The Neuroprotective Marine Compound Psammaplysene A Binds the RNA-Binding Protein HNRNPK

 , ,
, ,
Abstract
:1. Introduction
2. Results
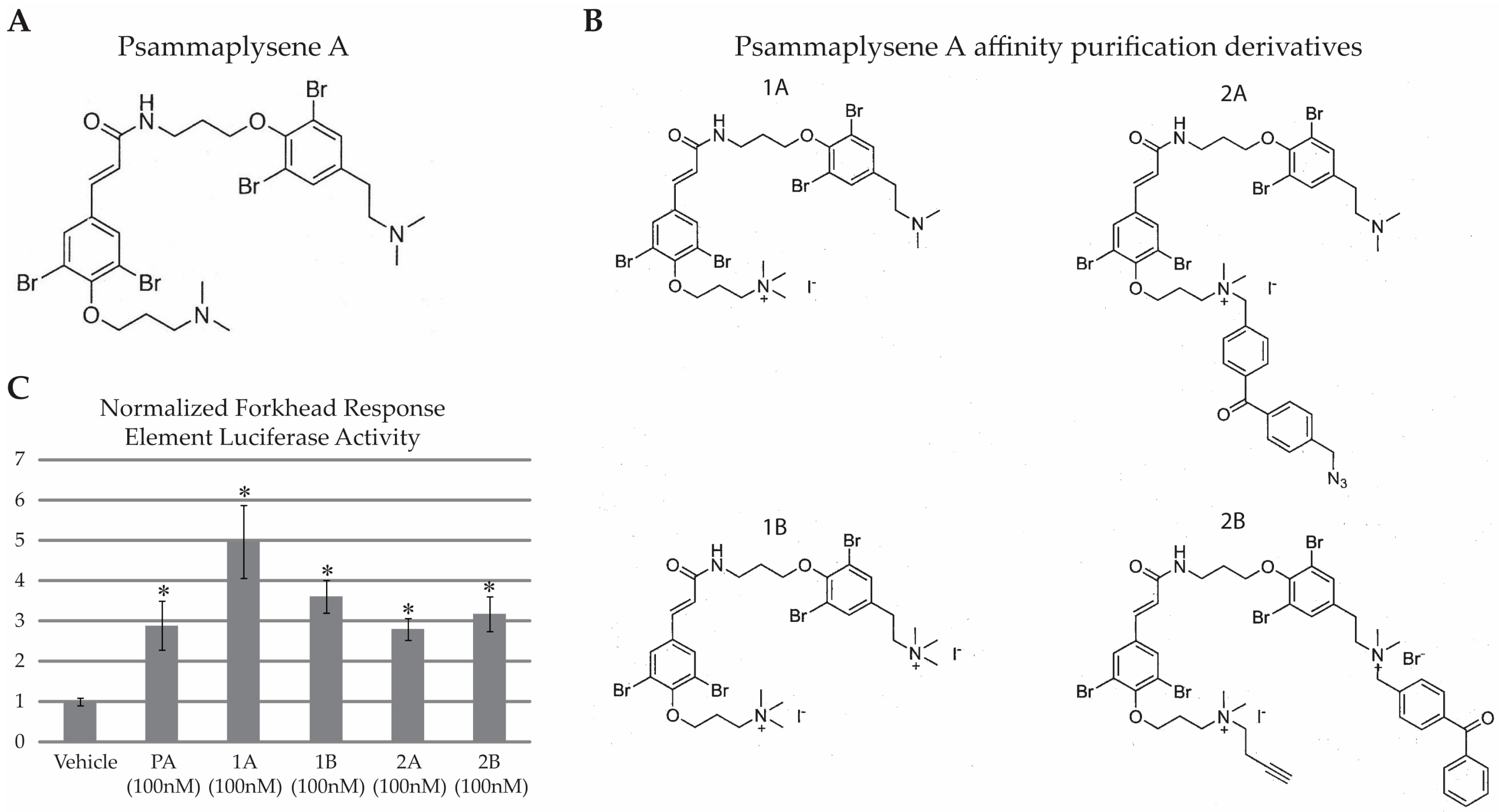
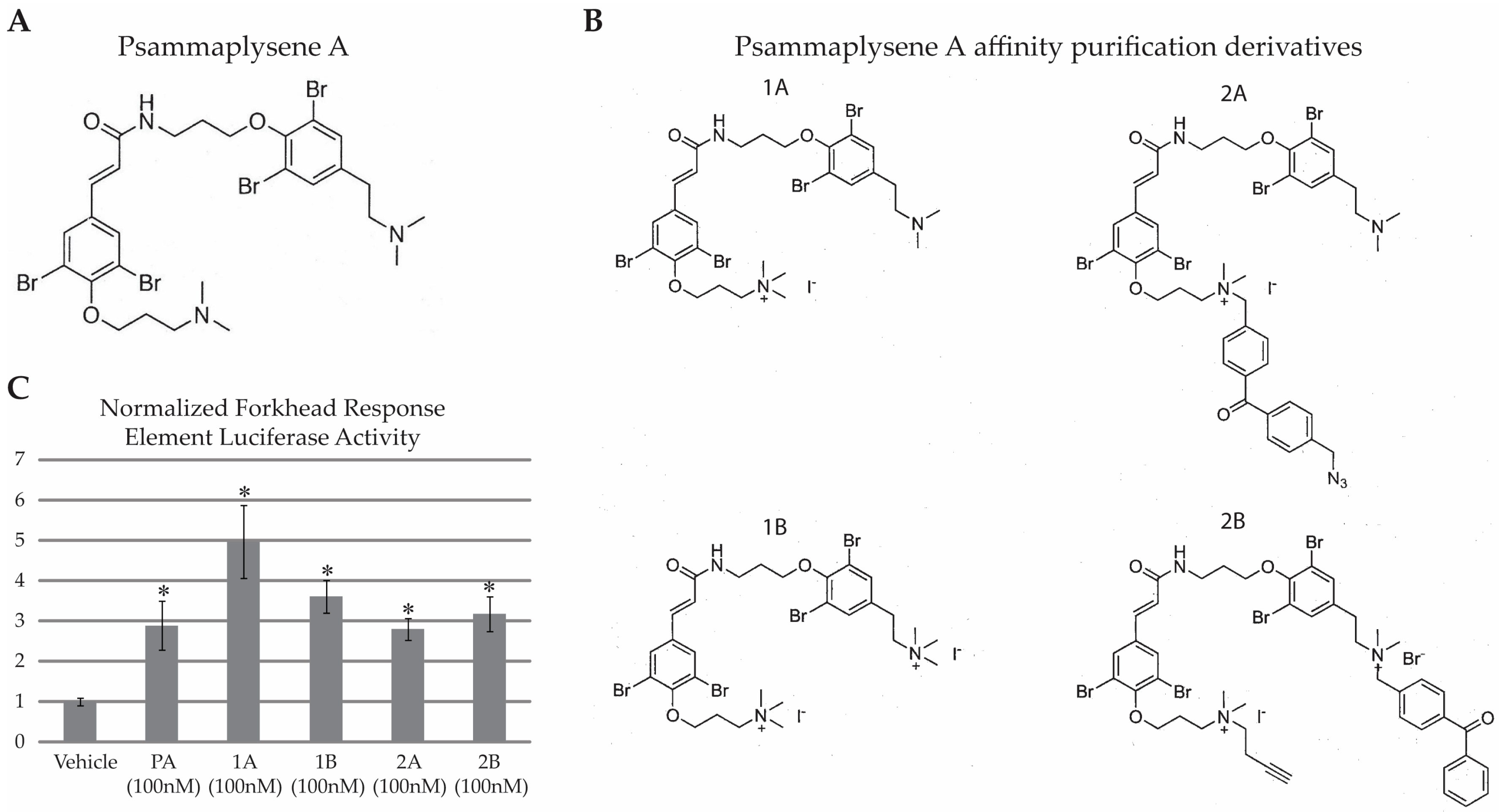
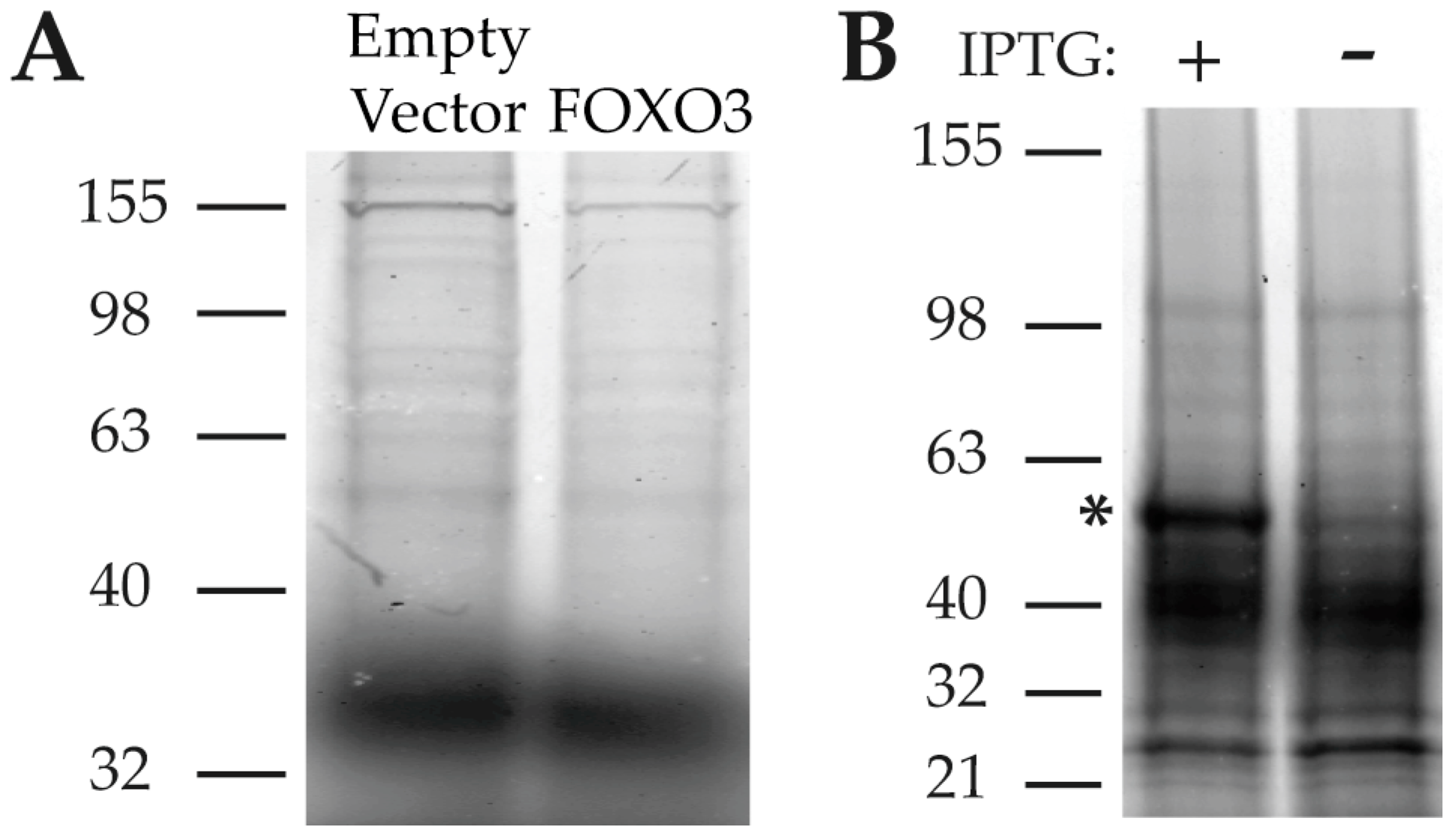
2.1. PA Is Not “Drug-Like”
2.2. HNRNPK Is a Target of PA Based on Two Parallel Target Purification Strategies
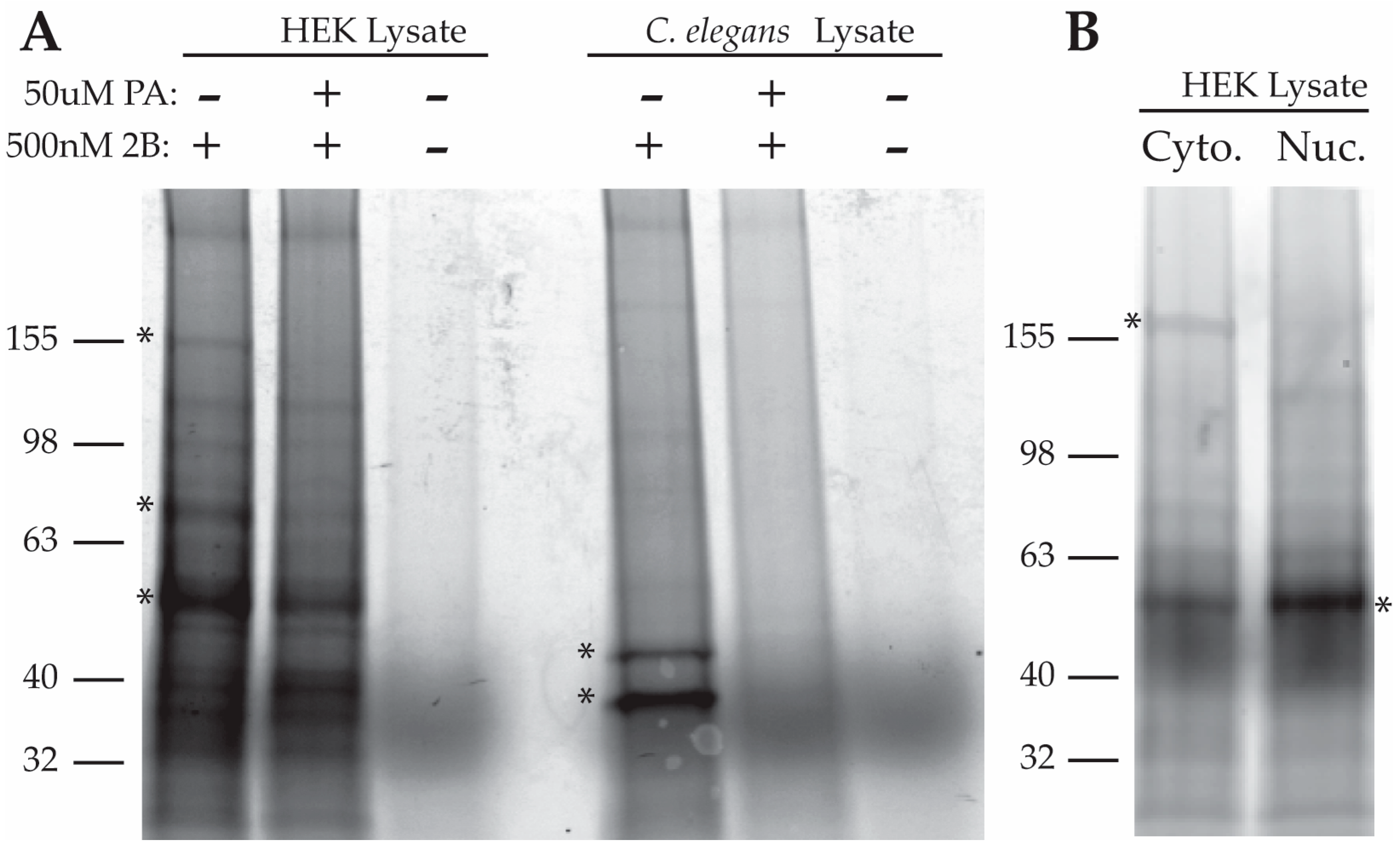
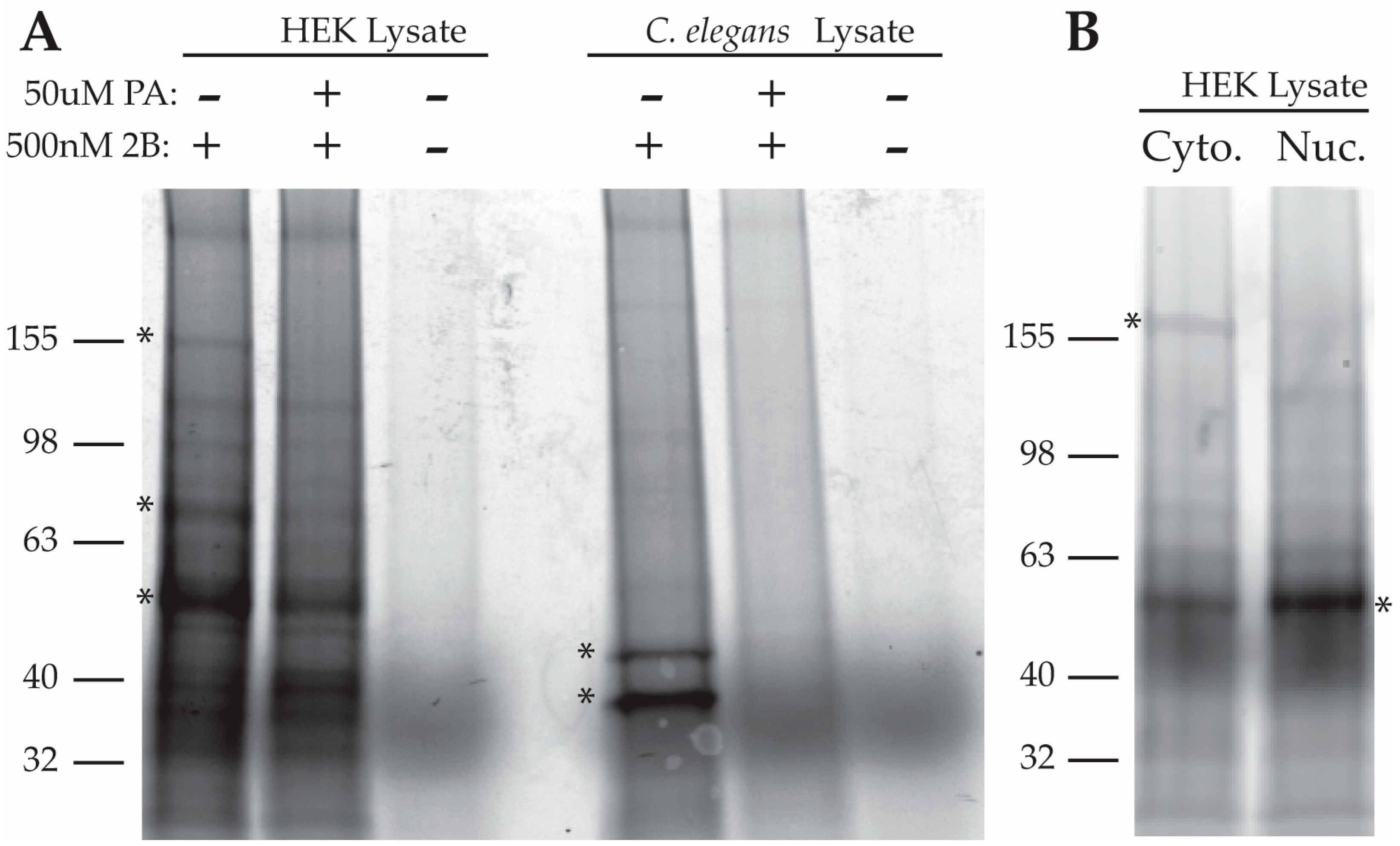
2.2.1. Purification of Proteins Associated with a Photo-Crosslinkable PA Derivative
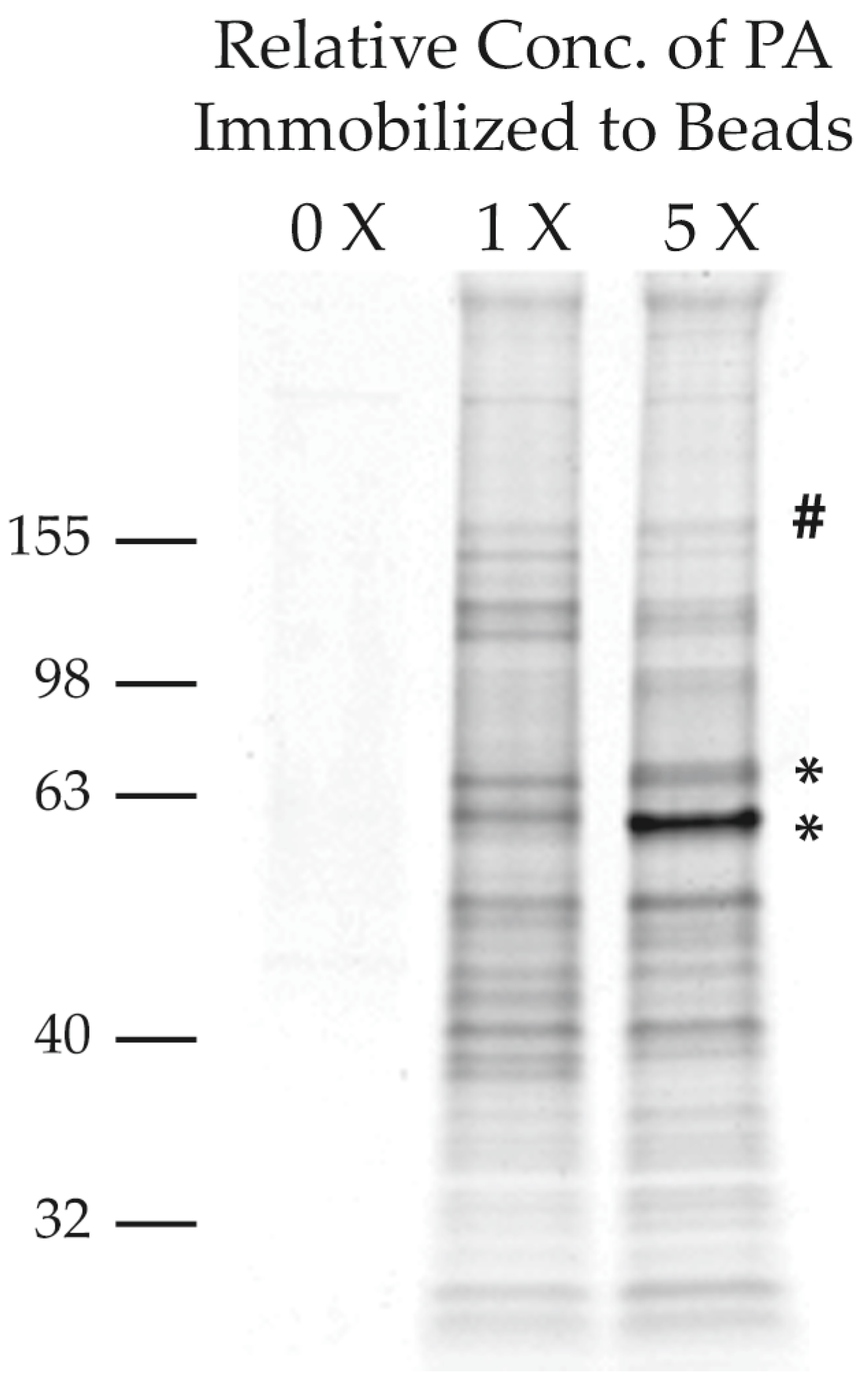
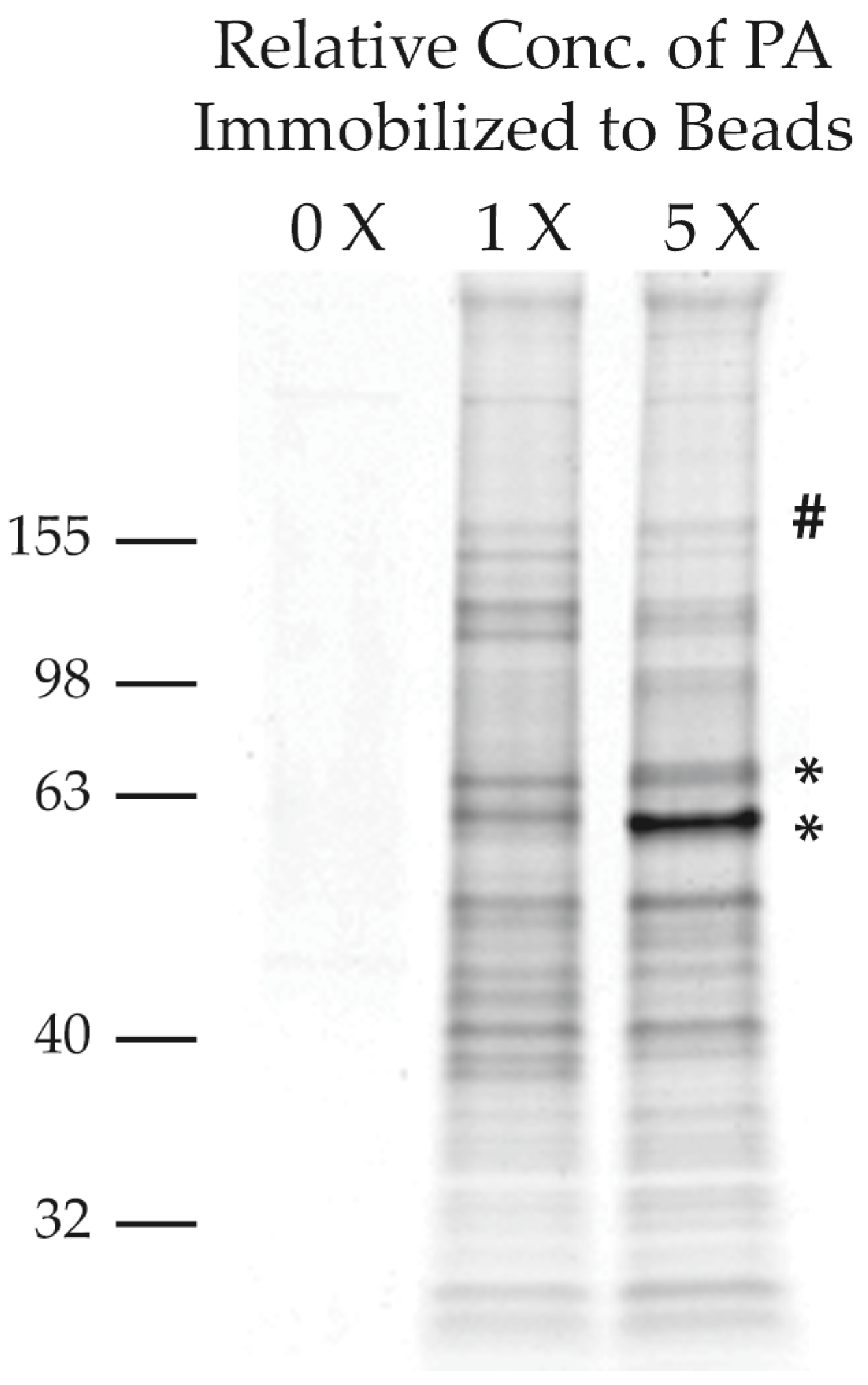
2.2.2. Purification of PA-Interacting Proteins with PA-Coupled Nano Beads
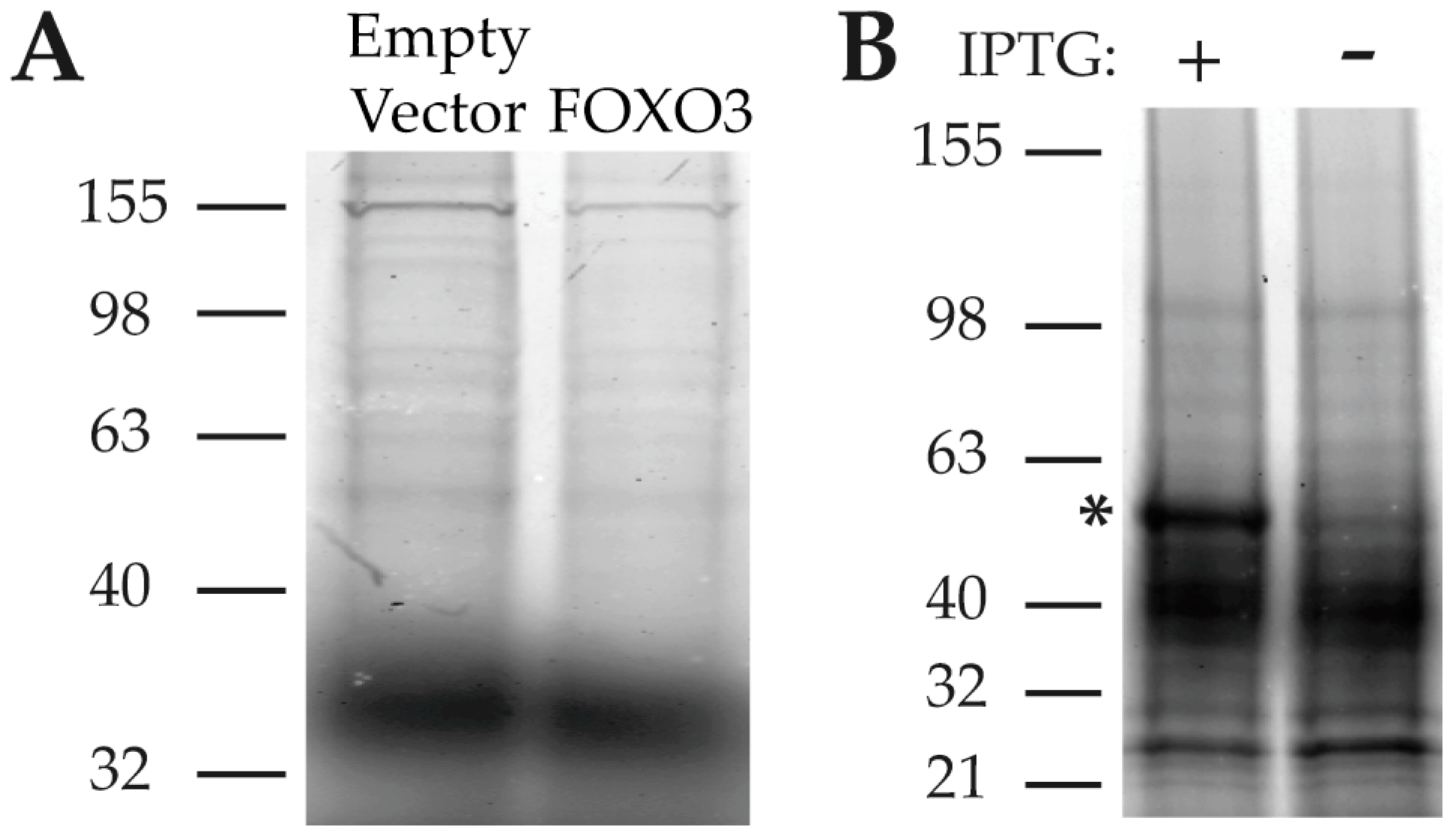
2.2.3. Analysis of 2B and PA-FG Interacting Proteins
2.2.4. Screening of PA-Interacting Candidate Proteins
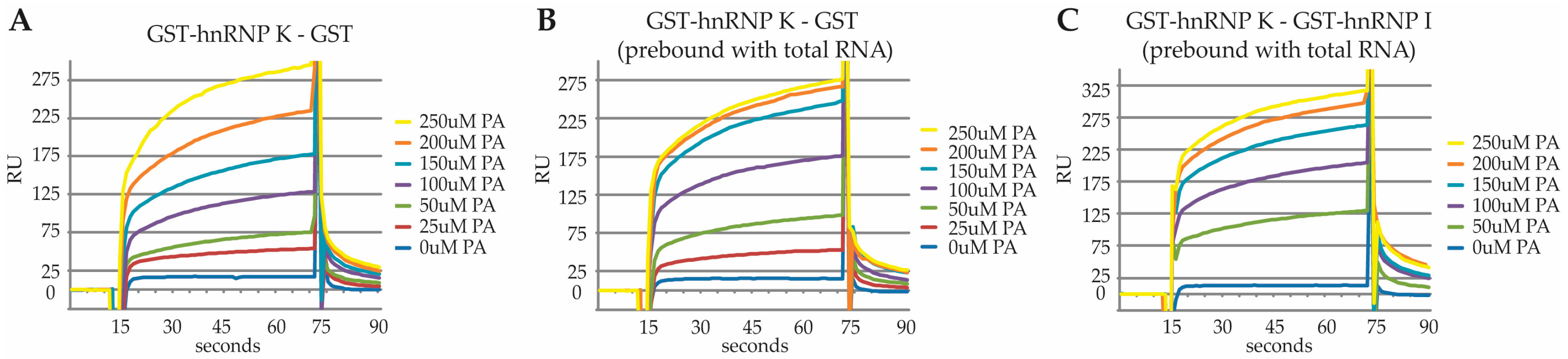
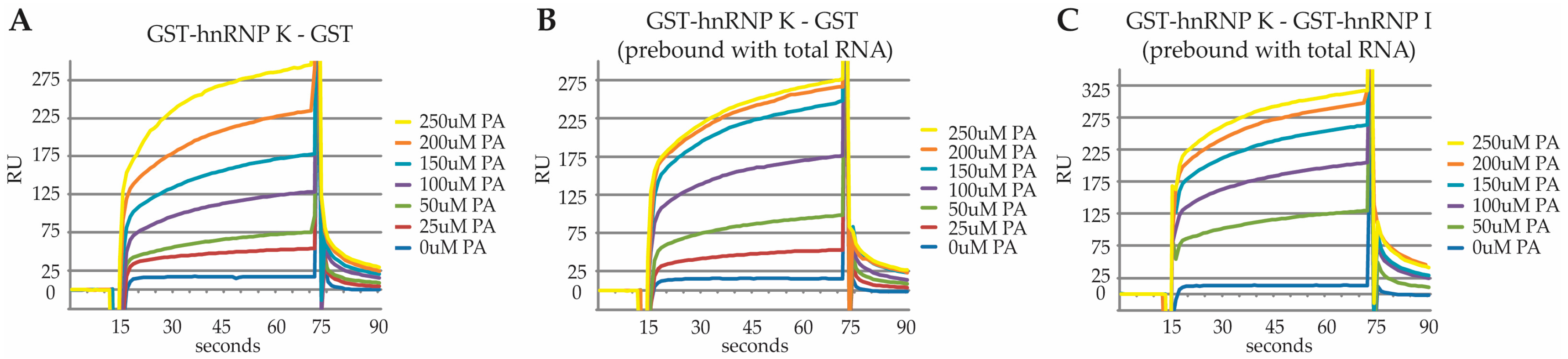
2.3. Characterization of the PA Interaction with HNRNPK by Surface Plasmon Resonance
3. Discussion
4. Materials and Methods
