A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism

 ,
, 
 and
and
Abstract
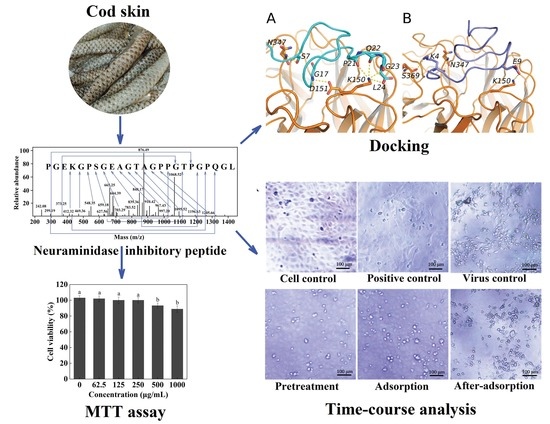
1. Introduction
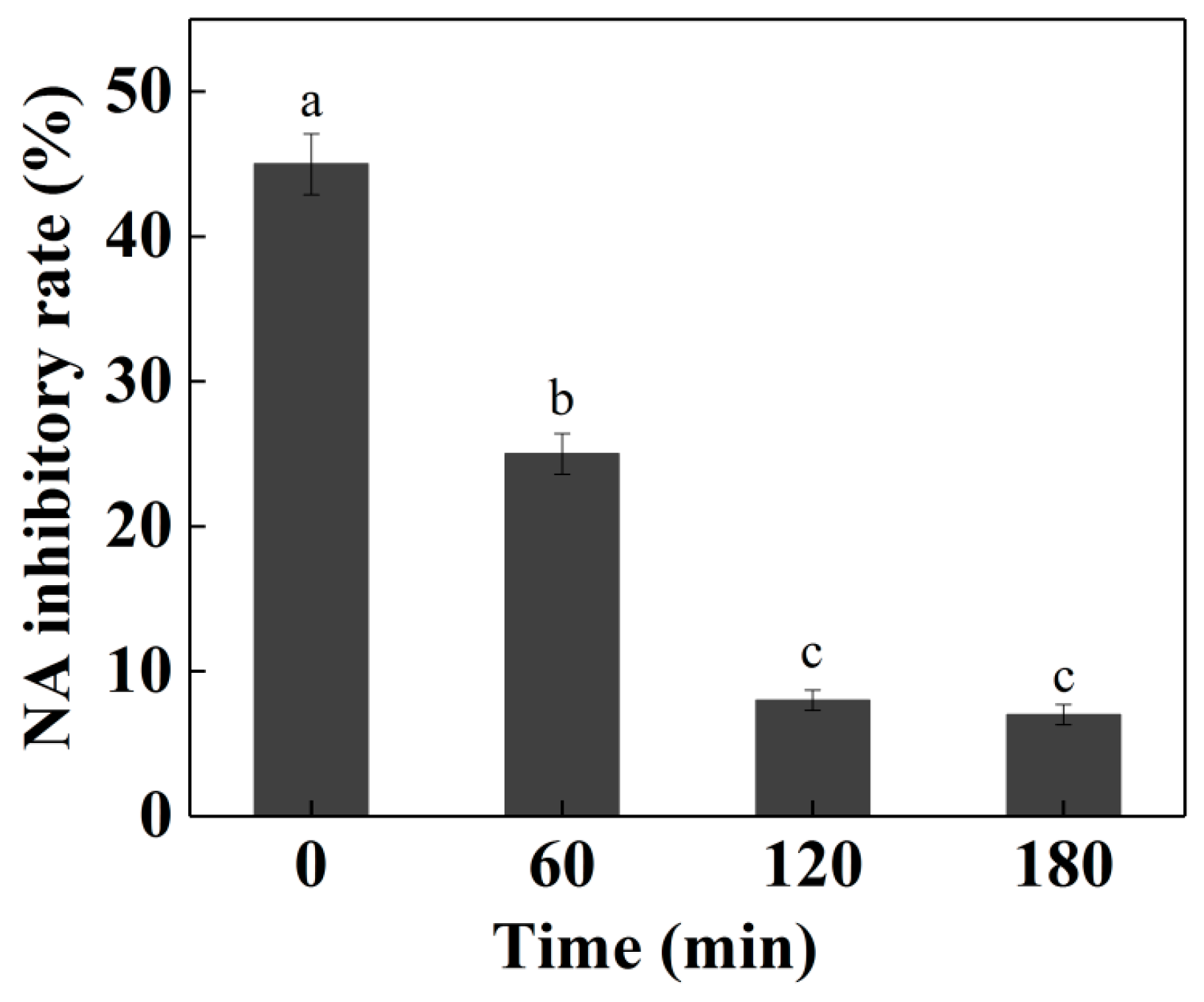
2. Results and Discussion
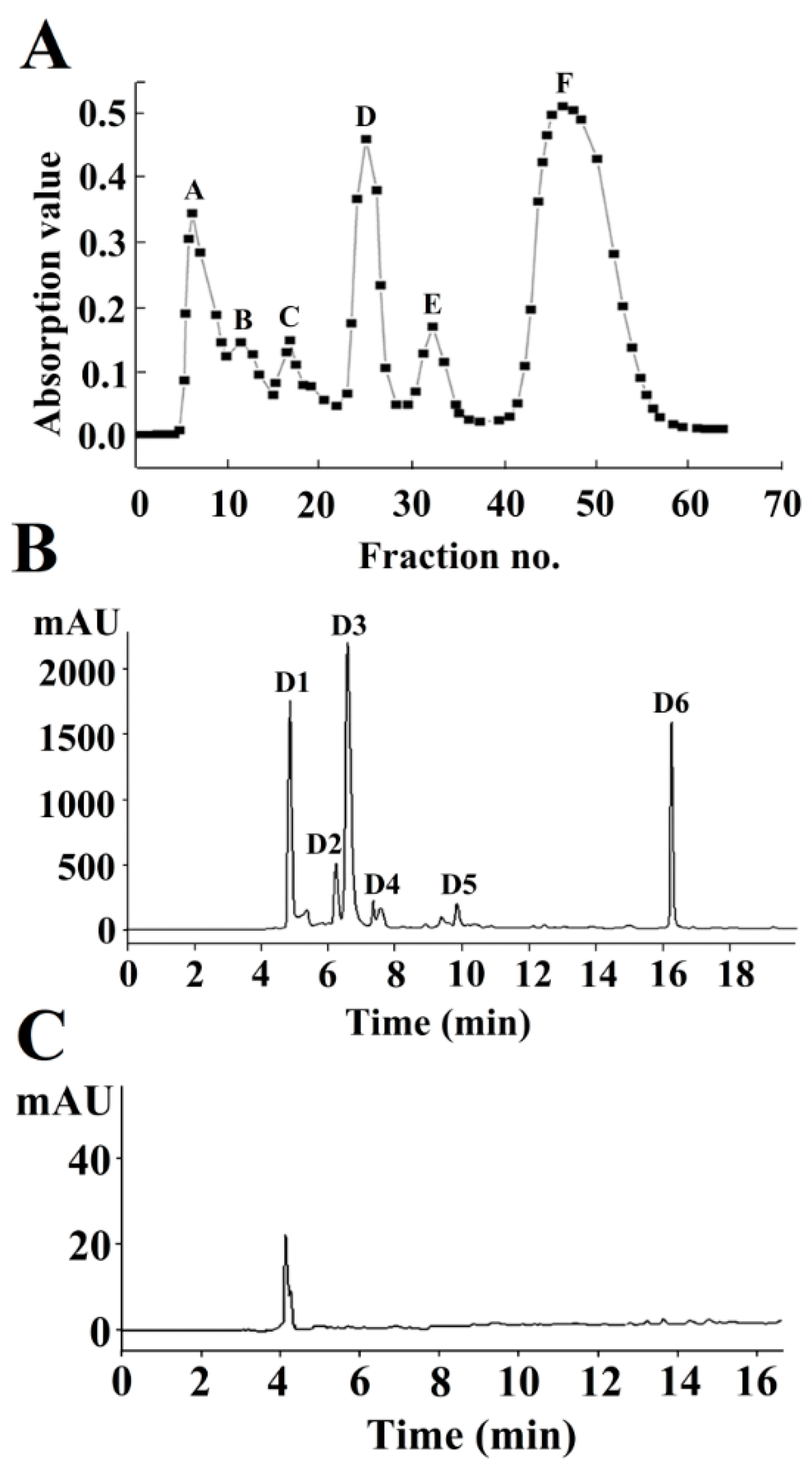
2.1. Isolation and Purification of Neuraminidase (NA)-Inhibitory Peptide
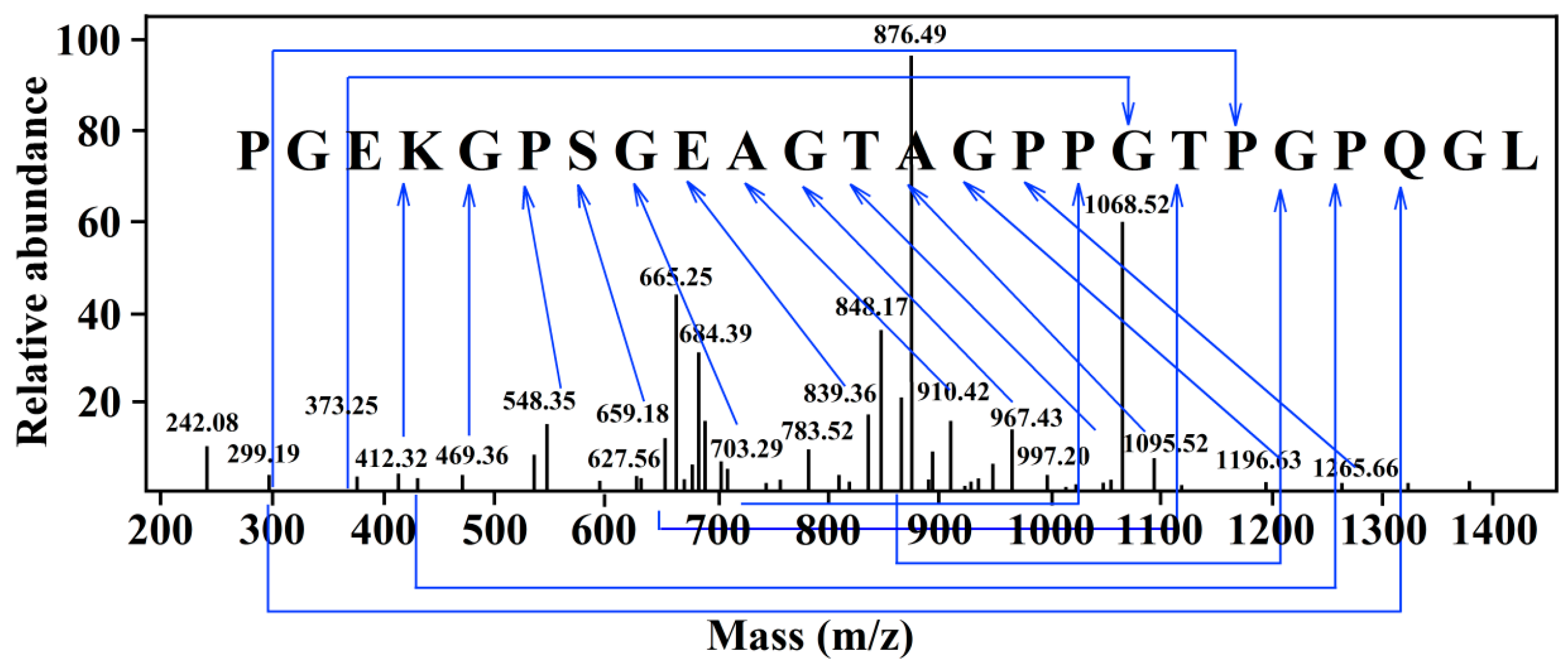
2.2. Identification of the NA-Inhibitory Peptide
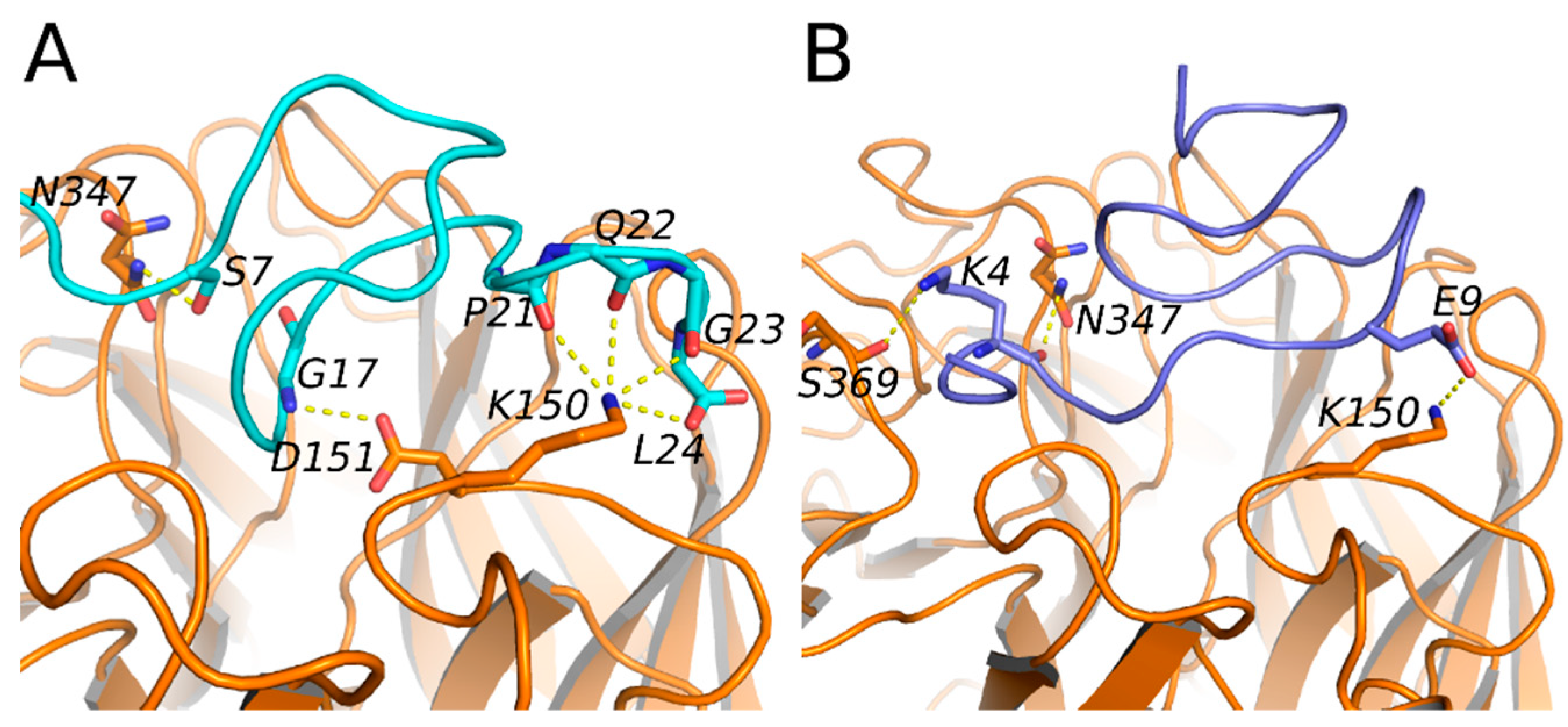
2.3. Mode of Action and Molecular Docking of PGEKGPSGEAGTAGPPGTPGPQGL
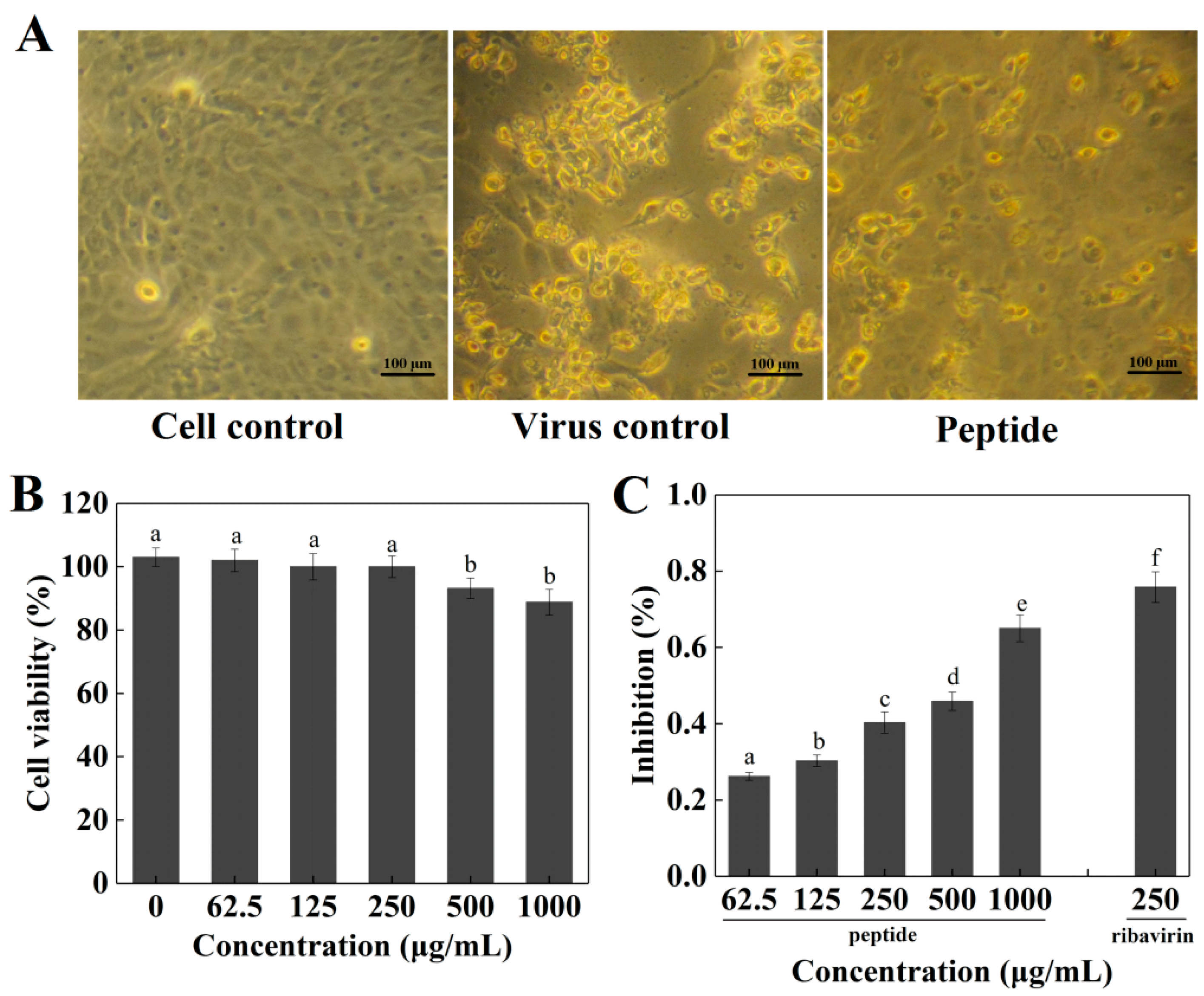
2.4. Cytotoxicity and Antiviral Activity of Peptide PGEKGPSGEAGTAGPPGTPGPQGL on Madin–Darby Canine Kidney (MDCK) Cells
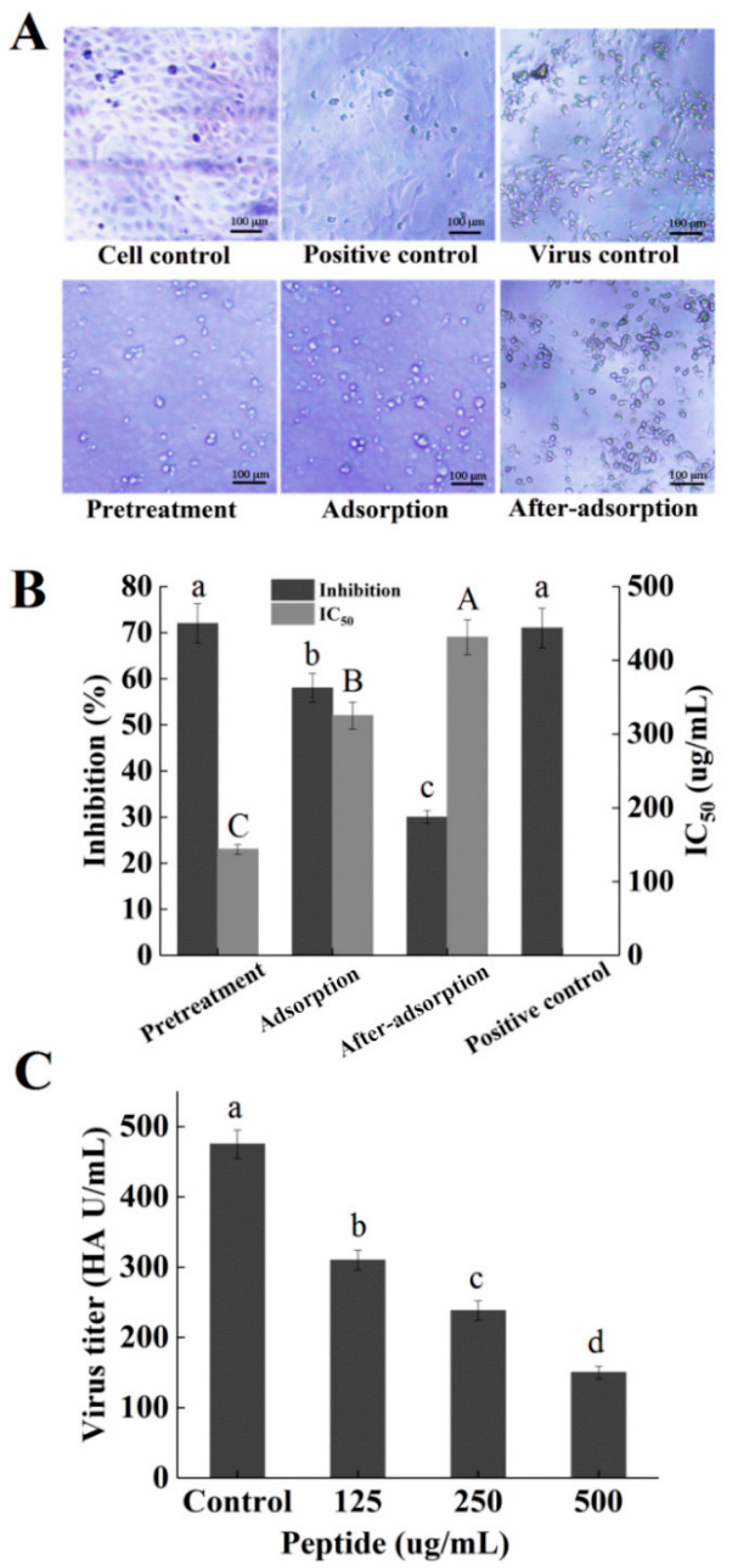
2.5. Inhibitory Effects of Peptide PGEKGPSGEAGTAGPPGTPGPQGL on Different Stages of Viral Replication
2.6. Hemagglutination (HA) Assay
2.7. Simulated Digestion Test on NA-Inhibitory Peptide
3. Materials and Methods
3.1. Reagents
3.2. Cells and Viruses
3.3. Protein Extraction of Cod Skins
3.4. Preparation of Cod Skin Protein Hydrolysates
3.5. Purification of NA-Inhibitory Peptide
3.6. NA-Inhibitory Activity Assay
3.7. Amino Acid Sequence Analysis
3.8. NA Inhibition Mode
3.9. Molecular Dynamics Simulation
3.10. Docking
3.11. Cytotoxicity Test by MTT Assay
3.12. Cytopathic Effect (CPE) Reduction Assay
3.13. Acting Mode of Inhibitory Peptide
3.14. HA Assay
3.15. Simulated Digestion Assay
