A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. 2D Ligand-Based Filter
2.2. 3D Ligand-Based Filter
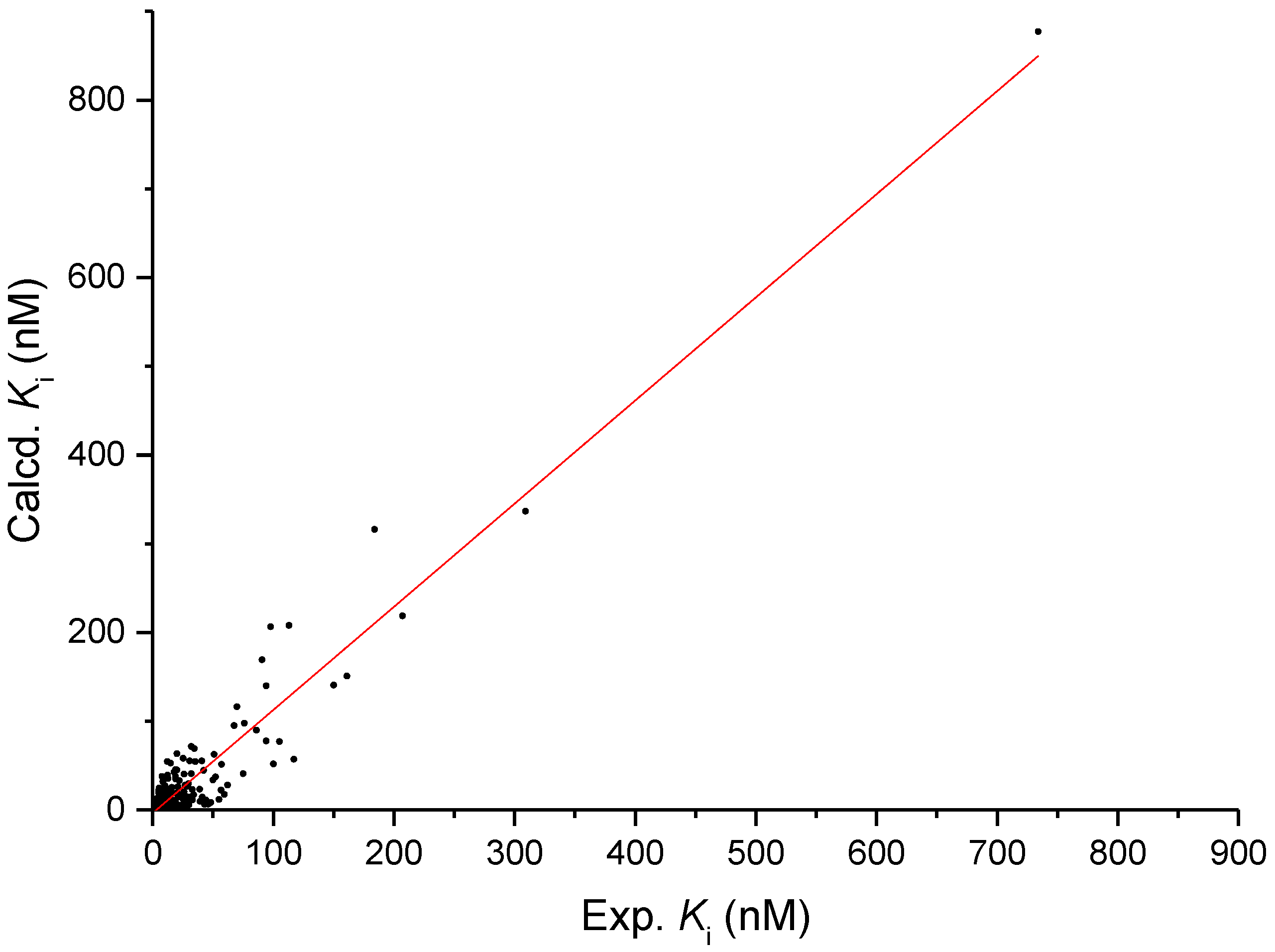
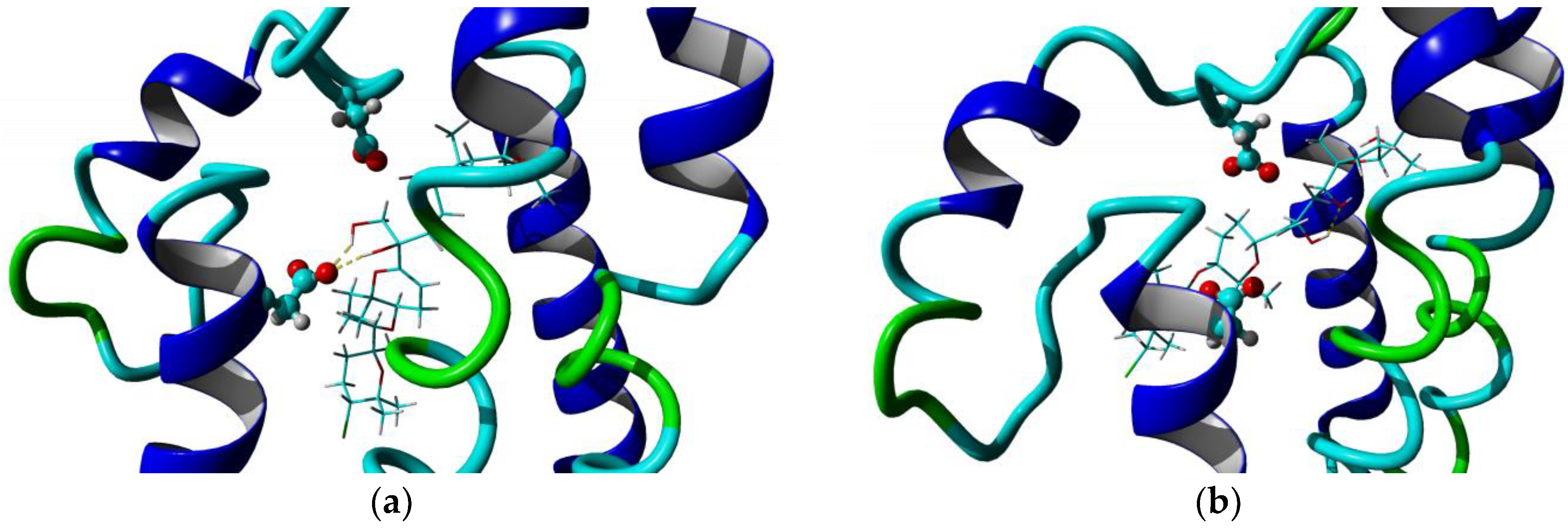
2.3. Homology Model and Molecular Docking
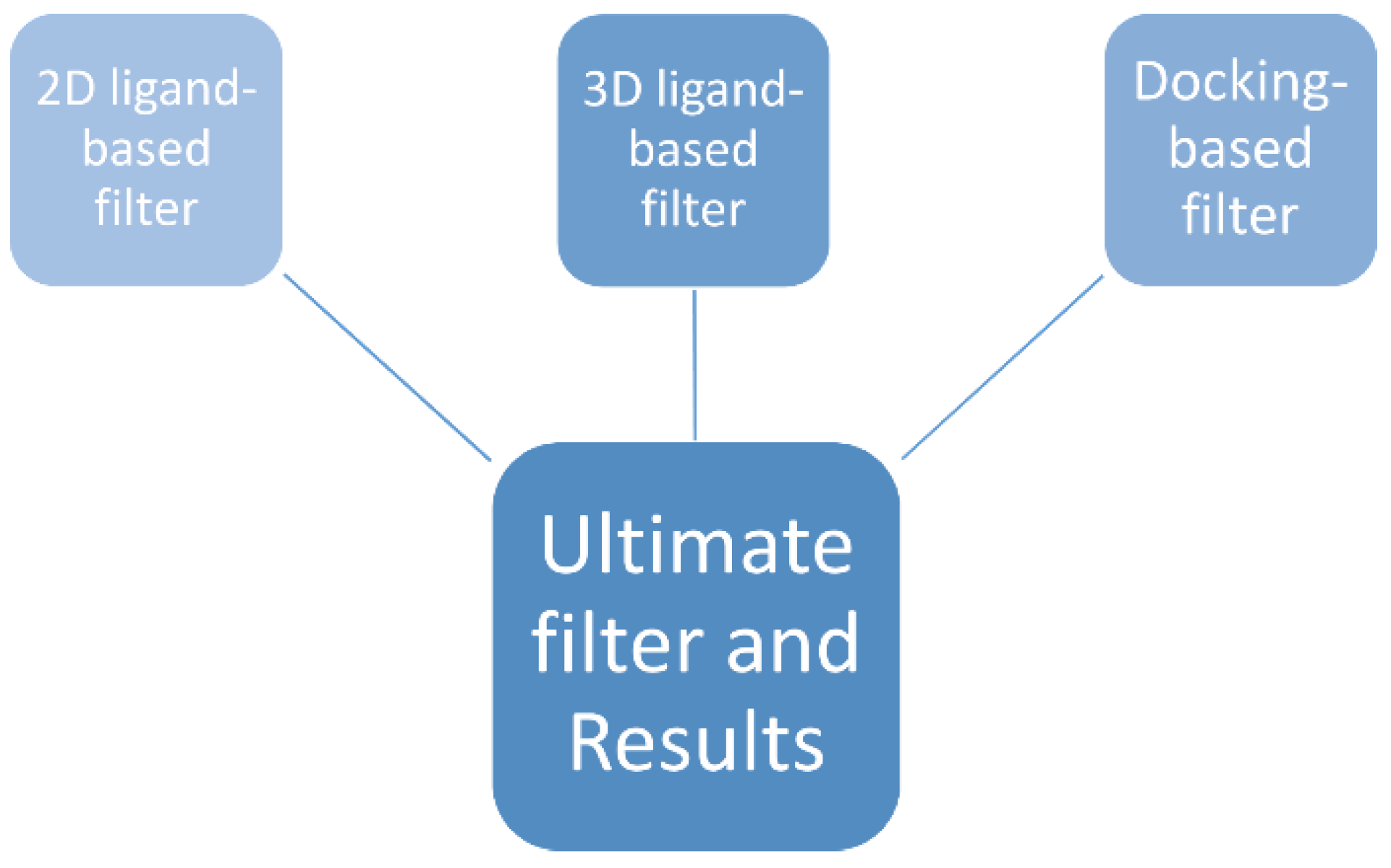
2.4. The Ultimate Filter
3. Materials and Methods
3.1. Dataset of Marine Compounds
3.2. Structures 2D to 3D Building and Minimization
3.3. 2D-QSAR
3.4. Compound Alignment for the 3D-Ligand Based Filter
3.5. Homology Model and Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Hellewell, S.B.; Bruce, A.; Feinstein, G.; Orringer, J.; Williams, W.; Bowen, W.D. Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: Characterization by ligand binding and photoaffinity labeling. Eur. J. Pharmacol. 1994, 268, 9–18. [Google Scholar] [CrossRef]
- Su, T.P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Sigma-1 receptor chaperone and brain-derived neurotrophic factor: Emerging links between cardiovascular disease and depression. Prog. Neurobiol. 2013, 100, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T. Improving Alzheimer’s Disease-Related Cognitive Deficits with sigma1 Receptor Agonists. Drug News Perspect. 2002, 15, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Mesangeau, C.; Amata, E.; Alsharif, W.; Seminerio, M.J.; Robson, M.J.; Matsumoto, R.R.; Poupaert, J.H.; McCurdy, C.R. Synthesis and pharmacological evaluation of indole-based sigma receptor ligands. Eur. J. Med. Chem. 2011, 46, 5154–5161. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Amata, E.; Vinciguerra, S.; Fiorito, J.; Giurdanella, G.; Drago, F.; Caporarello, N.; Prezzavento, O.; Arena, E.; Salerno, L.; et al. Antiangiogenic Effect of (+/−)-Haloperidol Metabolite II Valproate Ester [(+/−)-MRJF22] in Human Microvascular Retinal Endothelial Cells. J. Med. Chem. 2016, 59, 9960–9966. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Sanchez-Fernandez, C.; Turnaturi, R.; Parenti, C.; Marrazzo, A.; Catalano, R.; Amata, E.; Pasquinucci, L.; Cobos, E.J. (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/sigma1 antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017, 125, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Dichiara, M.; Arena, E.; Pittalà, V.; Pistarà, V.; Cardile, V.; Graziano, A.C.E.; Fraix, A.; Marrazzo, A.; Sortino, S.; et al. Novel Sigma Receptor Ligand-Nitric Oxide Photodonors: Molecular Hybrids for Double-Targeted Antiproliferative Effect. J. Med. Chem. 2017, 60, 9531–9544. