Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor


Abstract
:1. Introduction
2. Results and Discussion
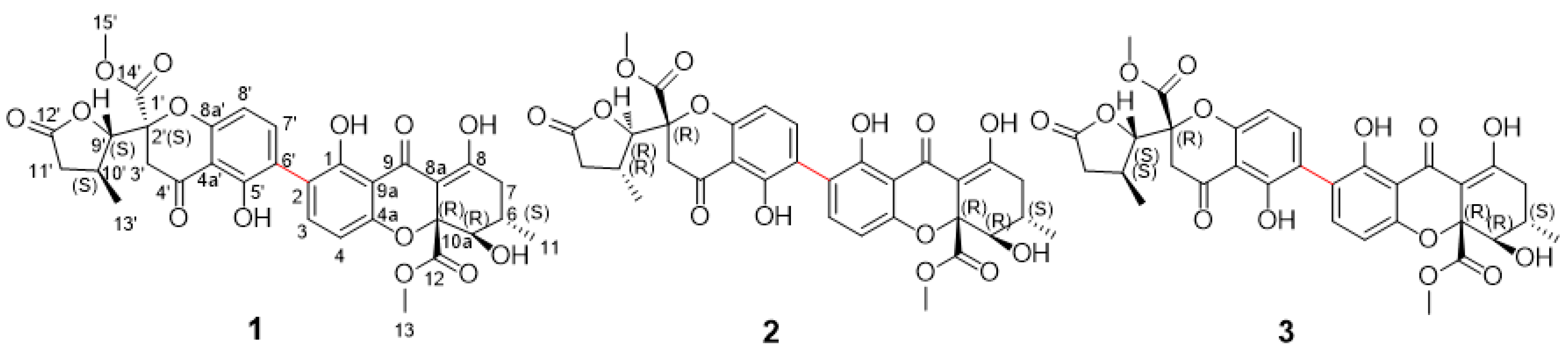
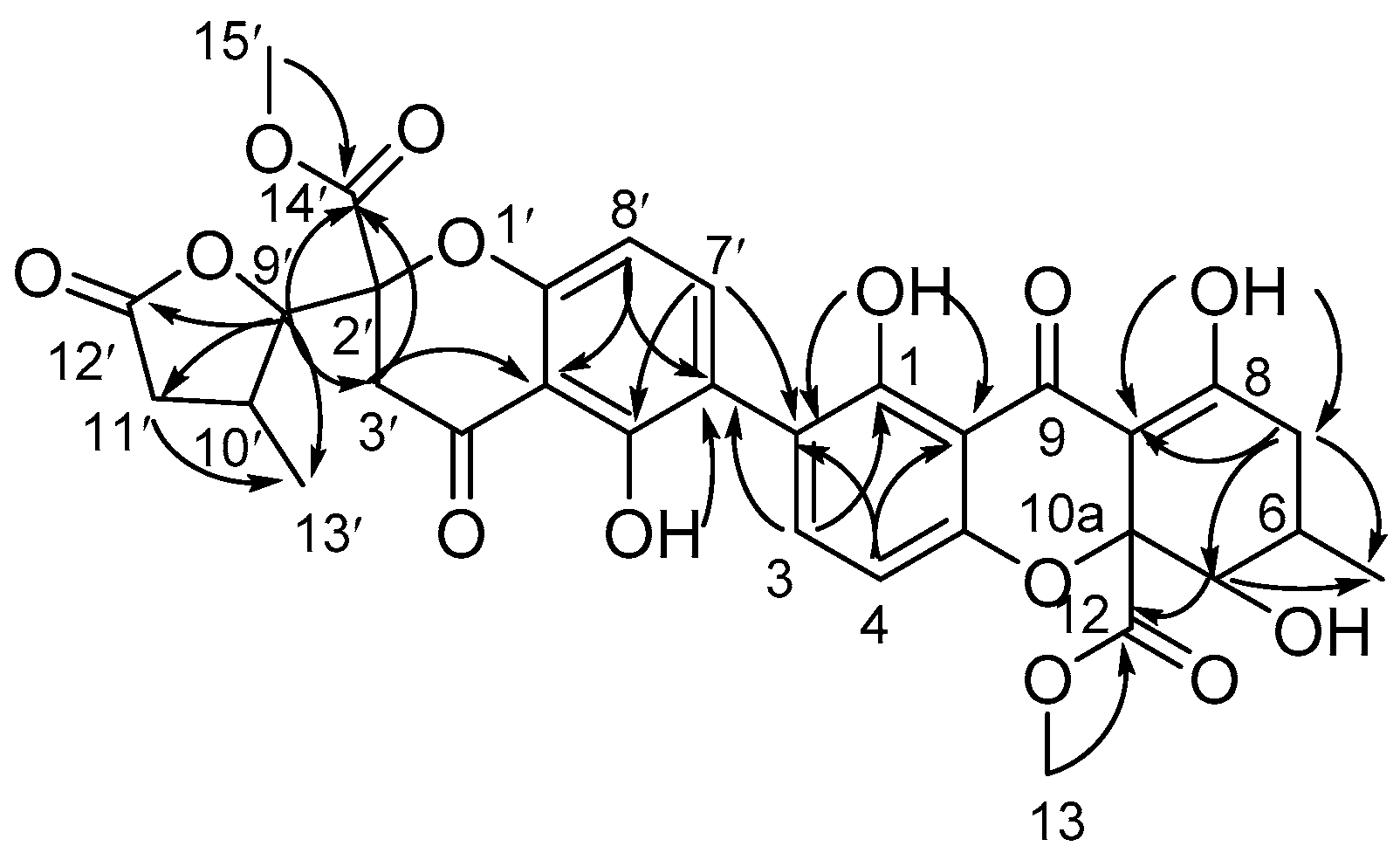
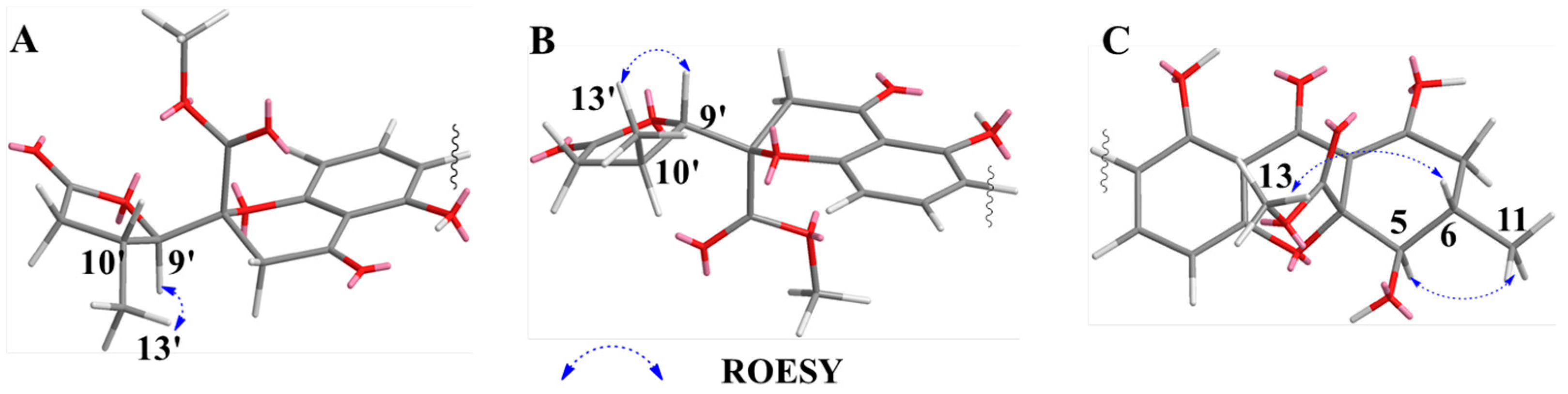
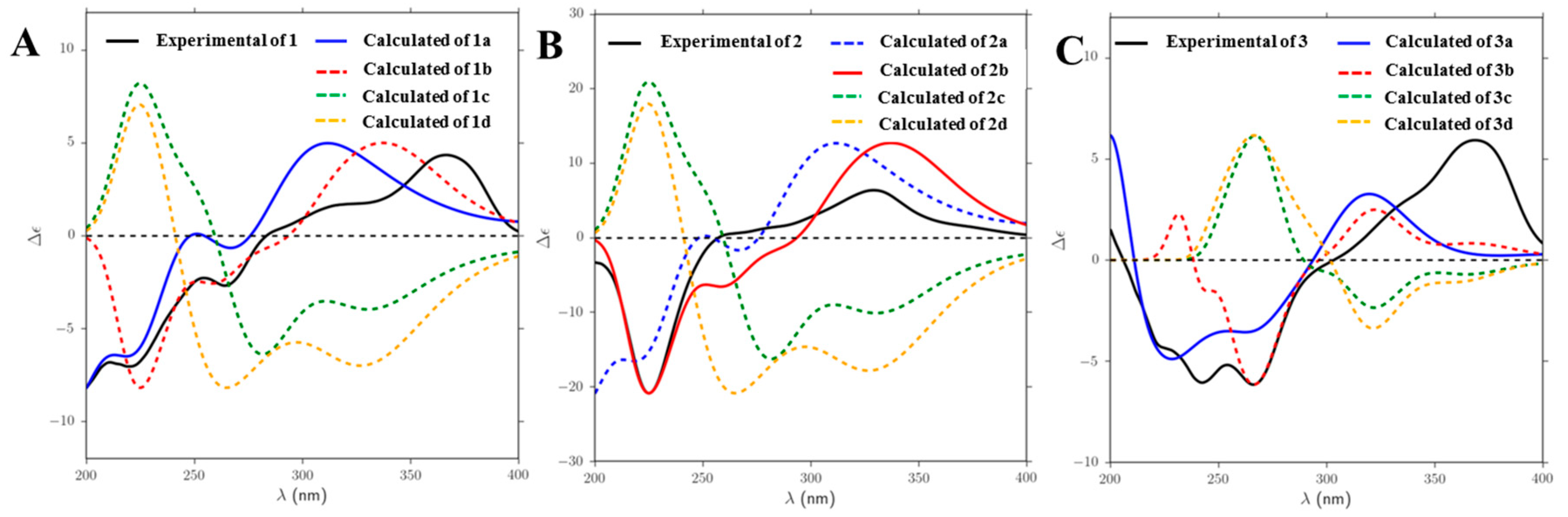
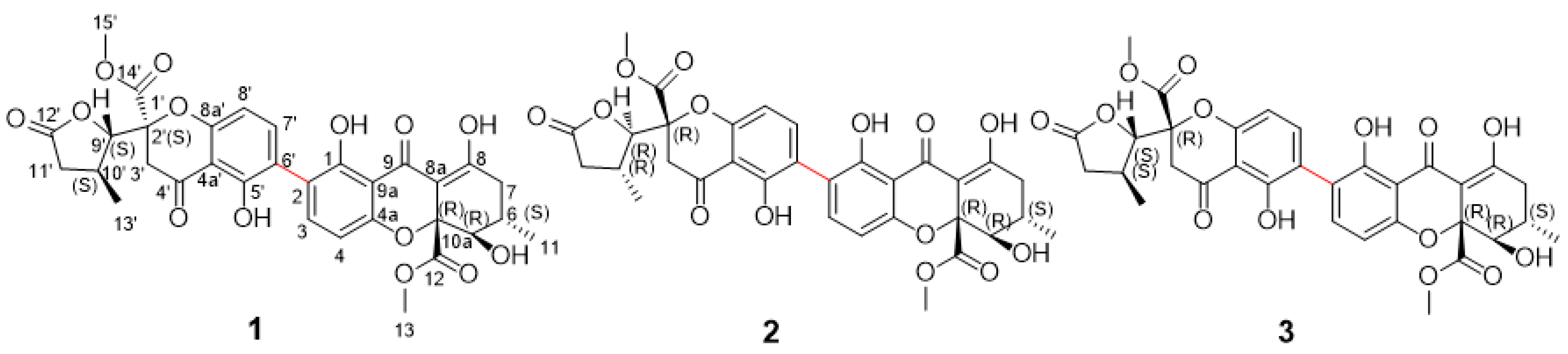
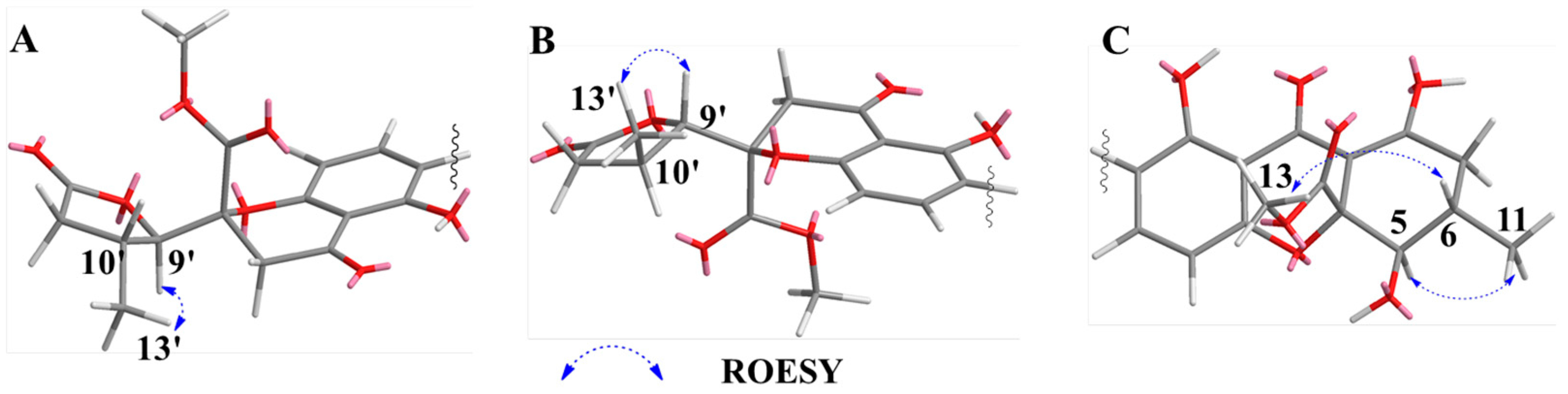
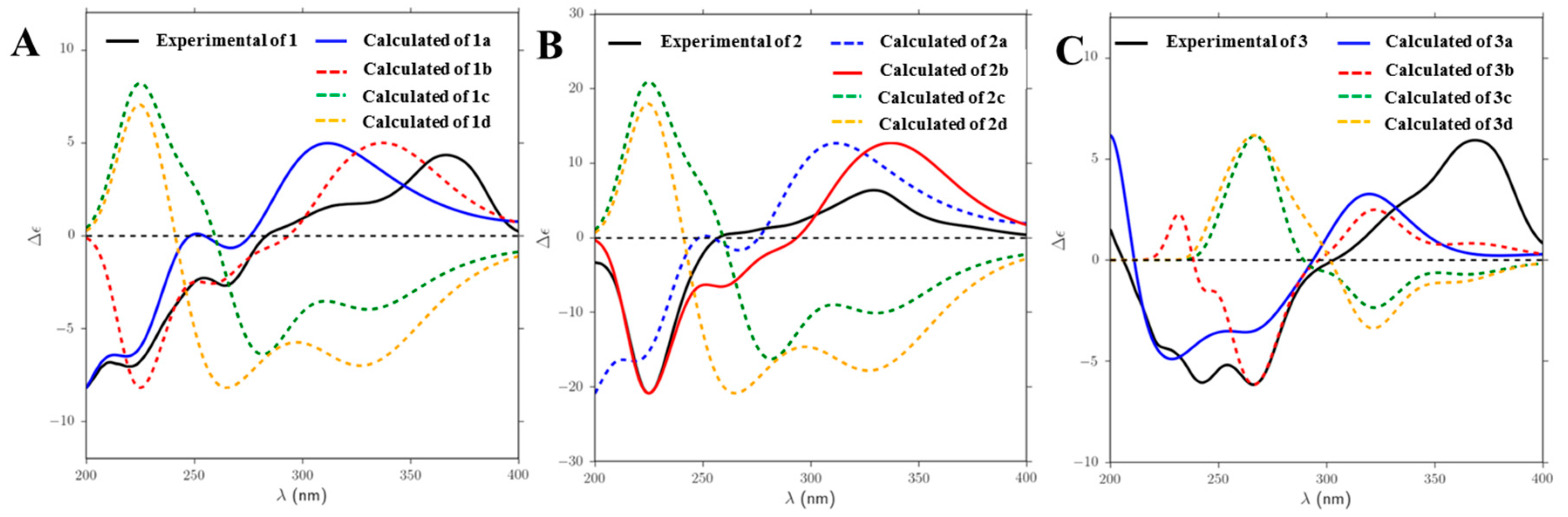
2.1. Structure Elucidation of Compounds
2.2. Biological Assays
2.3. Plausible Biosynthetic Gene Cluster and Pathway of Chrysoxanthones A–C
3. Materials and Methods
3.1. General
3.2. Fungal Material and Fermentation
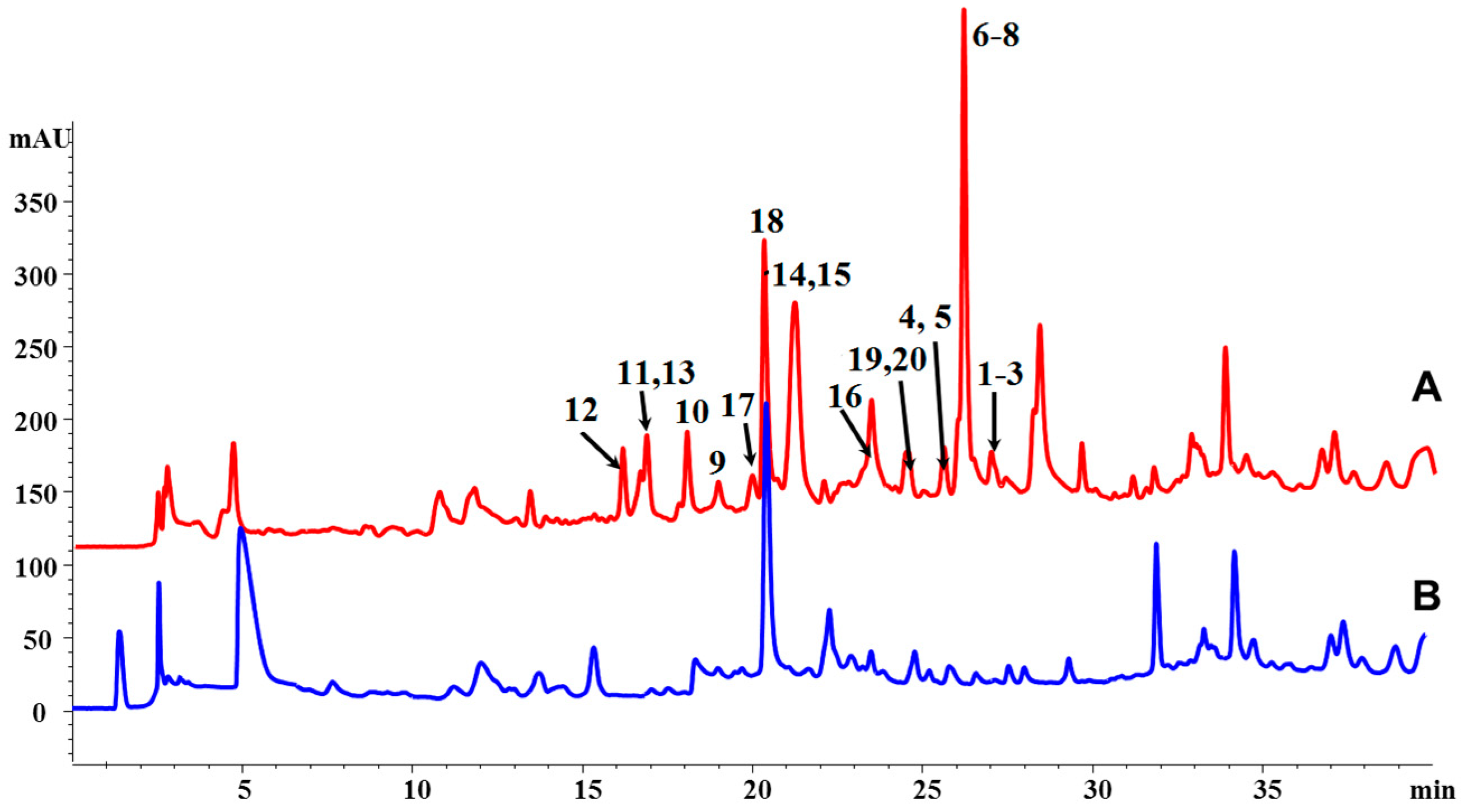
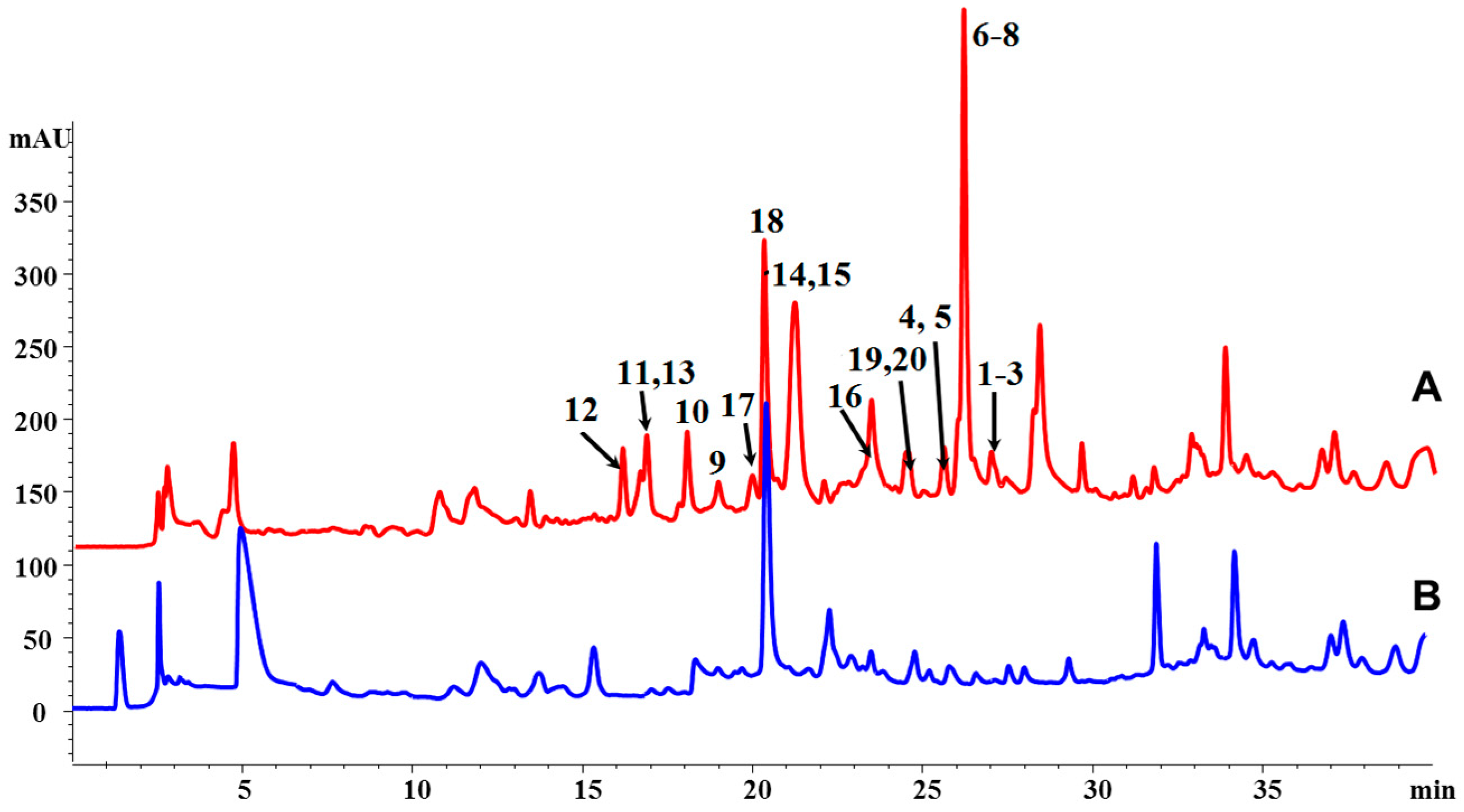
3.3. Extraction, HPLC Analysis, and Isolation
3.4. Spectroscopic Data
3.5. ECD Calculation
3.6. Biological Assay
3.6.1. Antibacterial Activity
3.6.2. Antitumor Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van den