3.16. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Baron, Y.; Glasner, A.; Meningher, T.; Achdout, H.; Gur, C.; Lankry, D.; Vitenshtein, A.; Meyers, A.F.A.; Mandelboim, M.; Mandelboim, O. Neuraminidase-mediated, NKp46-dependent immune-evasion mechanism of influenza viruses. Cell Rep. 2013, 3, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, Z.; Guo, M.; Xia, Y.; Zhao, M.; Wang, C.; Xu, T.; Chen, T.; Zhu, B. Inhibitory activity of selenium nanoparticles functionalized with oseltamivir on H1N1 influenza virus. Int. J. Nanomed. 2017, 12, 5733–5743. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 2004, 78, 12665–12667. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Yen, H.L. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antivir. Res. 2012, 96, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cui, Z.Q.; Zhang, P.; Hao, C.; Zhang, X.E.; Guan, H.S. In vitro inhibitory effect of carrageenan oligosaccharide on influenza A H1N1 virus. Antivir. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fantoni, G.; Couzens, L.; Gao, J.; Plant, E.; Ye, Z.; Eichelberger, M.C.; Wan, H. Comparative efficacy of monoclonal antibodies that bind to different epitopes of the 2009 pandemic H1N1 influenza virus neuraminidase. J. Virol. 2016, 90, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Pavia, A.T. Antiviral management of seasonal and pandemic influenza. J. Infect. Dis. 2006, 194, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.L.; Mckimm-Breschkin, J.L.; Choy, K.T.; Wong, D.D.Y.; Cheung, P.P.H.; Zhou, J.; Ng, I.H.; Zhu, H.; Webby, R.J.; Guan, Y.; et al. Resistance to neuraminidase inhibitors conferred by an R292K mutation in a human influenza virus H7N9 isolate can be masked by a mixed R/K viral population. MBio 2013, 4, e00396-13. [Google Scholar] [CrossRef] [PubMed]