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Rescifina, A.; Prezzavento, O.; Arena, E.; Dichiara, M.; Pittalà, V.; Montilla-García, Á.; Punzo, F.; Merino, P.; Cobos, E.J.; et al. (+)-Methyl (1R,2S)-2-{[4-(4-Chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}-1-phenylcyclopropanecarboxylate [(+)-MR200] Derivatives as Potent and Selective Sigma Receptor Ligands: Stereochemistry and Pharmacological Properties. J. Med. Chem. 2018, 61, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Nakane, T.; Koike, I.; Yamamoto, T.; Sugiura, Y.; Harada, E.; Sugase, K.; Shimamura, T.; Ohmura, M.; Muraoka, K.; et al. Haem-dependent dimerization of PGRMC1/Sigma-2 receptor facilitates cancer proliferation and chemoresistance. Nat. Commun. 2016, 7, 11030. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Mavlyutov, T.A.; Chu, M.L.; Yang, H.; Schulman, A.; Mesangeau, C.; McCurdy, C.R.; Guo, L.W.; Ruoho, A.E. The sigma-2 receptor and progesterone receptor membrane component 1 are different binding sites derived from independent genes. Ebiomedicine 2015, 2, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.R.; Bown, W.D.; Su, T.P. Sigma Receptors: Chemistry, Cell Biology and Clinical Implications; Springer: Berlin, Germany, 2007. [Google Scholar]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce, J.T.; et al. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Mousseau, D.D.; Dai, Y.; Cao, X.; Li, X.M. Haloperidol induces apoptosis via the sigma2 receptor system and Bcl-XS. Pharmacogenom. J. 2006, 6, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, M.S.; Fehrenbacher, N.; Hoyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jaattela, M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Rothfuss, J.; Zhang, J.; Chu, W.; Vangveravong, S.; Tu, Z.; Pan, F.; Chang, K.C.; Hotchkiss, R.; Mach, R.H. Sigma-2 ligands induce tumour cell death by multiple signalling pathways. Br. J. Cancer 2012, 106, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Jonhede, S.; Petersen, A.; Zetterberg, M.; Karlsson, J.O. Acute effects of the sigma-2 receptor agonist siramesine on lysosomal and extra-lysosomal proteolytic systems in lens epithelial cells. Mol. Vis. 2010, 16, 819–827. [Google Scholar] [PubMed]