Berg, M.A.; Albang, R.; Albermann, K.; Badger, J.H.; Daran, J.M.; Driessen, A.J.; Garcia-Estrada, C.; Fedorova, N.D.; Harris, D.M.; Heijne, W.H.; et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 2008, 26, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Chung, Y.-M.; Sakurai, H.; Ozeki, T.; Chang, F.-R.; Yamashita, K.; Oshima, Y. Tenuipyrone, a novel skeletal polyketide from the entomopathogenic fungus, Isaria tenuipes, cultivated in the presence of epigenetic modifiers. Org. Lett. 2011, 14, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Morita, S.; Shirata, N.; Taniguchi, T.; Monde, K.; Sakurai, H.; Ozeki, T.; Oshima, Y. Structural diversity of new C13-polyketides produced by Chaetomium mollipilium cultivated in the presence of a NAD+-dependent histone deacetylase Inhibitor. Org. Lett. 2012, 14, 5456–5459. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Mu, R.; Dou, M.; Yan, D.; Liu, B.; Wei, Q.; Wan, J.; Tang, Y.; Hu, Y. Epigenetic modification in histone deacetylase deletion strain of Calcarisporium arbuscula leads to diverse diterpenoids. Acta Pharm. Sin. B 2018, 8, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.M.; Xu, W.; Li, D.; Yin, W.B.; Chooi, Y.H.; Li, Y.Q.; Tang, Y.; Hu, Y.