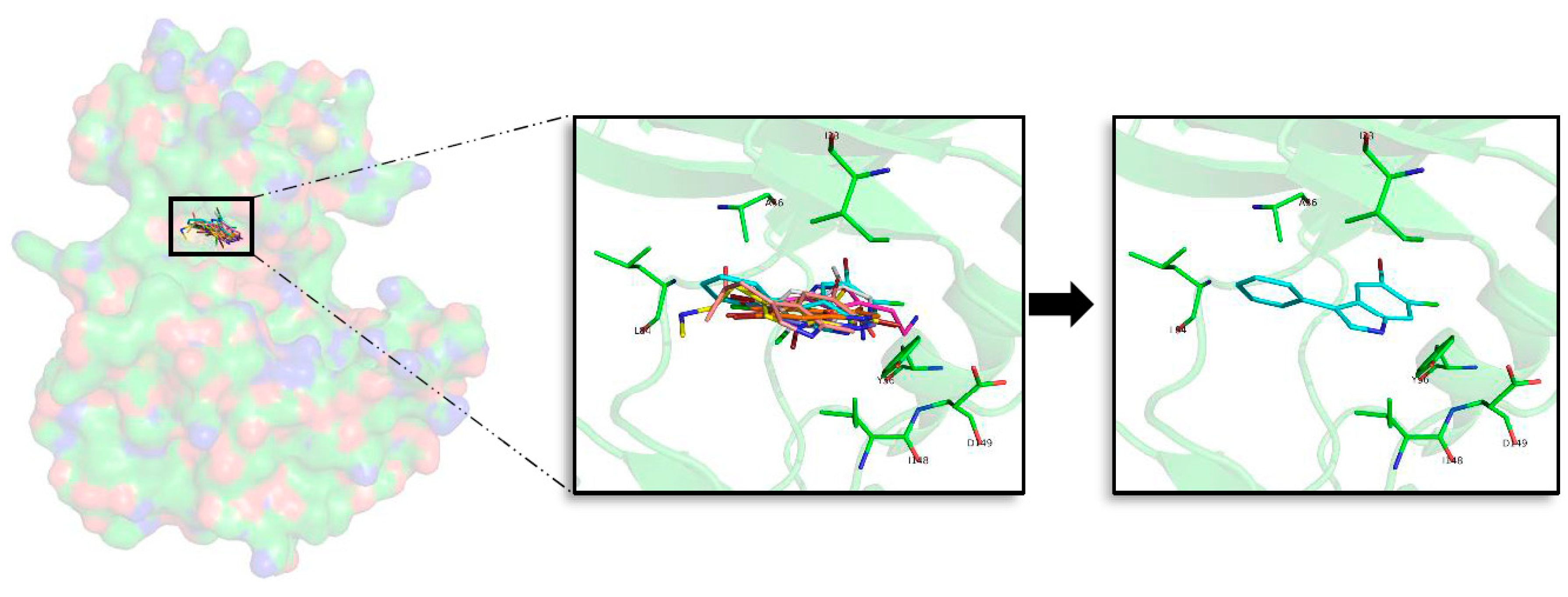
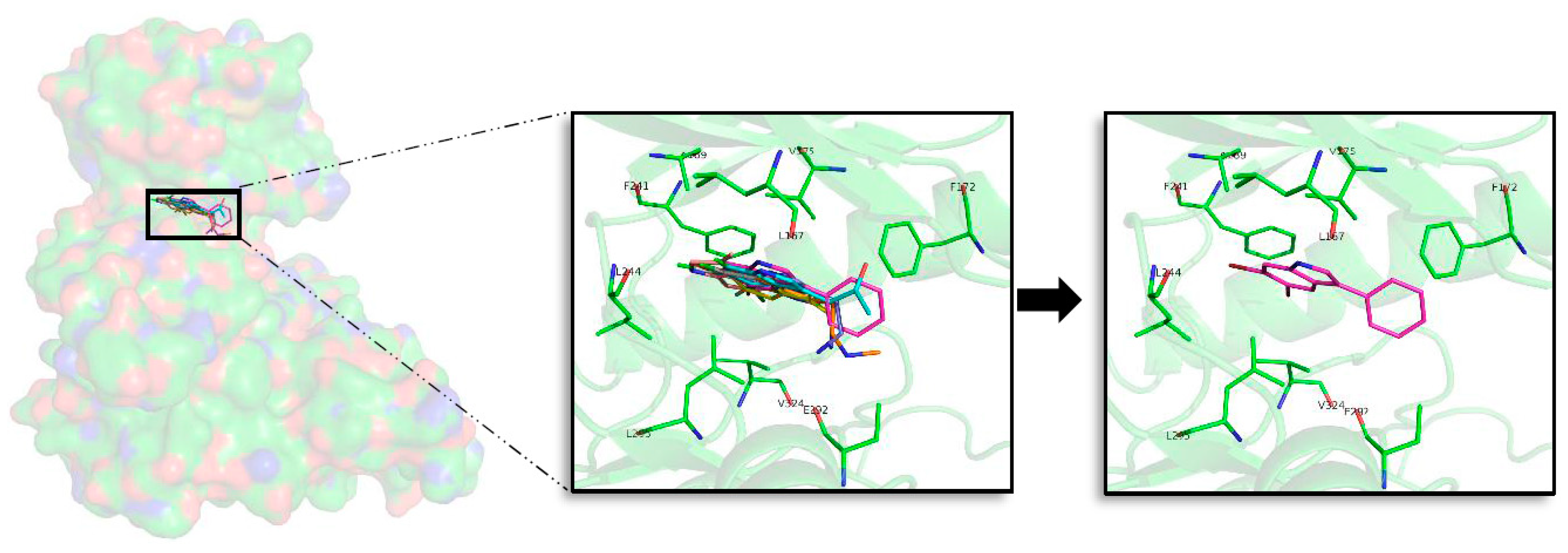
2.5. In Silico Binding and Binding Mode Analysis of Indole Derivatives
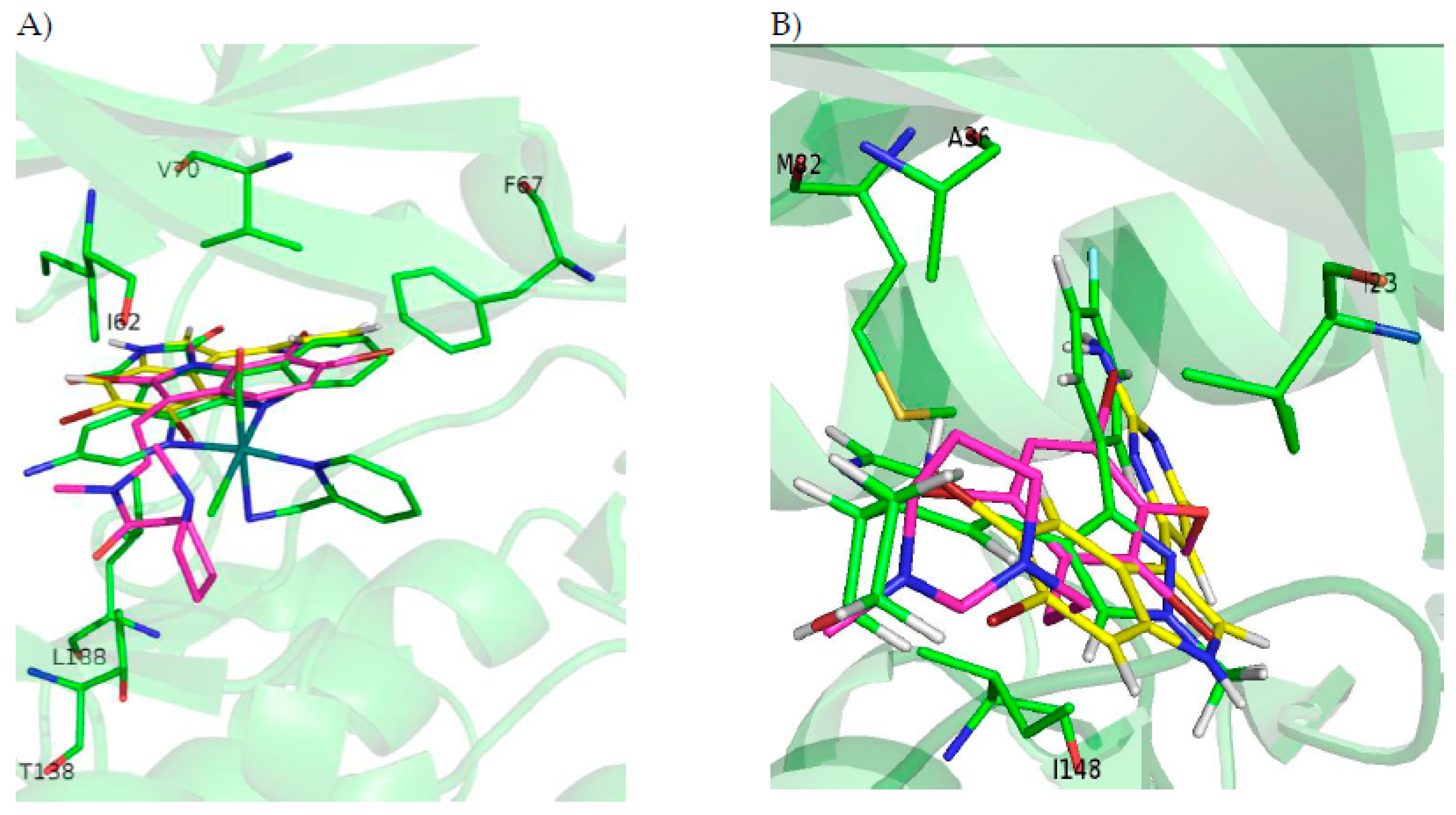
2.5.1. GSK3β
2.5.2. CK1δ
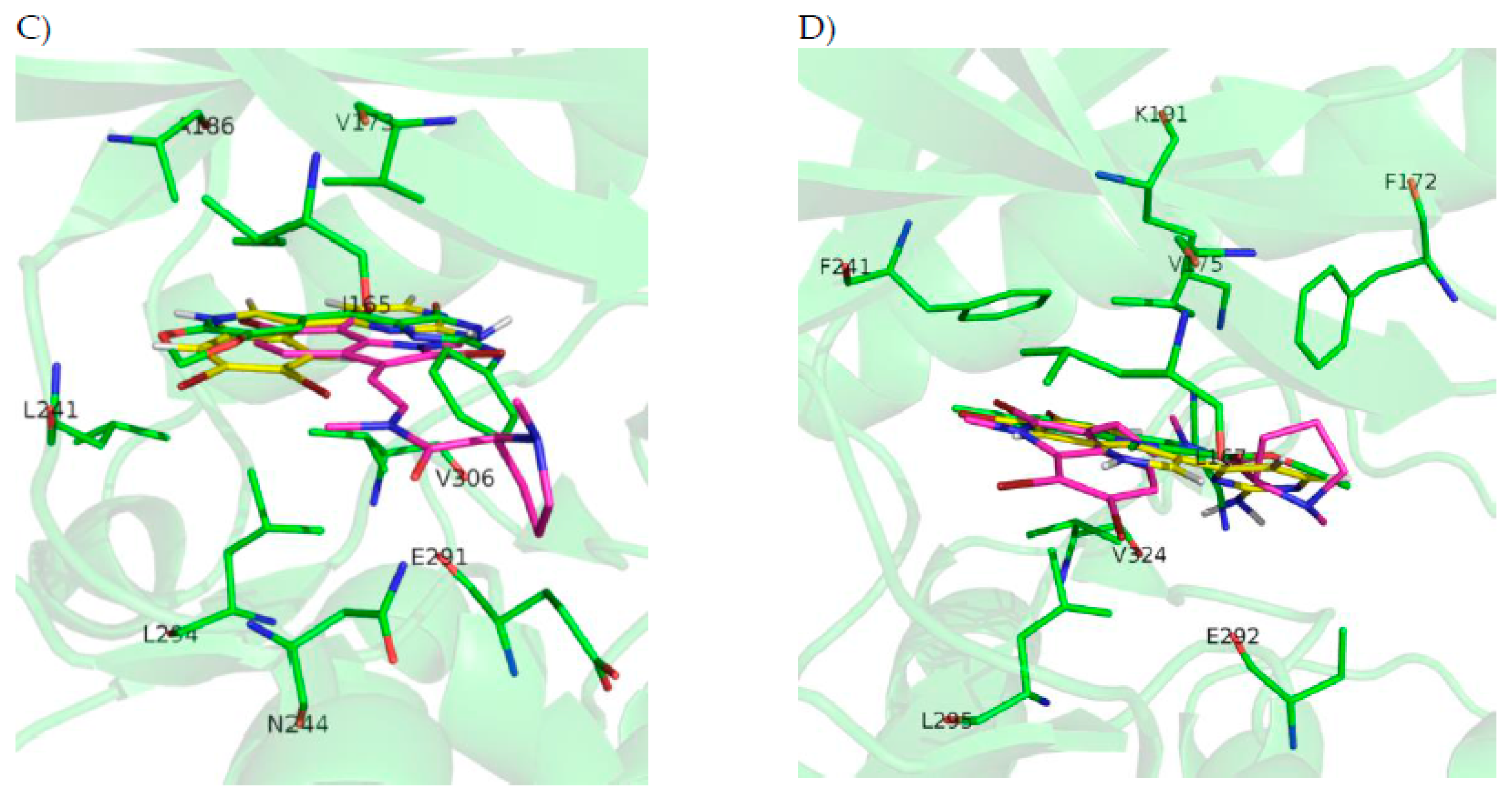
2.5.3. DYRK1A
2.5.4. CLK1
2.6. Selectivity
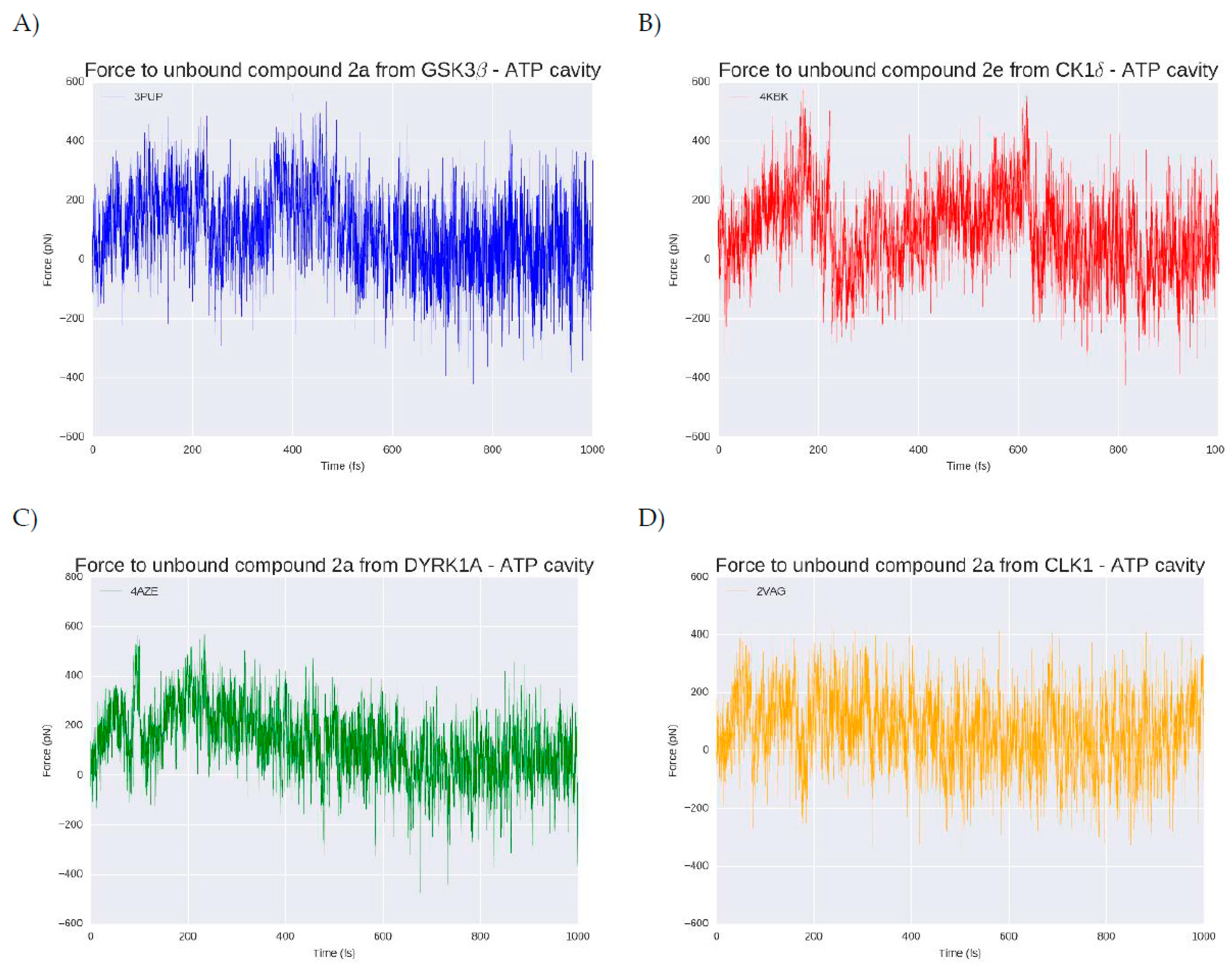
2.7. 2a and 2e Unbinding
2.8. Pharmacokinetic Properties of Kororamide A–B, Convolutamine I–J, and the Designed Derivatives
2.8.1. Absorption Properties
2.8.2. Distribution Properties
2.8.3. Metabolism Properties
2.8.4. Excretion Properties
2.8.5. Toxicity Properties
3. Materials and Methods
3.1. Computational Virtual Screening
3.2. Structure Modelling
3.3. Docking Calculations
3.4. Molecular Dynamics Simulations
3.5. Molecular Mechanics/Generalized Born Surface Area
3.6. Steered Molecular Dynamics
3.7. Interaction Analysis
3.8. ADME/Tox Properties Prediction
3.9. Graphical Representations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSK3β | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Binding Energy | R0/R1 | a | −6.9/−6.9 | −8.1/−8.1 | −5.9/−5.9 | −6.5/−6.5 | −6.3/−6.3 | −6.2/−6.2 | −6.6/−6.6 |
| MM/GBSA | −30.3141 | −31.2458 | −13.8779 | −27.6481 | −27.6534 | −18.5779 | −18.8955 | ||
| Binding Energy | R0/R1 | b | −6.8/−6.8 | −7.9/−7.9 | −5.1/−5.1 | −6.6/−6.6 | −5.6/−5.6 | −5.8/−5.8 | −5.9/−5.9 |
| MM/GBSA | −22.0902 | −23.9910 | −7.0321 | −17.6371 | −19.3959 | −9.2248 | −20.6117 | ||
| Binding Energy | R0/R1 | c | −6.8/−6.8 | −8.1/−8.1 | −5.8/−5.8 | −6.3/−6.3 | −6.3/−6.3 | −6.2/−6.2 | −6.7/−6.7 |
| MM/GBSA | −26.1345 | −28.4927 | −10.3167 | −24.8857 | −23.8207 | −14.7674 | −17.0307 | ||
| Binding Energy | R0/R1 | d | −7/−7 | −8.1/−8.1 | −5.2/−5.2 | −6.8/−6.8 | −6.5/−6.5 | −6.1/−6.1 | −6.7/−6.7 |
| MM/GBSA | −26.2805 | −29.6158 | −12.1225 | −22.7717 | −23.6754 | −14.4347 | −13.6539 | ||
| Binding Energy | R0/R1 | e | −7/−7 | −8.1/−8.1 | −6/−6 | −6.7/−6.7 | −5.8/−5.8 | −6.1/−6.1 | −6.7/−6.7 |
| MM/GBSA | −27.2898 | −19.5564 | −25.4497 | −26.4593 | −17.3314 | −18.9577 | |||
| Binding Energy | R0/R1 | f | −6/−6 | −8.2/−8.2 | −5.8/−5.8 | −6.3/−6.3 | −6.2/−6.2 | −6/−6 | −6.4/−6.4 |
| MM/GBSA | −26.4517 | −6.2475 | −23.7315 | −19.8909 | −13.3104 | ||||
| Binding Energy | R0/R1 | g | −6.2/−6.2 | −8.2/−8.2 | −5.8/−5.8 | −6.5/−6.5 | −6.2/−6.2 | −6.3/−6.3 | −5.9/−5.9 |
| MM/GBSA | −28.2864 | −14.2501 | −24.4026 | −24.7124 | −16.8986 | −20.7272 | |||
| CK1δ | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Binding Energy | R0/R1 | a | −5.1/−5.1 | −7.7/−7.7 | −5.2/−5.2 | −5.3/−5.3 | −5.6/−5.6 | −5.6/−5.6 | −5.3/−5.3 |
| MM/GBSA | −35.4499 | −3.7149 | −26.5327 | −26.4630 | −18.4901 | ||||
| Binding Energy | R0/R1 | b | −5.8/−5.8 | −7.7/−7.7 | −5.5/−5.5 | −5.3/−5.3 | −5.8/−5.8 | −5.6/−5.6 | −5.2/−5.2 |
| MM/GBSA | −24.0479 | −30.2266 | −21.2435 | −6.4142 | −11.6429 | ||||
| Binding Energy | R0/R1 | c | −5.6/−5.6 | −7.6/−7.6 | −4.8/−4.8 | −5.7/−5.7 | −5.2/−5.2 | −5.1/−5.1 | −5.6/−5.6 |
| MM/GBSA | −29.9803 | −19.4546 | −22.8644 | −26.4159 | −12.0871 | ||||
| Binding Energy | R0/R1 | d | −5.9/−5.9 | −7.5/−7.5 | −4.7/−4.7 | −5.5/−5.5 | −5.9/−5.9 | −5.1/−5.1 | −5.5/−5.5 |
| MM/GBSA | −19.8892 | −25.5694 | |||||||
| Binding Energy | R0/R1 | e | −6.1/−6.1 | −7.5/−7.5 | −5.4/−5.4 | −5.3/−5.3 | −5.1/−5.1 | −5.3/−5.3 | −5.3/−5.3 |