- Amri, N.; Parikesit, A.A.; Tambunan, U.S.F. In silico design of cyclic peptides as influenza virus, a subtype H1N1 neuraminidase inhibitor. Afr. J. Biotechnol. 2013, 11, 11474–11491. [Google Scholar]
- Upadhyay, A.; Chompoo, J.; Taira, N.; Fukuta, M.; Gima, S.; Tawata, S. Solid-phase synthesis of mimosine tetrapeptides and their inhibitory activities on neuraminidase and tyrosinase. J. Agric. Food Chem. 2011, 59, 12858–12863. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Zeng, M.Y.; Gao, F.Z.; Guo, X.M.; Wang, H.T.; Zhao, Y.H.; Wang, W. Preparation of active peptide from cod skins with inhibitory activity on influenza virus neuraminidase. Chin. J. Mar. Drugs 2012, 31, 1–7. [Google Scholar]
- Nagai, T.; Suzuki, N. Preparation and partial characterization of collagen from paper nautilus (Argonauta argo, Linnaeus) outer skin. Food Chem. 2002, 76, 149–153. [Google Scholar] [CrossRef]
- Mattice, W.L.; Mandelkern, L. Ordered structures in sequential copolypeptides containing l-proline or 4-hydroxy-l-proline. J. Am. Chem. Soc. 1970, 92, 5285–5287. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.T.; Chen, Y.; Wang, S.; Gentles, R.; Sowin, T.; Kati, W.; Muchmore, S.; Giranda, V.; Stewart, K.; Sham, H.; et al. Design, synthesis, and structural analysis of influenza neuraminidase inhibitors containing pyrrolidine cores. J. Med. Chem. 2001, 44, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Bayrakdarian, M.; Luo, X. Total synthesis of A-315675: A potent inhibitor of influenza neuraminidase. J. Am. Chem. Soc. 2002, 124, 4716–4721. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.W.A.; Ngo, D.H.; Ryu, B.M.; Kim, S.K. An active peptide purified from gastrointestinal enzyme hydrolysate of Pacific cod skin gelatin attenuates angiotensin-1 converting enzyme (ACE) activity and cellular oxidative stress. Food Chem. 2012, 132, 1872–1882. [Google Scholar] [CrossRef]