LC–MS/MS Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Georgiades, S.N.; Clardy, J. Total synthesis of psammaplysenes A and B, naturally occurring inhibitors of FOXO1a nuclear export. Org. Lett. 2005, 7, 4091–4094. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Pincus, Z.; Slack, F.J. Longevity and stress in Caenorhabditis elegans. Aging 2011, 3, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Boccitto, M.; Lamitina, T.; Kalb, R.G. Daf-2 Signaling Modifies Mutant SOD1 Toxicity in C. elegans. PLoS ONE 2012, 7, e33494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Mullane, P.C.; Periz, G.; Wang, J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet. 2011, 20, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Mojsilovic-Petrovic, J.; Nedelsky, N.; Boccitto, M.; Mano, I.; Georgiades, S.N.; Zhou, W.; Liu, Y.; Neve, R.L.; Taylor, J.P.; Driscoll, M.; et al. FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 8236–8247. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Petit, J.; Meurice, N.; Kaiser, C.; Maggiora, G. Softening the Rule of Five-where to draw the line? Bioorg. Med. Chem. 2012, 20, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Kabe, Y.; Hatakeyama, M.; Yamaguchi, Y.; Handa, H. Development and application of high-performance affinity beads: Toward chemical biology and drug discovery. Chem. Rec. 2009, 9, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Boccitto, M.; Kalb, R.G. Regulation of Foxo-dependent transcription by post-translational modifications. Curr. Drug Targets 2011, 12, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, G.; Philipson, L.; Mattaj, I.W. Ribonucleoprotein particles in cellular processes. J. Cell Biol. 1988, 106, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Bomsztyk, K.; Denisenko, O.; Ostrowski, J. hnRNP K: One protein multiple processes. BioEssays 2004, 26, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Mikula, M.; Dzwonek, A.; Karczmarski, J.; Rubel, T.; Dadlez, M.; Wyrwicz, L.S.; Bomsztyk, K.; Ostrowski, J. Landscape of the hnRNP K protein-protein interactome. Proteomics 2006, 6, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, C.; Kirby, J.; Highley, J.R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; McDermott, C.J.; Shaw, P