| MM/GBSA | −37.8982 | −28.6573 | −28.5831 | −16.2323 | |||||
| Binding Energy | R0/R1 | f | −6.2/−6.2 | −7.5/−7.5 | −5.4/−5.4 | −5.2/−5.2 | −5.1/−5.1 | −5.5/−5.5 | −5.4/−5.4 |
| MM/GBSA | −34.6944 | −22.6616 | −26.3915 | −13.6562 | −15.4050 | ||||
| Binding Energy | R0/R1 | g | −6.1/−6.1 | −7.3/−7.3 | −4.8/−4.8 | −5.2/−5.2 | −5.1/−5.1 | −5.6/−5.6 | −5.5/−5.5 |
| MM/GBSA | −33.2393 | −28.7631 | −26.0731 | −28.0238 | |||||
| DYRK1A | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Binding Energy | R0/R1 | a | −6.2/−6.2 | −8.6/−8.6 | −5.9/−5.9 | −6.8/−6.8 | −6.7/−6.7 | −5.7/−5.7 | −6/−6 |
| MM/GBSA | −37.8422 | −15.2733 | −30.7518 | −31.2535 | −18.9387 | −20.8203 | |||
| Binding Energy | R0/R1 | b | −6.5/−6.5 | −8.5/−8.5 | −7/−7 | −7.2/−7.2 | −6.6/−6.6 | −7/−7 | −7.3/−7.3 |
| MM/GBSA | −23.7829 | −28.0642 | −8.4887 | −19.4730 | −21.8802 | −10.2503 | −11.8981 | ||
| Binding Energy | R0/R1 | c | −6.3/−6.3 | −8.6/−8.6 | −5.9/−5.9 | −7.2/−7.2 | −6.5/−6.5 | −6.9/−6.9 | −7.3/−7.3 |
| MM/GBSA | −30.2004 | −34.1231 | −12.1852 | −26.5473 | −28.3000 | −18.7332 | −13.2158 | ||
| Binding Energy | R0/R1 | d | −5.9/−5.9 | −8.6/−8.6 | −6.7/−6.7 | −6.6/−6.6 | −6.3/−6.3 | −6.5/−6.5 | −6.6/−6.6 |
| MM/GBSA | −30.8597 | −36.1125 | −10.3667 | −26.3748 | −26.9602 | −17.1653 | −16.4510 | ||
| Binding Energy | R0/R1 | e | −6.4/−6.4 | −8.6/−8.6 | −6.5/−6.5 | −6.6/−6.6 | −6.4/−6.4 | −6.6/−6.6 | −6.4/−6.4 |
| MM/GBSA | −32.8862 | −13.5197 | −28.9038 | −29.6780 | −20.0635 | ||||
| Binding Energy | R0/R1 | f | −6.2/−6.2 | −8.6/−8.6 | −5.8/−5.8 | −6.8/−6.8 | −6.3/−6.3 | −6.4/−6.4 | −6.9/−6.9 |
| MM/GBSA | −28.9419 | −33.0823 | −14.5027 | −25.4579 | −28.4081 | −15.8583 | −17.0209 | ||
| Binding Energy | R0/R1 | g | −6.7/−6.7 | −8.7/−8.7 | −5.8/−5.8 | −6.8/−6.8 | −6.7/−6.7 | −6.1/−6.1 | −6.9/−6.9 |
| MM/GBSA | −30.3186 | −35.5805 | −13.4928 | −27.1510 | −29.6010 | −18.0146 | −17.3973 | ||
| CLK1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Binding Energy | R0/R1 | a | −7.5/−7.5 | −8.8/−8.8 | −6.4/−6.4 | −6.8/−6.8 | −6.2/−6.2 | −6.4/−6.4 | −6.9/−6.9 |
| MM/GBSA | −29.8546 | −34.1041 | −15.2943 | −26.7584 | −25.8120 | −27.7489 | −24.0832 | ||
| Binding Energy | R0/R1 | b | −7.1/−7.1 | −8.4/−8.4 | −7/−7 | −5.6/−5.6 | −7.5/−7.5 | −6.6/−6.6 | −7.2/−7.2 |
| MM/GBSA | −25.9089 | −27.3711 | −13.7919 | −26.2326 | −22.1539 | −16.6984 | −20.9149 | ||
| Binding Energy | R0/R1 | c | −7.6/−7.6 | −9.1/−9.1 | −7/−7 | −7.3/−7.3 | −6.4/−6.4 | −6.6/−6.6 | −6.4/−6.4 |
| MM/GBSA | −34.0221 | −16.2165 | −24.6708 | −29.4190 | −25.8368 | −20.9436 | |||
| Binding Energy | R0/R1 | d | −7.6/−7.6 | −8.4/−8.4 | −6.8/−6.8 | −7.8/−7.8 | −6.3/−6.3 | −6.9/−6.9 | −7/−7 |
| MM/GBSA | −26.9398 | −30.7361 | −25.6581 | −25.1797 | −19.3712 | −17.5727 | |||
| Binding Energy | R0/R1 | e | −6.8/−6.8 | −8.9/−8.9 | −6.9/−6.9 | −7.5/−7.5 | −5.9/−5.9 | −7.1/−7.1 | −7/−7 |
| MM/GBSA | −30.0891 | −16.2097 | −28.3695 | −28.0697 | −27.0478 | −17.4985 | |||
| Binding Energy | R0/R1 | f | −7.5/−7.5 | −8.6/−8.6 | −6.7/−6.7 | −7.5/−7.5 | −6.2/−6.2 | −6.4/−6.4 | −6.7/−6.7 |
| MM/GBSA | −28.1471 | −20.4786 | −23.9274 | −27.3596 | −15.4231 | −21.2829 | |||
| Binding Energy | R0/R1 | g | −7/−7 | −8.9/−8.9 | −6/−6 | −7.4/−7.4 | −6.8/−6.8 | −6.4/−6.4 | −6.8/−6.8 |