- Dehdashti, F.; Laforest, R.; Gao, F.; Shoghi, K.I.; Aft, R.L.; Nussenbaum, B.; Kreisel, F.H.; Bartlett, N.L.; Cashen, A.; Wagner-Johnston, N.; et al. Assessment of cellular proliferation in tumors by PET using 18F-ISO-1. J. Nucl. Med. 2013, 54, 350–357. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. In [18F]ISO-1 PET/CT in Breast Cancer; 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02284919 (accessed on 11 October 2018).
- ClinicalTrials.gov. In Imaging of In Vivo Sigma-2 Receptor Expression with 18F-ISO-1 Positron Emission Tomography in Metastatic Breast Cancer; 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT03057743 (accessed on 11 October 2018).
- Mach, R.H.; Zeng, C.; Hawkins, W.G. The sigma2 receptor: A novel protein for the imaging and treatment of cancer. J. Med. Chem. 2013, 56, 7137–7160. [Google Scholar] [CrossRef] [PubMed]
- Schininà, B.; Martorana, A.; Colabufo, N.A.; Contino, M.; Niso, M.; Perrone, M.G.; De Guidi, G.; Catalfo, A.; Rappazzo, G.; Zuccarello, E.; et al. 4-Nitro-2,1,3-benzoxadiazole derivatives as potential fluorescent sigma receptor probes. RSC Adv. 2015, 5, 47108–47116. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Sigma-2 receptor ligands QSAR model dataset. Data Brief 2017, 13, 514–535. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistarà, V.; Pittalà, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, G.; Miceli, C.; Pittala, V.; Modica, M.N.; Prezzavento, O.; Romeo, G.; Rescifina, A.; Marrazzo, A.; Amata, E. S2RSLDB: A comprehensive manually curated, internet-accessible database of the sigma-2 receptor selective ligands. J. Cheminform. 2017, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittalà, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Salerno, L.; Romeo, G.; Prezzavento, O.; Pittala, V.; Rescifina, A. Identification of Potentially Potent Heme Oxygenase1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem 2018, 13, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Pistara, V.; Amata, E.; Dichiara, M.; Damigella, A.; Marrazzo, A.; Prezzavento, O.; Punzo, F.; Rescifina, A. Molecular modeling studies of pseudouridine isoxazolidinyl nucleoside analogues as potential inhibitors of the pseudouridine 5’-monophosphate glycosidase. Chem. Biol. Drug Des. 2018, 91, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Pittala, V.; Sorrenti, V.; Romeo, G.; Salerno, L.; Rescifina, A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018, 81, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.D.; Vasanthi, A.H. Seaweed