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Troakes, C.; Maekawa, S.; Wijesekera, L.; Rogelj, B.; Siklos, L.; Bell, C.; Smith, B.; Newhouse, S.; Vance, C.; Johnson, L.; et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 2012, 32, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, G.; Matunis, M.J.; Pinol-Roma, S.; Burd, C.G. hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef] [PubMed]
- Thisted, T.; Lyakhov, D.L.; Liebhaber, S.A. Optimized RNA targets of two closely related triple KH domain proteins, heterogeneous nuclear ribonucleoprotein K and alphaCP-2KL, suggest Distinct modes of RNA recognition. J. Biol. Chem. 2001, 276, 17484–17496. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.Y.; Lin, W.J.; Fu, S.L.; Lin, C.H. Direct mass-spectrometric identification of Arg296 and Arg299 as the methylation sites of hnRNP K protein for methyltransferase PRMT1. Protein J. 2007, 26, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Nagata, K.; Suzuki, N.; Yokoyama, R.; Yamanaka, Y.; Kitamura, H.; Hirano, H.; Ohara, O. Characterization of multiple alternative forms of heterogeneous nuclear ribonucleoprotein K by phosphate-affinity electrophoresis. Proteomics 2010, 10, 3884–3895. [Google Scholar] [CrossRef] [PubMed]