| MM/GBSA | −30.3541 | −33.9082 | −16.3122 | −25.0002 | −30.7737 | −25.4765 | |||
| Absorption | Distribution | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mol Weight | LogS | P-Glycoprotein | Caco-2 permeability | Intestinal Absorption | LogP | BBB | PPB | VDss | CNS Permeability |
| Compound 1a | 331 | −4.8 | inactive | High | 92.328 | 2.8 | 0.298 | High | 0.231 | −1.832 |
| Compound 1b | 209.2 | −3.4 | inactive | Moderate | 94.522 | 2.6 | 0.186 | Medium | 0.131 | −1.888 |
| Compound 1c | 242.1 | −4.3 | inactive | High | 92.462 | 2.9 | 0.279 | High | 0.37 | −1.832 |
| Compound 1d | 270.1 | −4 | inactive | High | 93.463 | 2.6 | 0.152 | High | 0.249 | −1.866 |
| Compound 1e | 286.6 | −4.5 | inactive | High | 92.395 | 2.8 | 0.278 | High | 0.385 | −1.832 |
| Compound 1f | 270.1 | −4 | inactive | High | 93.49 | 2.6 | 0.152 | High | 0.256 | −1.87 |
| Compound 1g | 286.6 | −4.5 | inactive | High | 92.395 | 2.8 | 0.278 | High | 0.385 | −1.832 |
| Compound 2a | 351 | −6.1 | inactive | Moderate | 90.067 | 4.1 | 0.477 | High | 0.234 | −0.894 |
| Compound 2b | 229.2 | −5.1 | inactive | Moderate | 92.006 | 3.4 | 0.539 | High | −0.081 | −0.946 |
| Compound 2c | 262.1 | −5.9 | inactive | Moderate | 90.201 | 4.6 | 0.482 | High | 0.197 | −0.894 |
| Compound 2d | 306.6 | −6.1 | inactive | Moderate | 90.134 | 4.4 | 0.48 | High | 0.215 | −0.894 |
| Compound 2e | 290.1 | −5.7 | inactive | Moderate | 91.036 | 3.8 | 0.508 | High | 0.076 | −0.92 |
| Compound 2f | 290.1 | −5.7 | inactive | Moderate | 91.721 | 3.8 | 0.708 | High | 0.051 | −1.339 |
| Compound 2g | 306.6 | −6.1 | inactive | Moderate | 90.819 | 4.4 | 0.68 | High | 0.196 | −1.313 |
| Compound 3a | 304 | −4.7 | inactive | High | 89.848 | 2.8 | 0.227 | High | 0.95 | −1.961 |
| Compound 3b | 182.2 | −3.4 | inactive | Moderate | 91.82 | 2.7 | 0.375 | Low | 0.764 | −2.017 |
| Compound 3c | 215.1 | −4.2 | inactive | High | 89.982 | 2.9 | 0.23 | High | 0.919 | −1.961 |
| Compound 3d | 243.1 | −4 | inactive | High | 90.899 | 2.7 | 0.222 | Medium | 0.868 | −1.999 |
| Compound 3e | 259.5 | −4.4 | inactive | High | 89.915 | 2.8 | 0.228 | High | 0.934 | −1.961 |
| Compound 3f | 243.1 | −4 | inactive | High | 90.872 | 2.7 | 0.222 | Medium | 0.845 | −1.995 |
| Compound 3g | 259.5 | −4.4 | inactive | High | 89.915 | 2.8 | 0.228 | High | 0.934 | −1.961 |
| Compound 4a | 289 | −4.9 | inactive | High | 91.487 | 3.3 | 0.351 | High | 0.432 | −1.66 |
| Compound 4b | 167.2 | −3.4 | inactive | Moderate | 93.459 | 3.2 | 0.437 | Medium | 0.248 | −1.715 |
| Compound 4c | 200.1 | −4.5 | inactive | High | 91.621 | 3.6 | 0.357 | High | 0.401 | −1.66 |
| Compound 4d | 228.1 | −4.1 | inactive | High | 92.538 | 3.2 | 0.382 | Medium | 0.344 | −1.697 |
| Compound 4e | 244.5 | −4.7 | inactive | High | 91.554 | 3.5 | 0.354 | High | 0.416 | −1.66 |
| Compound 4f | 228.1 | −4.1 | inactive | High | 92.511 | 3.2 | 0.382 | Medium | 0.32 | −1.693 |
| Compound 4g | 244.5 | −4.7 | inactive | High | 91.554 | 3.5 | 0.354 | High | 0.416 | −1.66 |
| Compound 5a | 305 | −4.4 | inactive | High | 89.763 | 3 | 0.284 | High | 0.253 | −1.98 |
| Compound 5b | 183.2 | −2.9 | inactive | Moderate | 91.734 | 2.6 | 0.432 | Low | 0.086 | −2.036 |
| Compound 5c | 216.1 | −3.6 | inactive | High | 89.897 | 2.7 | 0.287 | High | 0.223 | −1.98 |