- Ngo, D.H.; Vo, T.S.; Ryu, B.M.; Kim, S.K. Angiotensin-I-converting enzyme (ACE) inhibitory peptides from Pacific cod skin gelatin using ultrafiltration membranes. Process Biochem. 2016, 51, 1622–1628. [Google Scholar] [CrossRef]
- Madrahimov, A.; Helikar, T.; Kowal, B.; Lu, G.; Rogers, J. Dynamics of influenza virus and human host interactions during infection and replication cycle. Bull. Math. Biol. 2013, 75, 988–1011. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, Y.; Guo, M.; Xu, T.; Wang, C.; Zhao, M.; Wang, H.; Chen, T.; Zhu, B. The inhibition of H1N1 influenza virus-induced apoptosis by silver nanoparticles functionalized with zanamivir. RSC Adv. 2017, 7, 742–750. [Google Scholar] [CrossRef]
- Lauster, D.; Glanz, M.; Bardua, M.; Ludwig, K.; Hellmund, M.; Hoffmann, U.; Hamann, A.; Böttcher, C.; Haag, R.; Hackenberger, C.P.R.; et al. Multivalent peptide-nanoparticle conjugates for influenza-virus inhibition. Angew. Chem. Int. Ed. 2017, 56, 5931–5936. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Si, L.; Tian, Z.; Jiao, P.; Fan, Z.; Meng, K.; Zhou, X.; Wang, H.; Xu, R.; Han, X.; et al. Pentacyclic triterpenes grafted on CD cores to interfere with influenza virus entry: A dramatic multivalent effect. Biomaterials 2016, 78, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Z.; Zhao, Y.; Zhu, X.; Yu, R.; Dong, S.; Wu, H. Novel natural angiotensin converting enzyme (ACE)-inhibitory peptides derived from sea cucumber-modified hydrolysates by adding exogenous proline and a study of their structure–activity relationship. Mar. Drugs 2018, 16, 271. [Google Scholar] [CrossRef] [PubMed]