-C. Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. 2015, 54, 7592–7596. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Raja, H.A.; Graf, T.N.; Swanson, S.M.; Falkinham, J.O., 3rd; Wani, M.C.; Pearce, C.J.; Oberlies, N.H. Biosynthetically distinct cytotoxic polyketides from Setophoma terrestris. Eur. J. Org. Chem. 2015, 2015, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Arunpanichlert, J.; Rukachaisirikul, V.; Tadpetch, K.; Phongpaichit, S.; Hutadilok-Towatana, N.; Supaphon, O.; Sakayaroj, J. A dimeric chromanone and a phthalide: Metabolites from the seagrass-derived fungus Bipolaris sp. PSU-ES64. Phytochem. Lett. 2012, 5, 604–608. [Google Scholar] [CrossRef]
- Wu, G.; Yu, G.; Kurtan, T.; Mandi, A.; Peng, J.; Mo, X.; Liu, M.; Li, H.; Sun, X.; Li, J.; et al. Versixanthones A–F, cytotoxic xanthone-chromanone dimers from the marine-derived fungus Aspergillus versicolor HDN1009. J. Nat. Prod. 2015, 78, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Sunazuka, T.; Tanaka, H.; Iwai, Y.; Takayanagi, H.; Omura, S. Andrastins A–C, new protein farnesyltransferase inhibitors produced by Penidllium sp. FO-3929. J. Antibiot. 1996, 49, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Inokoshi, J.; Uchida, R.; Shiomi, K.; Masuma, R.; Kawakubo, T.; Tanaka, H.; Iwai, Y.; Kosemura, S.; Yamamura, S. Andrastins A–C, new protein farnesyltransferase inhibitors produced by Penicillium sp. FO-3929. I. Producing strain, fermentation, isolation, and biological activities. J. Antibiot. 1996, 49, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Steyn, P. The isolation, structure and absolute configuration of secalonic acid D, the toxic metabolite of Penicillium oxalicum. Tetrahedron 1970, 26, 51–57. [Google Scholar] [CrossRef]
- Andersen, R.; Buechi, G.; Kobbe, B.; Demain, A.L. Secalonic acids D and F are toxic metabolites of Aspergillus aculeatus. J. Org. Chem. 1977, 42, 352–353. [Google Scholar] [CrossRef] [PubMed]