| Compound 5d | 244.1 | −3.5 | inactive | High | 90.814 | 2.7 | 0.279 | Medium | 0.169 | −2.017 |
| Compound 5e | 260.5 | −3.9 | inactive | High | 89.83 | 2.9 | 0.286 | High | 0.238 | −1.98 |
| Compound 5f | 244.1 | −3.5 | inactive | High | 90.786 | 2.7 | 0.279 | Medium | 0.143 | −2.014 |
| Compound 5g | 260.5 | −3.9 | inactive | High | 89.83 | 2.9 | 0.286 | High | 0.238 | −1.98 |
| Compound 6a | 318 | −4.7 | inactive | High | 90.757 | 2.7 | 0.146 | High | 1.061 | −1.917 |
| Compound 6b | 196.2 | −3.2 | inactive | Moderate | 92.728 | 2.7 | 0.293 | Low | 0.867 | −1.973 |
| Compound 6c | 229.1 | −4.3 | inactive | High | 90.891 | 3 | 0.148 | Medium | 1.031 | −1.917 |
| Compound 6d | 257.1 | −3.9 | inactive | High | 91.78 | 2.6 | 0.14 | Medium | 0.956 | −1.951 |
| Compound 6e | 273.6 | −4.4 | inactive | High | 90.824 | 2.8 | 0.147 | Medium | 1.046 | −1.917 |
| Compound 6f | 257.1 | −3.9 | inactive | High | 91.808 | 2.6 | 0.14 | Medium | 0.978 | −1.954 |
| Compound 6g | 273.6 | −4.4 | inactive | High | 90.824 | 2.8 | 0.147 | Medium | 1.046 | −1.917 |
| Compound 7a | 332 | −4.9 | inactive | High | 92.246 | 3 | 0.191 | High | 1.141 | −1.487 |
| Compound 7b | 210.2 | −3.4 | inactive | Moderate | 94.218 | 2.9 | 0.349 | Low | 0.994 | −1.543 |
| Compound 7c | 243.1 | −4.6 | inactive | High | 92.38 | 3.3 | 0.225 | Medium | 1.11 | −1.487 |
| Compound 7d | 271.1 | −4.1 | inactive | High | 93.297 | 2.9 | 0.258 | Medium | 1.084 | −1.525 |
| Compound 7e | 287.6 | −4.7 | inactive | High | 92.313 | 3.2 | 0.208 | High | 1.125 | −1.487 |
| Compound 7f | 271.1 | −4.1 | inactive | High | 93.27 | 2.9 | 0.258 | Medium | 1.055 | −1.521 |
| Compound 7g | 287.6 | −4.7 | inactive | High | 92.313 | 3.2 | 0.208 | High | 1.125 | −1.487 |
| J | 470 | −4.4 | inactive | Moderate | 90.483 | 4.4 | 0.386 | High | 0.868 | −2.215 |
| I | 473 | −4.3 | inactive | Moderate | 91.515 | 3.9 | 0.193 | High | 1.474 | −2.024 |
| A | 534.1 | −4.3 | inactive | Low | 90.979 | 4.6 | 0.316 | Low | 1.112 | −2.449 |
| B | 535.1 | −3.9 | inactive | Moderate | 100 | 3.4 | 0.184 | High | 0.002 | −2.93 |
| Metabolism | Excretion | Toxicity | ||||
|---|---|---|---|---|---|---|
| Compound | CYP450 | OCT2 Substrate | hERG | MRTD | AMES Toxicity | Hepatotoxicity |
| Compound 1a | Yes | No | <4.0 | 0.482 | No | No |
| Compound 1b | Yes | No | <4.0 | 0.666 | No | No |
| Compound 1c | Yes | No | <4.0 | 0.503 | No | No |
| Compound 1d | Yes | No | <4.0 | 0.45 | No | No |
| Compound 1e | Yes | No | <4.0 | 0.492 | No | No |
| Compound 1f | Yes | No | <4.0 | 0.574 | No | No |
| Compound 1g | Yes | No | <4.0 | 0.492 | No | No |
| Compound 2a | Yes | No | <4.0 | 0.673 | Yes | No |
| Compound 2b | Yes | No | <4.0 | 0.608 | Yes | No |
| Compound 2c | Yes | No | <4.0 | 0.671 | Yes | No |
| Compound 2d | Yes | No | <4.0 | 0.672 | Yes | No |
| Compound 2e | Yes | No | <4.0 | 0.641 | Yes | No |
| Compound 2f | Yes | No | <4.0 | 0.585 | Yes | No |
| Compound 2g | Yes | No | <4.0 | 0.616 | Yes | No |
| Compound 3a | Yes | No | <4.0 | 0.381 | No | No |
| Compound 3b | Yes | No | <4.0 | 0.512 | No | No |
| Compound 3c | Yes | No | <4.0 | 0.402 | No | No |
| Compound 3d | Yes | No | <4.0 | 0.455 | No | No |
| Compound 3e | Yes | No | <4.0 | 0.391 | No | No |
| Compound 3f | Yes | No | <4.0 | 0.303 | No | No |
| Compound 3g | Yes | No | <4.0 | 0.391 | No | No |