metabolite database (SWMD): A database of natural compounds from marine algae. Bioinformation 2011, 5, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcantara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.; Gaudencio, S.P. QSAR-assisted virtual screening of lead-like molecules from marine and microbial natural sources for antitumor and antibiotic drug discovery. Molecules 2015, 20, 4848–4873. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittala, V. Heme Oxygenase Database (HemeOxDB) and QSAR analysis of isoform 1 inhibitors. ChemMedChem 2017. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittala, V. Comprehensive data on a 2D-QSAR model for Heme Oxygenase isoform 1 inhibitors. Data Brief 2017, 15, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Toropova, M.A.; Toropov, A.A.; Raska, I., Jr.; Raskova, M. Searching therapeutic agents for treatment of Alzheimer disease using the Monte Carlo method. Comput. Biol. Med. 2015, 64, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Toropova, A.P.; Toropov, A.A.; Martyanov, S.E.; Benfenati, E.; Gini, G.; Leszczynska, D.; Leszczynski, J. CORAL: Monte Carlo Method as a Tool for the Prediction of the Bioconcentration Factor of Industrial Pollutants. Mol. Inform. 2013, 32, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology Modeling a Fast Tool for Drug Discovery: Current Perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Scharfe, C.P.I.; Rodrigues, J.P.G.L.M.; Green, A.G.; Kohlbacher, O.; Sander, C.; Bonvin, A.M.J.J.; Marks, D.S. Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife 2014, 3, e03430. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the gene that codes for the σ2 receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.H.; Huang, S.Y.; Wang, G.H.; Duh, C.Y. Study on cytotoxic oxygenated desmosterols isolated from the red alga Galaxaura marginata. J. Nat. Prod. 1997, 60, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Liu, G.; Qiu, L.; Lin, X.; Liu, M. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, represses angiogenesis in HUVEC cells and in zebrafish embryos via inhibiting the VEGF signal systems. Biomed. Pharmacother. 2015, 75, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, W.; Wei, J.; Qiu, L.; Lin, X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol. Lett. 2012, 211, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Y.; Li, J.; Guo, S.J.; Su, H.; Fan, X. The antitumor effect of bromophenol derivatives in vitro and Leathesia nana extract in vivo. Chin. J. Oceanol. Limnol. 2009, 27, 277–282. [Google Scholar] [CrossRef]