- Kurobane, I.; Vining, L.C.; MCINNES, A.G. Biosynthetic relationships among the secalonic acids. J. Antibiot. 1979, 32, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, B.; Krohn, K.; Flörke, U.; Root, N.; Aust, H.J.; Draeger, S.; Schulz, B.; Antus, S.; Kurtán, T. X-ray structure determination, absolute configuration and biological activity of phomoxanthone A. Eur. J. Org. Chem. 2005, 2005, 4563–4570. [Google Scholar] [CrossRef]
- Gong, T.; Sun, C.-Y.; Zhen, X.; Qiu, J.-Z.; Yang, J.-L.; Zhu, P. Bioactive metabolites isolated from the fungus Penicillium chrysogenum HLS111 associated with the marine sponge Gelliodes carnosa. Chin. Med. Biotechnol. 2014, 9, 174–179. [Google Scholar]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Wang, Q.X.; Bao, L.; Yang, X.L.; Guo, H.; Yang, R.N.; Ren, B.; Zhang, L.X.; Dai, H.Q.; Guo, L.D.; Liu, H.W. Polyketides with antimicrobial activity from the solid culture of an endolichenic fungus Ulocladium sp. Fitoterapia 2012, 83, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Li, X.-M.; Wang, B.-G. Penicisimpins A–C, three new dihydroisocoumarins from Penicillium simplicissimum MA-332, a marine fungus derived from the rhizosphere of the mangrove plant Bruguiera sexangula var. rhynchopetala. Phytochem. Lett. 2016, 17, 114–118. [Google Scholar] [CrossRef]
- Nozawa, K.; Nakajima, S. Isolation of radicicol from Penicillium luteo-aurantium, and meleagrin, a new metabolite from Penicillium meleagrinum. J. Nat. Prod. 1979, 42, 374–377. [Google Scholar] [CrossRef]
- Nagel, D.W.; Pachler, K.G.; Steyn, P.S.; Vleggaar, R.; Wessels, P.L. The chemistry and 13C NMR assignments of oxaline, a novel alkaloid from Penicillium oxalicum. Tetrahedron 1976, 32, 2625–2631. [Google Scholar] [CrossRef]
- Ohmomo, S.; Utagawa, T.; Abe, M. Identification of roquefortine C produced by Penicillium roqueforti. Agric. Biol. Chem. 1977, 41, 2097–2098. [Google Scholar] [CrossRef]
- Song, F.; He, H.; Ma, R.; Xiao, X.; Wei, Q.; Wang, Q.; Ji, Z.; Dai, H.; Zhang, L.; Capon, R.J. Structure revision of the penicillium alkaloids haenamindole and citreoindole. Tetrahedron Lett. 2016, 57, 3851–3852. [Google Scholar] [CrossRef]
- Du, L.; Li, D.; Zhu, T.; Cai, S.; Wang, F.; Xiao, X.; Gu, Q. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron 2009, 65, 1033–1039. [Google Scholar] [CrossRef]
- Neubauer, L.; Dopstadt, J.; Humpf, H.-U.; Tudzynski, P. Identification and characterization of the ergochrome gene cluster in the plant pathogenic fungus Claviceps purpurea. Fungal Biol. Biotechnol. 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Li, C.Q.; Zhu, P.; Yang, J.L.; Cheng, K.D. Phylogenetic diversity of culturable fungi associated with two marine sponges: Haliclona simulans and Gelliodes carnosa, collected from the Hainan Island coastal waters of the South China Sea. Fungal Divers. 2010, 42, 1–15. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09, R.A. 1, Gaussian Inc.: Wallingford, CT, USA, 2009.