- Irungu, J.; Go, E.P.; Zhang, Y.; Dalpathado, D.S.; Liao, H.X.; Haynes, B.F.; Desaire, H. Comparison of HPLC/ESI-FTICR MS versus MALDI-TOF/TOF MS for glycopeptide analysis of a highly glycosylated HIV envelope glycoprotein. J. Am. Soc. Mass Spectr. 2008, 19, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Sangsawad, P.; Roytrakul, S.; Choowongkomon, K.; Kitts, D.D.; Chen, X.M.; Meng, G.; Lichan, E.; Yongsawatdigul, J. Transepithelial transport across Caco-2 cell monolayers of angiotensin converting enzyme (ACE) inhibitory peptides derived from simulated in vitro gastrointestinal digestion of cooked chicken muscles. Food Chem. 2018, 251, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, T.J.; Zhao, X.H. Ace inhibition and enzymatic resistance in vitro of a casein hydrolysate subjected to plastein reaction in the presence of extrinsic proline and ethanol- or methanol-water fractionation. Int. J. Food Prop. 2014, 17, 386–398. [Google Scholar] [CrossRef]
- Park, J.Y.; Jeong, H.J.; Kim, Y.M.; Park, S.J.; Rho, M.C.; Park, K.H.; Ryu, Y.B.; Lee, W.S. Characteristic of alkylated chalcones from Angelica keiskei on influenza virus neuraminidase inhibition. Bioorg. Med. Chem. Lett. 2011, 21, 5602–5604. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.A.; Dao, T.T.; Tung, B.T.; Hwanwon, C.; Eunhee, K.; Junsoo, P.; Seongil, L.; Wonkeun, O. Influenza A (H1N1) neuraminidase inhibitors from Vitis amurensis. Food Chem. 2011, 124, 437–443. [Google Scholar] [CrossRef]