- Fernandez, J.J.; Souto, M.L.; Norte, M. Evaluation of the cytotoxic activity of polyethers isolated from Laurencia. Bioorg. Med. Chem. 1998, 6, 2237–2243. [Google Scholar] [CrossRef]
- Norte, M.; Fernandez, J.J.; Souto, M.L.; GarciaGravalos, M.D. Two new antitumoral polyether squalene derivatives. Tetrahedron Lett. 1996, 37, 2671–2674. [Google Scholar] [CrossRef]
- Pacheco, F.C.; Villa-Pulgarin, J.A.; Mollinedo, F.; Martin, M.N.; Fernandez, J.J.; Daranas, A.H. New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation. Mar. Drugs 2011, 9, 2220–2235. [Google Scholar] [CrossRef] [PubMed]
- Pati, M.L.; Niso, M.; Ferorelli, S.; Abate, C.; Berardi, F. Novel metal chelators thiosemicarbazones with activity at the sigma(2) receptors and P-glycoprotein: An innovative strategy for resistant tumor treatment. RSC Adv. 2015, 5, 103131–103146. [Google Scholar] [CrossRef]
- Niso, M.; Abate, C.; Contino, M.; Ferorelli, S.; Azzariti, A.; Perrone, R.; Colabufo, N.A.; Berardi, F. Sigma-2 Receptor Agonists as Possible Antitumor Agents in Resistant Tumors: Hints for Collateral Sensitivity. ChemMedChem 2013, 8, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Azzariti, A.; Colabufo, N.A.; Berardi, F.; Porcelli, L.; Niso, M.; Simone, G.M.; Perrone, R.; Paradiso, A. Cyclohexylpiperazine derivative PB28, a sigma(2) agonist and sigma(1) antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol Cancer Ther 2006, 5, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Al-Nabulsi, I.; Mach, R.H.; Wang, L.M.; Wallen, C.A.; Keng, P.C.; Sten, K.; Childers, S.R.; Wheeler, K.T. Effect of ploidy, recruitment, environmental factors, and tamoxifen treatment on the expression of sigma-2 receptors in proliferating and quiescent tumour cells. Br. J. Cancer 1999, 81, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, M.; Fontanilla, D.; Mavlyutov, T.; Ruoho, A.E.; Jackson, M.B. Antagonist action of progesterone at sigma-receptors in the modulation of voltage-gated sodium channels. Am. J. Physiol.-Cell Physiol. 2011, 300, C328–C337. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Lehmann, F.; Hammer, K.; Haile, S.; Axen, E.; Medina, C.; Uppenberg, J.; Svensson, S.; Rondahl, L.; Lundback, T. N-Benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Qiao, F.; Luo, L.; Peng, H.; Luo, S.; Huang, W.; Cui, J.; Li, X.; Kong, L.; Jiang, D.; Chitwood, D.J.; et al. Characterization of Three Novel Fatty Acid- and Retinoid-Binding Protein Genes (Ha-far-1, Ha-far-2 and Hf-far-1) from the Cereal Cyst Nematodes Heterodera avenae and H. filipjevi. PLoS ONE 2016, 11, e0160003. [Google Scholar] [CrossRef] [PubMed]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| BDB ID | Structure | Predicted pKi |
|---|---|---|
| 621 |  | 10.9 |
| 1612 |  | 9 |
| 848 |  | 8.6 |
| 670 |  | 8.3 |
| 1703 |  | 8.3 |
| 1144 |  | 8.3 |
| 1342 |  | 8.3 |
| 55 |  | 8.2 |
| 972 |  | 8.2 |
| 933 |  | 8.1 |
| 1426 |  | 8.1 |
| 120 |  | 8.1 |
| 414 |  | 8.1 |
| 1453 |  | 8 |
| 84 |  | 8 |
| BDB ID | Structure | Calcd. pKi |
|---|---|---|
| 1169 |  | 10.26 |
| 1421 |  | 9.79 |
| 984 |  | 9.77 |
| 28 |  | 9.64 |
| 306 |  | 9.60 |
| 1333 |  | 9.45 |
| 45 |  | 9.42 |
| 524 |  | 9.31 |
| 1172 |  | 9.31 |
| 1123 |  | 9.23 |
| 279 |  | 9.18 |
| 1581 |  | 9.14 |
| 798 |  | 9.05 |
| 84 |  | 9.03 |
| 529 |  | 8.95 |
| BDB ID | Structure | Mean pKi | Mean Ki |
|---|---|---|---|
| 1169 |  | 9.24 | 0.6 |
| 28 |  | 8.91 | 1.2 |
| 45 |  | 8.89 | 1.3 |
| 1172 |  | 8.89 | 1.3 |
| 1421 |  | 8.70 | 2.0 |
| 246 |  | 8.60 | 2.5 |
| 14 |  | 8.57 | 2.7 |
| 298 |  | 8.55 | 2.8 |
| 798 |  | 8.54 | 2.9 |
| 984 |  | 8.44 | 3.6 |
| 1179 |  | 8.44 | 3.6 |
| 848 |  | 8.43 | 3.7 |
| 1333 |  | 8.40 | 4.0 |
| 420 |  | 8.33 | 4.6 |
| 272 |  | 8.27 | 5.3 |
| BDB ID | A-549 | HT-29 |
|---|---|---|
| 848 | 1.00 | 0.63 |
| 984 | 2.5 | 2.5 |
| 1169 | 10 | 10 |
| 1172 | 2.5 | 2.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. https://doi.org/10.3390/md16100384
Floresta G, Amata E, Barbaraci C, Gentile D, Turnaturi R, Marrazzo A, Rescifina A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Marine Drugs. 2018; 16(10):384. https://doi.org/10.3390/md16100384
Chicago/Turabian StyleFloresta, Giuseppe, Emanuele Amata, Carla Barbaraci, Davide Gentile, Rita Turnaturi, Agostino Marrazzo, and Antonio Rescifina. 2018. "A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands" Marine Drugs 16, no. 10: 384. https://doi.org/10.3390/md16100384
APA StyleFloresta, G., Amata, E., Barbaraci, C., Gentile, D., Turnaturi, R., Marrazzo, A., & Rescifina, A. (2018). A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Marine Drugs, 16(10), 384. https://doi.org/10.3390/md16100384