| Compound 4a | Yes | No | <4.0 | 0.525 | No | No |
| Compound 4b | Yes | No | <4.0 | 0.716 | No | No |
| Compound 4c | Yes | No | <4.0 | 0.544 | No | No |
| Compound 4d | Yes | No | <4.0 | 0.625 | No | No |
| Compound 4e | Yes | No | <4.0 | 0.534 | No | No |
| Compound 4f | Yes | No | <4.0 | 0.471 | No | No |
| Compound 4g | Yes | No | <4.0 | 0.534 | No | No |
| Compound 5a | Yes | No | <4.0 | 0.55 | No | No |
| Compound 5b | Yes | No | <4.0 | 0.678 | No | No |
| Compound 5c | Yes | No | <4.0 | 0.572 | No | No |
| Compound 5d | Yes | No | <4.0 | 0.627 | No | No |
| Compound 5e | Yes | No | <4.0 | 0.561 | No | No |
| Compound 5f | Yes | No | <4.0 | 0.47 | No | No |
| Compound 5g | Yes | No | <4.0 | 0.561 | No | No |
| Compound 6a | Yes | No | <4.0 | 0.376 | No | No |
| Compound 6b | Yes | No | <4.0 | 0.502 | No | No |
| Compound 6c | Yes | No | <4.0 | 0.397 | No | No |
| Compound 6d | Yes | No | <4.0 | 0.293 | No | No |
| Compound 6e | Yes | No | <4.0 | 0.387 | No | No |
| Compound 6f | Yes | No | <4.0 | 0.441 | No | No |
| Compound 6g | Yes | No | <4.0 | 0.387 | No | No |
| Compound 7a | Yes | No | <4.0 | 0.2 | No | No |
| Compound 7b | Yes | No | <4.0 | 0.311 | No | No |
| Compound 7c | Yes | No | <4.0 | 0.219 | No | No |
| Compound 7d | Yes | No | <4.0 | 0.259 | No | No |
| Compound 7e | Yes | No | <4.0 | 0.209 | No | No |
| Compound 7f | Yes | No | <4.0 | 0.117 | No | No |
| Compound 7g | Yes | No | <4.0 | 0.209 | No | No |
| I | Yes | Yes | <4.0 | 0.029 | No | Yes |
| J | Yes | Yes | <4.0 | −0.814 | No | Yes |
| A | Yes | No | <4.0 | −0.599 | No | Yes |
| B | No | No | <4.0 | 0.405 | Yes | No |
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Tell, V.; Hilgeroth, A. Recent developments of protein kinase inhibitors as potential AD therapeutics. Front Cell Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.J.; Johnson, G.V.W. The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Devel. 2010, 13, 595–603. [Google Scholar] [PubMed]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell 2017, 16, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, H.; Kuret, J. Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 33, S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Sahara, N.; Ishiguro, K.; Kersh, J.; Mcgowan, E.; Lewis, J.; Hutton, M.; Dickson, D.W.; Yen, S.-H. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am. J. Pathol. 2003, 163, 1057–1067. [Google Scholar] [CrossRef]
- Ryoo, S.-R.; Kyeong Jeong, H.; Radnaabazar, C.; Yoo, J.-J.; Cho, H.-J.; Lee, H.-W.; Kim, I.-S.; Cheon, Y.-H.; Ahn, Y.S.; Chung, S.-H.; et al. DYRK1A-mediated hyperphosphorylation of tau a functional link between down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.S.; Mignani, S.; Vishwakarma, R.A. Why are the majority of active compounds in the CNS domain natural products? A critical analysis. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New drugs from marine organisms in Alzheimer’s disease. Mar. Drugs 2015, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.; Khodagholi, F. Natural products as promising drug candidates for the treatment of Alzheimer’s disease: Molecular mechanism aspect. Curr. Neuropharmacol. 