- Van Seuningen, I.; Ostrowski, J.; Bustelo, X.R.; Sleath, P.R.; Bomsztyk, K. The K protein domain that recruits the interleukin 1-responsive K protein kinase lies adjacent to a cluster of c-Src and Vav SH3-binding sites. Implications that K protein acts as a docking platform. J. Biol. Chem. 1995, 270, 26976–26985. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef] [PubMed]
- Nishio, K.; Masaike, Y.; Ikeda, M.; Narimatsu, H.; Gokon, N.; Tsubouchi, S.; Hatakeyama, M.; Sakamoto, S.; Hanyu, N.; Sandhu, A.; et al. Development of novel magnetic nano-carriers for high-performance affinity purification. Coll. Surf. B Biointerfaces 2008, 64, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass Spectrometric Sequencing of Proteins from Silver-Stained Polyacrylamide Gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Ali-Khan, N.; Echan, L.A.; Levenkova, N.; Rux, J.J.; Speicher, D.W. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 2005, 5, 3329–3342. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biotinylated 2B Pulldown | |||||||
| Term | Count | % | List Total | Pop Hits | Pop Total | Fold Enrichment | FDR |
| GO: 0016071~mRNA metabolic process | 14 | 45.16 | 31 | 370 | 13,528 | 16.51 | 4.04 × 10−10 |
| GO: 0008380~RNA splicing | 13 | 41.94 | 31 | 284 | 13,528 | 19.98 | 4.74 × 10−10 |
| GO: 0006397~mRNA processing | 13 | 41.94 | 31 | 321 | 13,528 | 17.67 | 2.02 × 10−9 |
| GO: 0000377~RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 10 | 32.26 | 31 | 153 | 13,528 | 28.52 | 3.68 × 10−8 |
| GO: 0000398~nuclear mRNA splicing, via spliceosome | 10 | 32.26 | 31 | 153 | 13,528 | 28.52 | 3.68 × 10−8 |
| GO: 0000375~RNA splicing, via transesterification reactions | 10 | 32.26 | 31 | 153 | 13,528 | 28.52 | 3.68 × 10−8 |
| GO: 0006396~RNA processing | 14 | 45.16 | 31 | 547 | 13,528 | 11.17 | 5.63 × 10−8 |
| PA-FG Bead Pulldown | |||||||
| Term | Count | % | List Total | Pop Hits | Pop Total | Fold Enrichment | FDR |
| GO: 0006396~RNA processing | 43 | 27.22 | 132 | 547 | 13,528 | 8.06 | 1.23 × 10−23 |