- Jiang, H.L.; Wang, C.X.; Jiang, N.Y.; Chen, M.H.; Peng, F.; Xie, Y.; Jiang, H. Neuraminidase inhibitors produced by the marine derived Streptomyces sp. FIM090041. Chin. J. Antibiot. 2012, 37, 265–268. [Google Scholar]
- Wang, F.; Ai, H.; Lei, C. In vitro anti-influenza activity of a protein-enriched fraction from larvae of the housefly (Musca domestica). Pharm. Biol. 2013, 51, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Haruyama, T.; Nagata, K. Anti-influenza virus activity of Ginkgo biloba leaf extracts. J. Nat. Med. 2013, 67, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Zu, M.; Yang, F.; Zhou, W.; Liu, A.; Du, G.; Zheng, L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antivir. Res. 2012, 94, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Cao, Z.; Cao, L.; Ding, G.; Wang, Z.; Xiao, W. Antiviral activity of chlorogenic acid against influenza A (H1N1/H3N2) virus and its inhibition of neuraminidase. Sci. Rep. 2017, 7, 45723. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.B.; Chen, Q.Y.; Wu, X.M.; Che, Y.L.; Wang, C.Y.; Chen, R.J.; Zhou, L.J. Isolation of a reassortant H1N2 swine Flu strain of type “Swine-Human-Avian” and its genetic variability analysis. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Kuba, M.; Tanaka, K.; Tawata, S.; Takeda, Y.; Yasuda, M. Angiotensin I-converting enzyme inhibitory peptides isolated from tofuyo fermented soybean food. Biosci. Biotechnol. Biochem. 2003, 67, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.Y.; Liang, C.C.; Miao, J.L.; Li, G.Y. Preparation and composition analysis of cod skin collagen protein. Chin. J. Mar. Drugs 2005, 24, 30–32. [Google Scholar]