- O’Boyle, N.M.; Vandermeersch, T.; Hutchison, G.R. Confab—Generation of diverse low energy conformers. J Cheminform. 2011, 3, P32. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumloeffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Q.; Xu, S.; Zuo, L.; You, X.; Hu, H.-Y. Aminoglycoside-based novel probes for bacterial diagnostic and therapeutic applications. Chem. Commun. 2017, 53, 1366–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Gong, T.; Liu, F.; Zhang, P.-C.; Zhou, W.-Q.; Li, Y.; Zhu, P. A new analogue ofechinomycin and a new cyclic dipeptide from a marine-derived Streptomyces sp. LS298. Mar. Drugs 2015, 13, 6947–6961. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 and 2 | 3 | ||
|---|---|---|---|---|
| δH, mult, J in Hz | δC | δH, mult, J in Hz | δC | |
| 1 | 159.3 | 159.3 | ||
| 2 | 117.7 | 117.8 | ||
| 3 | 7.46, d, 8.5 | 140.1 | 7.46, d, 8.5 | 140.2 |
| 4 | 6.63, d, 8.5 | 107.5 | 6.62, d, 8.5 | 107.6 |
| 4a | 158.4 | 158.3 | ||
| 5 | 3.93, dd, 11.5, 2.5 | 76.9 | 3.93, dd, 11.0, 1.5 | 76.9 |
| 6 | 2.42, m | 29.2 | 2.42, m | 29.2 |
| 7a | 2.32, dd, 19.0, 10.5 | 36.2 | 2.31, dd, 19.0, 10.5 | 36.3 |
| 7b | 2.74, dd, 19.0, 6.5 | 2.74, dd, 19.0, 6.5 | ||
| 8 | 177.6 | 177.6 | ||
| 8a | 101.5 | 101.5 | ||
| 9 | 187.1 | 187.1 | ||
| 9a | 106.8 | 106.8 | ||
| 10a | 84.7 | 84.7 | ||
| 11 | 1.18, d, 6.5 | 18.0 | 1.17, d, 6.5 | 18.0 |
| 12 | 170.2 | 170.2 | ||
| 13 | 3.73, s | 53.3 | 3.73, s | 53.3 |
| 1-OH | 11.72, s | 11.72, s | ||
| 5-OH | 2.78, d, 2.5 | 2.80, br s | ||
| 8-OH | 13.76, s | 13.78, s | ||
| 2′ | 84.2 | 84.3 | ||
| 3a’ | 3.05, d, 17.0 | 39.6 | 3.10, d, 17.0 | 40.4 |
| 3b’ | 3.21, d, 17.0 | 3.51, d, 17.0 | ||
| 4′ | 194.0 | 195.0 | ||
| 4a’ | 107.6 | 107.3 | ||
| 5′ | 159.2 | 159.1 | ||
| 6′ | 118.1 | 118.0 | ||
| 7′ | 7.52, d, 8.5 | 141.3 | 7.51, d, 8.5 | 141.1 |
| 8′ | 6.62, d, 8.5 | 107.3 | 6.57, d, 8.5 | 107.0 |
| 8a’ | 158.6 | 158.6 | ||
| 9′ | 4.45, d, 4.0 | 87.5 | 4.38, d, 3.5 | 86.4 |
| 10′ | 2.84, m | 30.0 | 2.88, m | 29.6 |
| 11a’ | 2.92, dd, 17.5, 9.0 | 36.0 | 3.03, dd, 18.0,9.0 | 36.2 |
| 11b’ | 2.23, dd, 17.5, 4.0 | 2.23, dd, 18.0,4.0 | ||
| 12′ | 175.2 | 175.5 | ||
| 13′ | 1.29, d, 7.0 | 20.9 | 1.19, d, 7.0 | 20.6 |
| 14′ | 168.8 | 169.0 | ||
| 15′ | 3.77, s | 53.7 | 3.78, s | 53.7 |
| 5′-OH | 11.89, s | 11.92, s | ||
| Compounds | MICs (μg/mL) | ||||
|---|---|---|---|---|---|
| Staphylococcus epidermidis ATCC 12228 | Staphylococcus aureus ATCC 29213 | Bacillus subtilis ATCC 63501 | Enterococcus faecalis ATCC 29212 | Escherichia coli ATCC25922 | |
| 1 | --- | --- | 5 | --- | >100 |
| 2 | 10 | 20 | 5 | >100 | >100 |
| 3 | 20 | 80 | 10 | >100 | >100 |
| Ampicillin | 6.25 | 6.25 | 3.12 | 6.25 | 25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhen, X.; Gong, T.; Wen, Y.-H.; Yan, D.-J.; Chen, J.-J.; Zhu, P. Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor. Mar. Drugs 2018, 16, 357. https://doi.org/10.3390/md16100357
Zhen X, Gong T, Wen Y-H, Yan D-J, Chen J-J, Zhu P. Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor. Marine Drugs. 2018; 16(10):357. https://doi.org/10.3390/md16100357
Chicago/Turabian StyleZhen, Xin, Ting Gong, Yan-Hua Wen, Dao-Jiang Yan, Jing-Jing Chen, and Ping Zhu. 2018. "Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor" Marine Drugs 16, no. 10: 357. https://doi.org/10.3390/md16100357
APA StyleZhen, X., Gong, T., Wen, Y.-H., Yan, D.-J., Chen, J.-J., & Zhu, P. (2018). Chrysoxanthones A–C, Three New Xanthone–Chromanone Heterdimers from Sponge-Associated Penicillium chrysogenum HLS111 Treated with Histone Deacetylase Inhibitor. Marine Drugs, 16(10), 357. https://doi.org/10.3390/md16100357