2013, 11, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Bhattacharya, R.; Mukherjee, A.; Pandey, D.K. Natural products against Alzheimer’s disease: Pharmaco-therapeutics and biotechnological interventions. Biotechnol. Adv. 2017, 35, 178–216. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, R.A. Meridianins: Marine-derived potent kinase inhibitors. Mini-Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Meine, R.; Becker, W.; Falke, H.; Preu, L.; Loaëc, N.; Meijer, L.; Kunick, C. Indole-3-carbonitriles as DYRK1A inhibitors by fragment-based drug design. Molecules 2018, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Giraud, F.; Alves, G.; Debiton, E.; Nauton, L.; Thery, V.; Durieu, E.; Ferandin, Y.; Lozach, O.; Meijer, L.; Anizon, F.; et al. Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J. Med. Chem 2011, 54, 4474–4489. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.; Waiker, D.; Jain, A.; Trivedi, P. Human CDC2-like kinase 1 (CLK1): A novel target for Alzheimer’s disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Halekotte, J.; Witt, L.; Ianes, C.; Krüger, M.; Bührmann, M.; Rauh, D.; Pichlo, C.; Brunstein, E.; Luxenburger, A.; Baumann, U.; et al. Optimized 4,5-diarylimidazoles as potent/selective inhibitors of protein kinase CK1δ and their structural relation to p38α MAPK. Molecules 2017, 22, 522. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Gianoncelli, A.; Montopoli, M.; Caparrotta, L.; Venerando, A.; Meggio, F.; Pinna, L.A.; Zagotto, G.; Moro, S. Identification of novel protein kinase CK1 delta (CK1δ) inhibitors through structure-based virtual screening. Bioorg. Med. Chem. Lett. 2008, 18, 5672–5675. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.R.; Sharma, S.; Joshi, P.; Wani, A.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. Meridianin derivatives as potent Dyrk1A inhibitors and neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 2948–2952. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Geisselbrecht, Y.; Blanck, S.; Wilbuer, A.; Atilla-Gokcumen, G.E.; Filippakopoulos, P.; Kräling, K.; Celik, M.A.; Harms, K.; Maksimoska, J.; et al. Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors. J. Am. Chem. Soc. 2011, 133, 5976–5986. [Google Scholar] [CrossRef] [PubMed]
- Mente, S.; Arnold, E.; Butler, T.; Chakrapani, S.; Chandrasekaran, R.; Cherry, K.; Dirico, K.; Doran, A.; Fisher, K.; Galatsis, P.; et al. Ligand-protein interactions of selective casein kinase 1 delta inhibitors. J. Med. Chem. 2013. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-aided drug design applied to marine drug discovery: Meridianins as Alzheimer’s disease therapeutic agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Dashti, Y.; Vial, M.-L.; Wood, S.A.; Mellick, G.D.; Roullier, C.; Quinn, R.J. Kororamide B, a brominated alkaloid from the bryozoan Amathia tortuosa and its effects on Parkinson’s disease cells. Tetrahedron 2015, 71, 7879–7884. [Google Scholar] [CrossRef]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, P.M.; Cintra, L.S.; Braguine, C.G.; da Silva Filho, A.A.; Silva, M.L.A.E.; Cunha, W.R.; Januário, A.H. Halogenated indole alkaloids from marine invertebrates. Mar. Drugs 2010, 8, 1526–1549. [Google Scholar] [CrossRef] [PubMed]
- Munro, M.H.G.; Blunt, J.W. MarinLit: A database of the marine natural products literature. Available online: http://pubs.rsc.org/marinlit/ (accessed on 16 October 2018).
