2. Results and Discussion
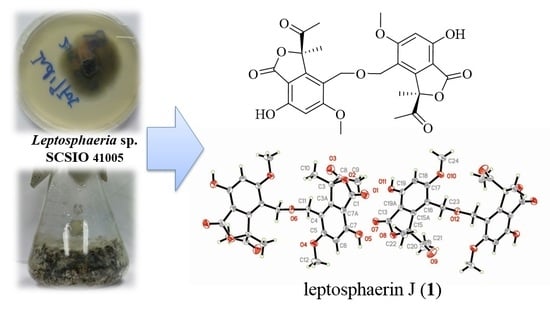
The culture broth of fungus Leptosphaeria sp. SCSIO 41005 was extracted with ethyl acetate at room temperature. The solvent was evaporated in vacuo to afford a crude extract, which was subjected to various chromatographic methods to yield compounds 1–16.
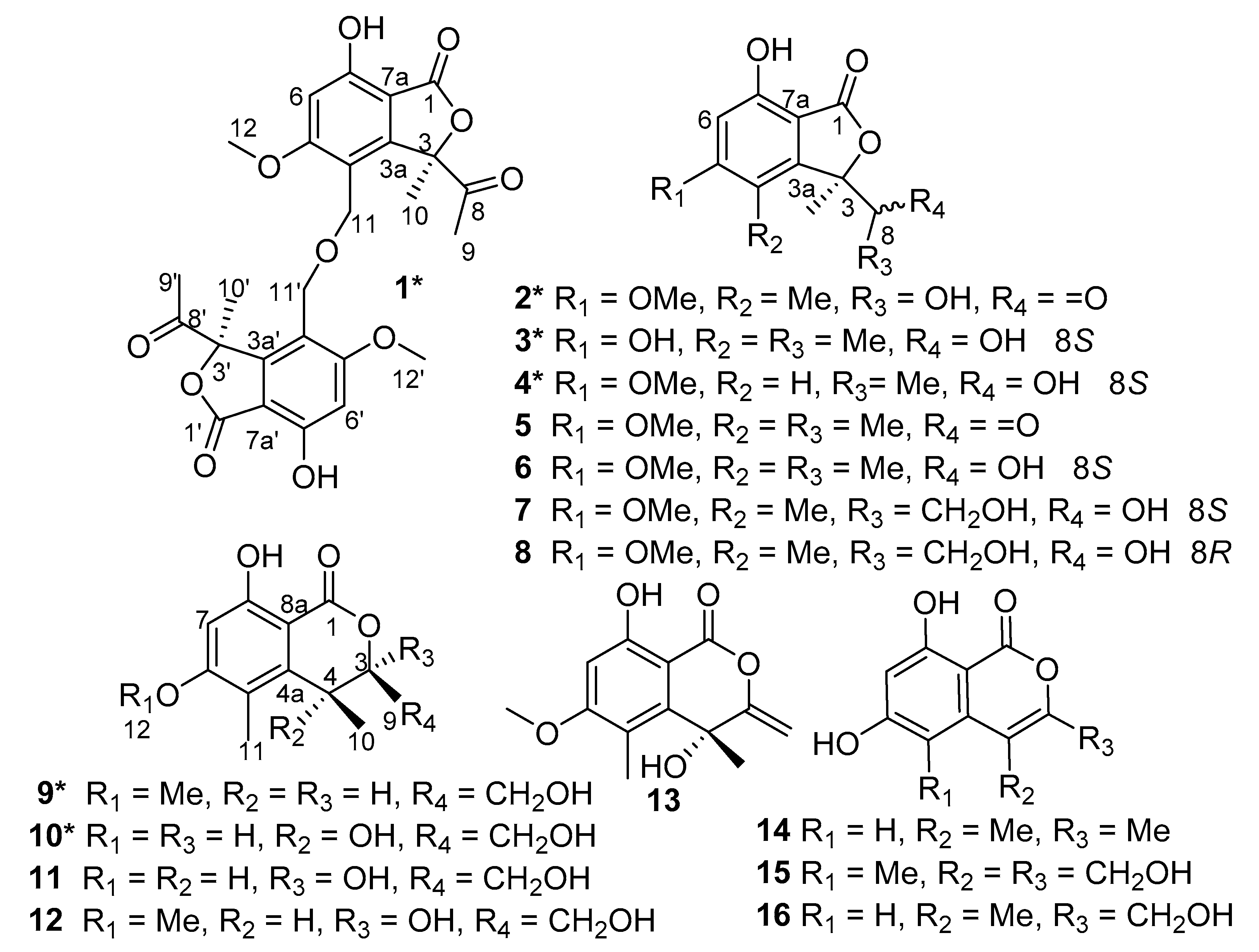
Leptosphaerin J (
1) was isolated as colorless crystals. A molecular formula of C
26H
26O
11 was derived from a sodium adduct ion peak at
m/
z 537.1361 [M + Na]
+ (calcd. for 537.1373) in the (+)-HRESIMS. The