- Cao, H.P.; Tao, P.Z.; Du, G.H. Establishment and application of high throughput screening model for influenza virus neuraminidase inhibitors in vitro. Acta Pharm. Sin. 2002, 37, 930–933. [Google Scholar]
- Jag, R.; Ramos, M.; Recio, I. Angiotensin converting enzyme-inhibitory activity of peptides isolated from Manchego cheese. Stability under simulated gastrointestinal digestion. Int. Dairy J. 2004, 14, 1075–1080. [Google Scholar]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins 2010, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, N.; Tae, H.S.; Jia, X.; Kaas, Q.; Jiang, T.; Adams, D.J.; Yu, R. Role of Cys I-Cys III disulfide bond on the structure and activity of α-conotoxins at human neuronal nicotinic acetylcholine receptors. ACS Omega 2017, 2, 4621–4631. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comp. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.F.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2010, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Talarico, L.B.; Damonte, E.B. Interference in dengue virus adsorption and uncoating by carrageenans. Virology 2007, 363, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, P.; Yu, G.L.; Li, C.X.; Hao, C.; Qi, X.; Zhang, L.J.; Guan, H.S. Preparation and anti-influenza A virus activity of k-carrageenan oligosaccharide and its sulphated derivatives. Food Chem. 2012, 133, 880–888. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Kadowaki, A.; Onishi, Y.; Gato, N.; Ujike, M.; Odagiri, T.; Tashiro, M.; Suzuki, Y. Mumefural and related HMF derivatives from Japanese apricot fruit juice concentrate show multiple inhibitory effects on pandemic influenza A (H1N1) virus. Food Chem. 2011, 127, 1–9. [Google Scholar] [CrossRef]