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aid. Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- McGregor, M.J. A pharmacophore map of small molecule protein kinase inhibitors. J. Chem. Inf. Model. 2007, 47, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhou, T.; Lafleur, K.; Nevado, C.; Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: A network analysis. Bioinformatics 2010, 26, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors as potential therapeutics. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Klein-Junior, L.; Santos Passos, C.; Moraes, A.; Wakui, V.; Konrath, E.; Nurisso, A.; Carrupt, P.-A.; Alves de Oliveira, C.; Kato, L.; Henriques, A. Indole alkaloids and semisynthetic indole derivatives as multifunctional scaffolds aiming the inhibition of enzymes related to neurodegenerative diseases—A focus on psychotria L. Genus. Curr. Top. Med. Chem. 2014, 14, 1056–1075. [Google Scholar] [CrossRef]
- Ren, P.; Liu, Y.; Li, L.; Chan, K.; Wilson, T.E. Heterocyclic Kinase Inhibitors. US Patent Application No. US20130095100A1, 18 April 2013. U.S. Patent Application No. US12331431, 9 December 2008. [Google Scholar]
- Almstetter, M.T.; Treml, A.; Koestler, R.; Yehia, N. Heterocyclic Compounds as Kinase Inhibitors. EP2699572B1, 10 August 2016. [Google Scholar]
- Castro, A.C.; Chan, K.; Evans, C.A.; Janardanannair, S.; Lescarbeau, A.; Li, L.; Liu, T.; Liu, Y.; Ren, P.; Snyder, D.A.; et al. Heterocyclic Compounds and Uses Thereof. WO2013154878A1, 17 October 2013. [Google Scholar]
- An, W.F.; Germain, A.R.; Bishop, J.A.; Nag, P.P.; Metkar, S.; Ketterman, J.; Walk, M.; Weiwer, M.; Liu, X.; Patnaik, D.; et al. Discovery of potent and highly selective inhibitors of GSK3b. In Probe Reports from the NIH Molecular Libraries Program; NCBI: Bethesda, MD, USA, 2010. [Google Scholar]
- Benek, O.; Hroch, L.; Aitken, L.; Gunn-Moore, F.; Vinklarova, L.; Kuca, K.; Perez, D.I.; Perez, C.; Martinez, A.; Fisar, Z.; et al. 1-(Benzo[d]thiazol-2-yl)-3-phenylureas as dual inhibitors of casein kinase 1 and ABAD enzymes for treatment of neurodegenerative disorders. J. Enzyme Inhib. Med. Chem. 2018, 33, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Coombs, T.C.; Tanega, C.; Shen, M.; Wang, J.L.; Auld, D.S.; Gerritz, S.W.; Schoenen, F.J.; Thomas, C.J.; Aubé, J. Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: Development of chemical probe ML315. Bioorg. Med. Chem. Lett. 2013, 23, 3654–3661. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W. Natural halogenated organics. Elsevier Oceanogr. Ser. 1981, 31, 375–393. [Google Scholar] [CrossRef]
- Hernandes, M.; Cavalcanti, S.M.; Moreira, D.R.; de Azevedo Junior, W.; Leite, A.C. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein–ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Chemistry. Halogen versus hydrogen. Science 2008, 321, 918–919. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-molecule inhibitors of GSK-3: Structural insights and Their application to Alzheimer’s disease models. Int. J. Alzheimers Dis. 2012, 2012, 381029. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Bodles-Brakhop, A.M.; Barger, S.W. A role for P-glycoprotein in clearance of Alzheimer amyloid β-peptide from the brain HHS public access. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-deposition in an Alzheimer disease mouse model. J. Clin. Invest. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Bibi, Z. Role of cytochrome P450 in drug interactions. Nutr. Metab. 2008, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front. Chem. 2018, 6, 1–12. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide protein data bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, 2006–2008. [Google Scholar] [CrossRef] [PubMed]
- Asdssss Felix, E.; Santamaría-Navarro, E.; Sanchez-Martinez, M.; Nonell-Canals, A. Itzamna. Available online: https://www.mindthebyte.com/ (accessed on 15 October 2018).
- Yu, W.; Mackerell, A.D. Computer-aided drug design methods. Methods Mol. Biol. 2017, 1520, 93–94. [Google Scholar] [CrossRef]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry 2011, 50, 6157–6169. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- de Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. The role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Martín-García, F.; Papaleo, E.; Gomez-Puertas, P.; Boomsma, W.; Lindorff-Larsen, K. Comparing molecular dynamics force fields in the essential subspace. PLoS ONE 2015, 10, e0121114. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS ONE 2012, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber, an accessory software package for molecular mechanical calculations. J. Chem. Inf. Comp. Sci. 2005, 25, 25–1174. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Jenson, C. Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: Seeking temperatures of maximum density. J. Comput. Chem. 1998, 19, 1179–1186. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 0441, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Degliesposti, G.; Del Rio, A.; Sgobba, M. Binding estimation after refinement, a new automated procedure for the refinement and rescoring of docked ligands in virtual screening. Chem. Biol. Drug Des. 2009, 73, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.L.; Binh, K.M. Steered molecular dynamics-A promising tool for drug design. Curr. Bioinform. 2012, 7, 342–351. [Google Scholar] [CrossRef]
- Patel, J.S.; Berteotti, A.; Ronsisvalle, S.; Rocchia, W.; Cavalli, A. Steered molecular dynamics simulations for studying protein–Ligand interaction in cyclin-dependent kinase 5. J. Chem. Inf. Model. 2014, 54, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered molecular dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas; Deuflhard, P., Hermans, J., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 39–65. [Google Scholar]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- The UniProt consortium update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [CrossRef]
- Vidal, D.; Nonell-Canals, A. ADMETer. Available online: https://www.mindthebyte.com/ (accessed on 15 October 2018).
- Poongavanam, V.; Haider, N.; Ecker, G.F. Fingerprint-based in silico models for the prediction of P-glycoprotein substrates and inhibitors. Bioorg. Med. Chem. 2012, 20, 5388–5395. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, A.; Fourches, D.; Duan, J.; Hucke, O.; Garneau, M.; Zhu, H.; Bonneau, P.; Tropsha, A. Human intestinal transporter database: QSAR modeling and virtual profiling of drug uptake, efflux and interactions. Pharm. Res. 2013, 30, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-source cheminformatics. Available online: http://rdkit.org/ (accessed on 15 October 2018).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Ostblom, J.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; et al. Mwaskom/Seaborn: V0.9.0 (July 2018). Zenodo. Available online: http://doi.org/10.5281/zenodo.1313201 (accessed on 15 October 2018).