| GO: 0016071~mRNA metabolic process | 36 | 22.78 | 132 | 370 | 13,528 | 9.97 | 4.51 × 10−22 |
| GO: 0006397~mRNA processing | 33 | 20.89 | 132 | 321 | 13,528 | 10.54 | 1.35 × 10−20 |
| GO: 0008380~RNA splicing | 31 | 19.62 | 132 | 284 | 13,528 | 11.19 | 7.54 × 10−20 |
| GO: 0000375~RNA splicing, via transesterification reactions | 22 | 13.92 | 132 | 153 | 13,528 | 14.74 | 1.78 × 10−15 |
| GO: 0000398~nuclear mRNA splicing, via spliceosome | 22 | 13.92 | 132 | 153 | 13,528 | 14.74 | 1.78 × 10−15 |
| GO: 0000377~RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 22 | 13.92 | 132 | 153 | 13528 | 14.74 | 1.78 × 10−15 |
| GO: 0010608~posttranscriptional regulation of gene expression | 14 | 8.86 | 132 | 211 | 13,528 | 6.80 | 0.000203 |
| GO: 0043489~RNA stabilization | 6 | 3.80 | 132 | 15 | 13,528 | 40.99 | 0.000345 |
| GO: 0048255~mRNA stabilization | 6 | 3.80 | 132 | 15 | 13,528 | 40.99 | 0.000345 |
| GO: 0043488~regulation of mRNA stability | 6 | 3.80 | 132 | 22 | 13,528 | 27.95 | 0.002864 |