- Sangsawad, P.; Roytrakul, S.; Yongsawatdigul, J. Angiotensin converting enzyme (ACE) inhibitory peptides derived from the simulated in vitro gastrointestinal digestion of cooked chicken breast. J. Funct. Foods 2017, 29, 77–83. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Purification | IC50 (mg/mL) | Purification Fold |
|---|---|---|---|
| Hydrolysates (<5000 Da) | Ultrafiltration | 6.40 ± 0.13 a | 1.00 |
| D | Sephadex G-15 | 3.50 ± 0.11 b | 1.83 |
| D1 | RP-HPLC | 0.89 ± 0.07 c | 7.19 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, Y.; Yuan, N.; Zeng, M.; Zhao, Y.; Yu, R.; Liu, Z.; Wu, H.; Dong, S. A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism. Mar. Drugs 2018, 16, 377. https://doi.org/10.3390/md16100377
Li J, Chen Y, Yuan N, Zeng M, Zhao Y, Yu R, Liu Z, Wu H, Dong S. A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism. Marine Drugs. 2018; 16(10):377. https://doi.org/10.3390/md16100377
Chicago/Turabian StyleLi, Jianpeng, Yiping Chen, Ning Yuan, Mingyong Zeng, Yuanhui Zhao, Rilei Yu, Zunying Liu, Haohao Wu, and Shiyuan Dong. 2018. "A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism" Marine Drugs 16, no. 10: 377. https://doi.org/10.3390/md16100377
APA StyleLi, J., Chen, Y., Yuan, N., Zeng, M., Zhao, Y., Yu, R., Liu, Z., Wu, H., & Dong, S. (2018). A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism. Marine Drugs, 16(10), 377. https://doi.org/10.3390/md16100377