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, P.; D’Auria, M.; Muller, C.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring marine resources for bioactive compounds. Planta Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Valentão, P.; Ferreres, F.; Andrade, P.B. Review: Bioactive marine drugs and marine biomaterials for brain diseases. Mar. Drugs 2014, 12, 2539–2589. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]









| Glycine-Rich Region | Hydrophobic Pocket | Adenine Region | Sugar Pocket | Phosphate Binding Pocket | |
|---|---|---|---|---|---|
| GSK3β | GNGSFG 63-68 | VAIK 82-85 | LDYV 132-135 | PQNLL 184-188 | LKLCD 196-200 |
| CK1δ | GSGSFG 16-21 | VAIK 35-38 | MELL 82-85 | PDNFL 131-135 | VYIID 145-149 |
| DYRK1A | GKGSFG 166-171 | VAIK 184-187 | FEML 238-241 | PENIL 290-294 | IKIVD 303-307 |
| CLK1 | GEGAFG 168-173 | VAVK 188-191 | FELL 241-244 | PENIL 291-295 | IKVVD 312-325 |
| GSK3β | CK1δ | DYRK1A | CLK1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | Binding Energy (kcal/mol) | ||||
| R0/R1 | R0/R1 | R0/R1 | R0/R1 | ||||||||
| F | −7.9/−7.9 | −35.18 | F | −7.2/−7.3 | −38.55 | F | −8.0/−7.8 | −39.99 | F | −8.7/−8.7 | −37.71 |
| −7.7/−7.9 | −34.73 | −7.1/−7.1 | −38.93 | −7.8/−7.7 | −39.91 | −8.5/−8.5 | −37.61 | ||||
| I | −5.6/−5.6 | −23.08 | I | −5.0/−5.0 | −3.19 | I | −5.6/−5.6 | −26.52 | I | −5.8/−5.5 | −33.23 |
| −6.3/−6.3 | −18.38 | −5.4/−5.4 | −11.26 | −4.8/−4.8 | −11.02 | −5.8/−5.8 | −31.93 | ||||
| J | −6.7/−6.7 | −31.58 | J | −6.2/−6.2 | −37.76 | J | −7.4/−7.4 | −31.35 | J | −6.0/−6.0 | −21.47 |
| −5.9/−5.9 | −31.61 | −5.8/−5.8 | −28.91 | −7.0/−7.0 | −32.27 | −4.6/−4.6 | −24.37 | ||||
| A | −8.3/−8.3 | −34.88 | A | −8.0/−8.0 | −35.48 | A | −8.2/−8.2 | −32.94 | A | −6.7/−6.7 | −37.46 |
| −8.1/−8.1 | −31.02 | −7.4/−7.4 | −33.94 | −6.7/−6.7 | −14.61 | −2.9/−2.9 | −38.93 | ||||
| B | −9.1/−9.1 | −31.80 | B | −8.1/−8.1 | −28.68 | B | −7.7/−7.7 | −23.83 | B | −4.4/−4.4 | −28.71 |
| −8.3/−8.3 | −32.34 | −6.6/−6.6 | −35.53 | −7.3/−7.3 | −24.29 | −4.0/−4.0 | −22.96 | ||||
 |  | ||||||||
| GSK3β | CK1δ | DYRK1A | CLK1 | GSK3β | CK1δ | DYRK1A | CLK1 | ||
| Binding Energy | Binding Energy | Binding Energy | Binding Energy | Binding Energy | Binding Energy | Binding Energy | Binding Energy | ||
| R0/R1 | R0/R1 | R0/R1 | R0/R1 | R0/R1 | R0/R1 | R0/R1 | R0/R1 | ||
| L17640 | −6.4/−6.4 | −7.3/−7.3 | −7/−7 | −6/−6 | L4950 | −9.1/−9.1 | −9.1/−9.1 | −9.1/−9.1 | −9.1/−9.1 |
| L1189 | −6.8/−6.8 | −7.6/−7.6 | −7.2/−7.2 | −5.9/−5.9 | L4949 | −8.7/−8.7 | −8.7/−8.7 | −8.7/−8.7 | −8.7/−8.7 |
| L34 | −7.2/−7.2 | −8.1/−8.1 | −8.2/−8.2 | −6.9/−6.9 | L4951 | −9/−9 | −9/−9 | −9/−9 | −9/−9 |
| L4080 | −6.1/−6.1 | −6.9/−6.9 | −6.8/−6.8 | −6/−6 | |||||
| L28238 | −6.5/−6.5 | −7.8/−7.8 | −7.3/−7.3 | −5.8/−5.8 | |||||
| L7472 | −6.3/−6.3 | −7.1/−7.1 | −6.8/−6.8 | −6.2/−6.2 | |||||
| L10723 | −6.1/−6.1 | −6.7/−6.7 | −6.7/−6.7 | −5.2/−5.2 |  | ||||
| L17639 | −6.4/−6.4 | −7.1/−7.1 | −6.8/−6.8 | −5.6/−5.6 | |||||
| L1192 | −7.1/−7.1 | −7.6/−7.6 | −7.6/−7.6 | −5.9/−5.9 | |||||
| L17641 | −6.8/−6.8 | −7/−7 | −7/−7 | −5.7/−5.7 | |||||
| L11375 | −6.2/−6.2 | −6.7/−6.7 | −6.6/−6.6 | −5.4/−5.4 | |||||
| L35 | −7.3/−7.3 | −8/−8 | −8.1/−8.1 | −7.1/−7.1 | GSK3β | CK1δ | DYRK1A | CLK1 | |
| L28804 | −7.1/−7.1 | −7.6/−7.6 | −7.2/−7.2 | −5.8/−5.8 | Binding Energy | Binding Energy | Binding Energy | Binding Energy | |
| L4081 | −6.4/−6.4 | −7/−7 | −6.8/−6.8 | −6.4/−6.4 | |||||
| L29233 | −8.5/−8.5 | −8.5/−8.5 | −9.3/−9.3 | −8.2/−8.2 | R0/R1 | R0/R1 | R0/R1 | R0/R1 | |
| L24201 | −10.6/−10.6 | −8.8/−8.8 | −10.4/−10.4 | −9.3/−9.3 | L9830 | −7.4/−7.4 | −7.4/−7.4 | −7.4/−7.4 | −7.4/−7.4 |
| L25368 | −9.7/−9.7 | −8.8/−8.8 | −10.3/−10.3 | −8.9/−8.9 | L9831 | −7.8/−7.8 | −7.8/−7.8 | −7.8/−7.8 | −7.8/−7.8 |
| L7473 | −6.9/−6.9 | −7.3/−7.3 | −7.1/−7.1 | −6/−6 | L2330 | −8.7/−8.7 | −8.7/−8.7 | −8.7/−8.7 | −8.7/−8.7 |

| Compounds | R1 | R2 |
|---|---|---|
| a | Br | Br |
| b | F | F |
| c | Cl | Cl |
| d | Br | F |
| e | Br | Cl |
| f | F | Br |
| g | Cl | Br |
| GSK3β | CK1δ | DYRK1A | CLK1 | |||||
|---|---|---|---|---|---|---|---|---|
| Binding Energy | Binding Energy | Binding Energy | Binding Energy | |||||
| Compound 1 | a | −30.3141 | a | −35.4499 | e | −32.8862 | g | −30.3541 |
| Compound 2 | a | −31.2458 | e | −37.8982 | a | −37.8422 | a | −34.1041 |
| Compound 3 | a | −13.8779 | g | −28.7631 | a | −15.2733 | f | −20.4786 |
| Compound 4 | a | −27.6481 | e | −28.6573 | a | −30.7518 | e | −28.3695 |
| Compound 5 | a | −27.6534 | e | −28.5831 | a | −31.2535 | c | −29.4190 |
| Compound 6 | a | −18.5779 | a | −26.4630 | a | −18.9387 | g | −30.7737 |
| Compound 7 | a | −18.8955 | a | −18.4901 | a | −20.8203 | g | −25.4765 |
| Absorption | Distribution | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mol Weight | LogS | P-Glycoprotein | Caco-2 Permeability | Intestinal Absorption | LogP | BBB | PPB | VDss | CNS Permeability |
| Compound 2a | 351 | −6.1 | inactive | Moderate | 90.067 | 4.1 | 0.477 | High | 0.234 | −0.894 |
| Compound 2e | 290.1 | −5.7 | inactive | Moderate | 91.036 | 3.8 | 0.508 | High | 0.076 | −0.92 |
| Metabolism | Excretion | Toxicity | ||||
|---|---|---|---|---|---|---|
| Compound | CYP450 | OCT2 Substrate | hERG | MRTD | AMES Toxicity | Hepatotoxicity |
| Compound 2a | Yes | No | <4.0 | 0.673 | Yes | No |
| Compound 2e | Yes | No | <4.0 | 0.641 | Yes | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Mar. Drugs 2018, 16, 386. https://doi.org/10.3390/md16100386
Llorach-Pares L, Nonell-Canals A, Avila C, Sanchez-Martinez M. Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Marine Drugs. 2018; 16(10):386. https://doi.org/10.3390/md16100386
Chicago/Turabian StyleLlorach-Pares, Laura, Alfons Nonell-Canals, Conxita Avila, and Melchor Sanchez-Martinez. 2018. "Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study" Marine Drugs 16, no. 10: 386. https://doi.org/10.3390/md16100386
APA StyleLlorach-Pares, L., Nonell-Canals, A., Avila, C., & Sanchez-Martinez, M. (2018). Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Marine Drugs, 16(10), 386. https://doi.org/10.3390/md16100386