| GO: 0043487~regulation of RNA stability | 6 | 3.80 | 132 | 24 | 13,528 | 25.62 | 0.004552 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccitto, M.; Lee, N.; Sakamoto, S.; Spruce, L.A.; Handa, H.; Clardy, J.; Seeholzer, S.H.; Kalb, R.G. The Neuroprotective Marine Compound Psammaplysene A Binds the RNA-Binding Protein HNRNPK. Mar. Drugs 2017, 15, 246. https://doi.org/10.3390/md15080246
Boccitto M, Lee N, Sakamoto S, Spruce LA, Handa H, Clardy J, Seeholzer SH, Kalb RG. The Neuroprotective Marine Compound Psammaplysene A Binds the RNA-Binding Protein HNRNPK. Marine Drugs. 2017; 15(8):246. https://doi.org/10.3390/md15080246
Chicago/Turabian StyleBoccitto, Marco, Nayoung Lee, Satoshi Sakamoto, Lynn A. Spruce, Hiroshi Handa, Jon Clardy, Steven H. Seeholzer, and Robert G. Kalb. 2017. "The Neuroprotective Marine Compound Psammaplysene A Binds the RNA-Binding Protein HNRNPK" Marine Drugs 15, no. 8: 246. https://doi.org/10.3390/md15080246
APA StyleBoccitto, M., Lee, N., Sakamoto, S., Spruce, L. A., Handa, H., Clardy, J., Seeholzer, S. H., & Kalb, R. G. (2017). The Neuroprotective Marine Compound Psammaplysene A Binds the RNA-Binding Protein HNRNPK. Marine Drugs, 15(8), 246. https://doi.org/10.3390/md15080246