1H NMR spectrum (
Table 1) of
1 exhibited two primary methyls (
δH 1.69, 1.94), one oxygen-bearing methylene (
δH 4.25, 4.43), one methoxy (
δH 3.90), and one aromatic proton (
δH 6.59). The analysis of the
13C NMR and DEPT spectra (
Table 1) revealed thirteen carbons, ascribed to three methyls (one oxygenated), one oxygenated methylene, one aromatic methine and eight non-protonated carbons (one oxygenated, one carbonyl, one
α,
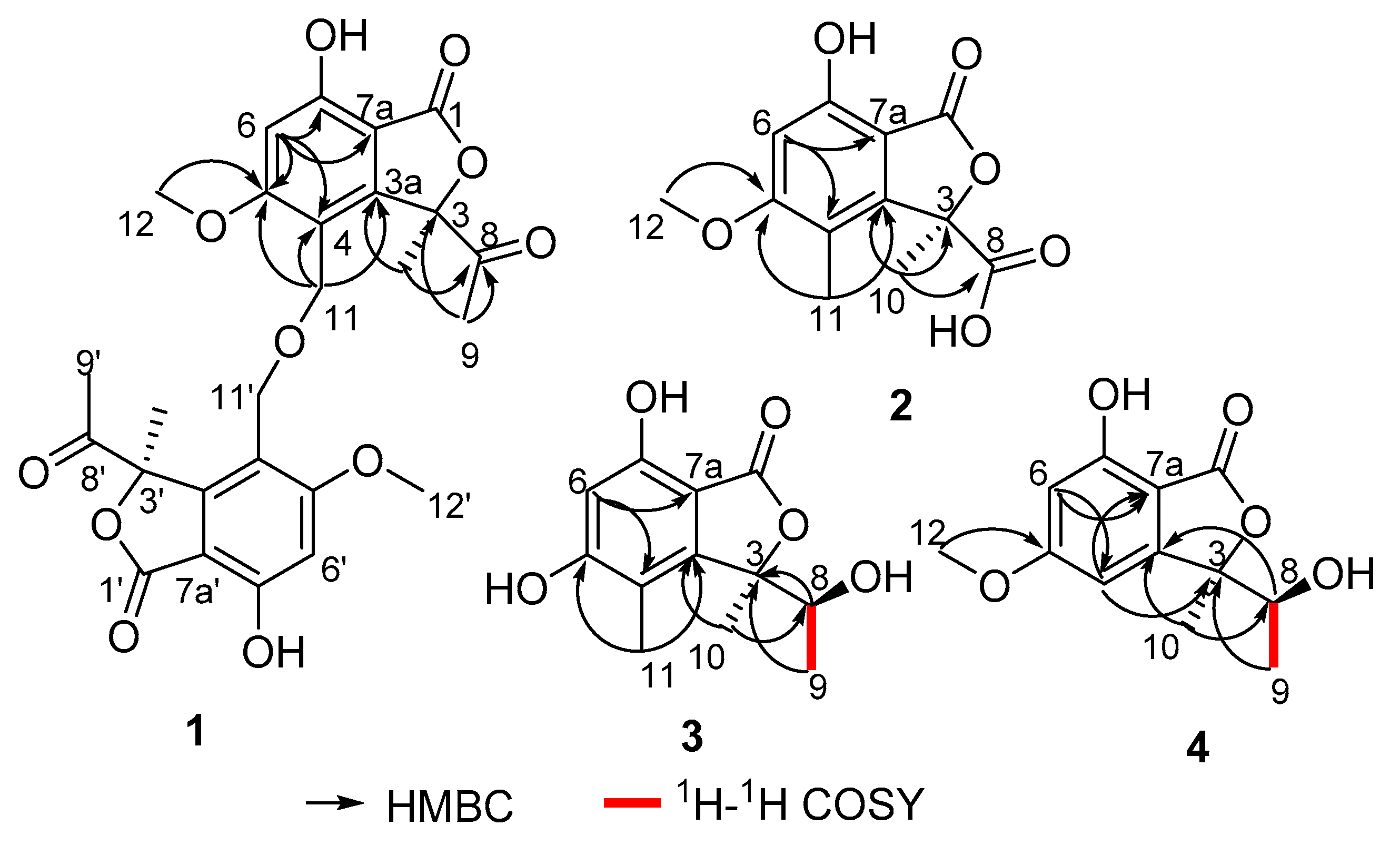
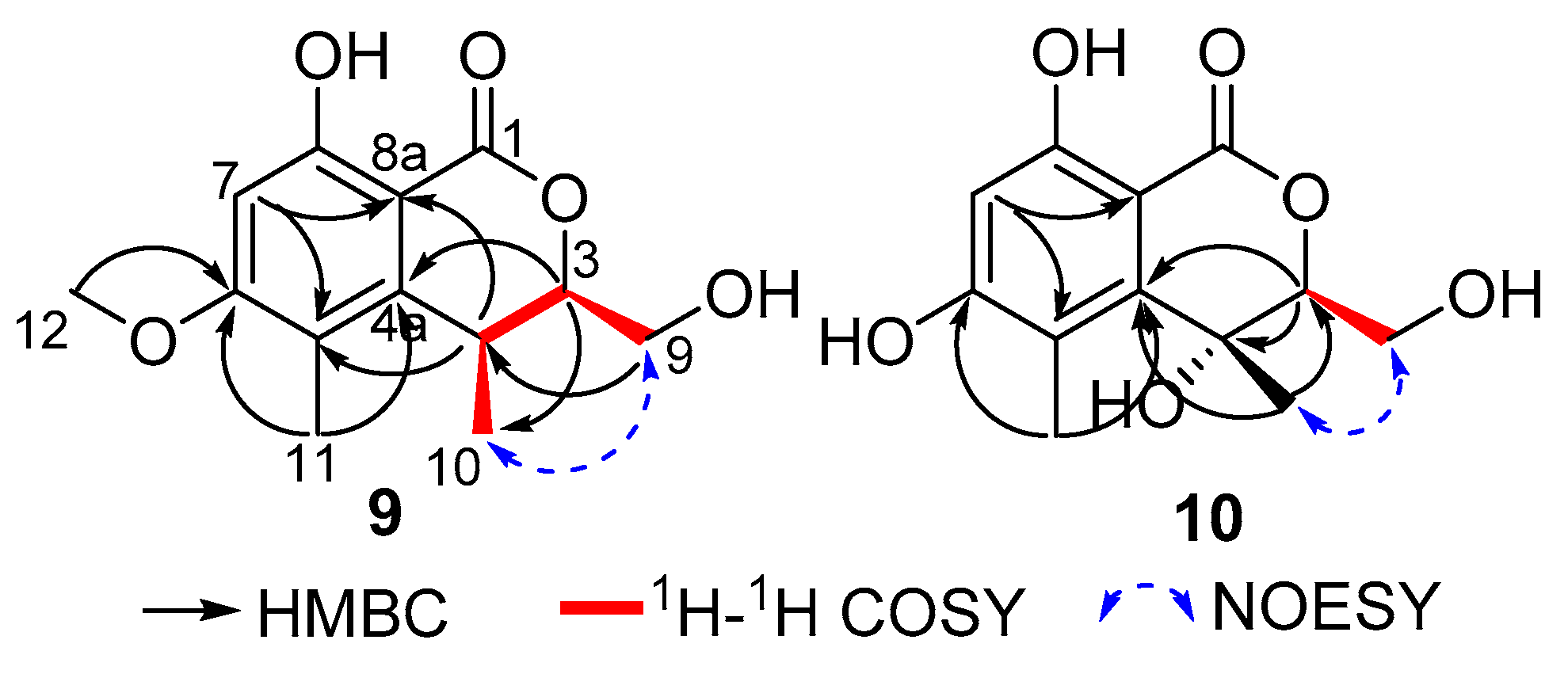
β-conjugated carbonyl and five aromatics). The HMBC spectrum of
1 showed the correlations from H-6 to C-4, C-5, C-7 and C-7a, from H
3-9 to C-3 and C-8, from H
3-10 to C-3, C-3a and C-8, in turn coupled with H
3-9 (
Figure 2). All the above data suggested that
1 presented an isobenzofuranone skeleton, which is similar to (
R)-3-acetyl-7-hydroxy-5-methoxy-3,4-dimethylisobenzofuran-1(3H)-one (
5) [
6], the known compound we also obtained, except the primary methyl in
5 was replaced with an oxygenated methylene in
1. Due to the molecular formula of C
26H
26O
11 given by HRESIMS, compound
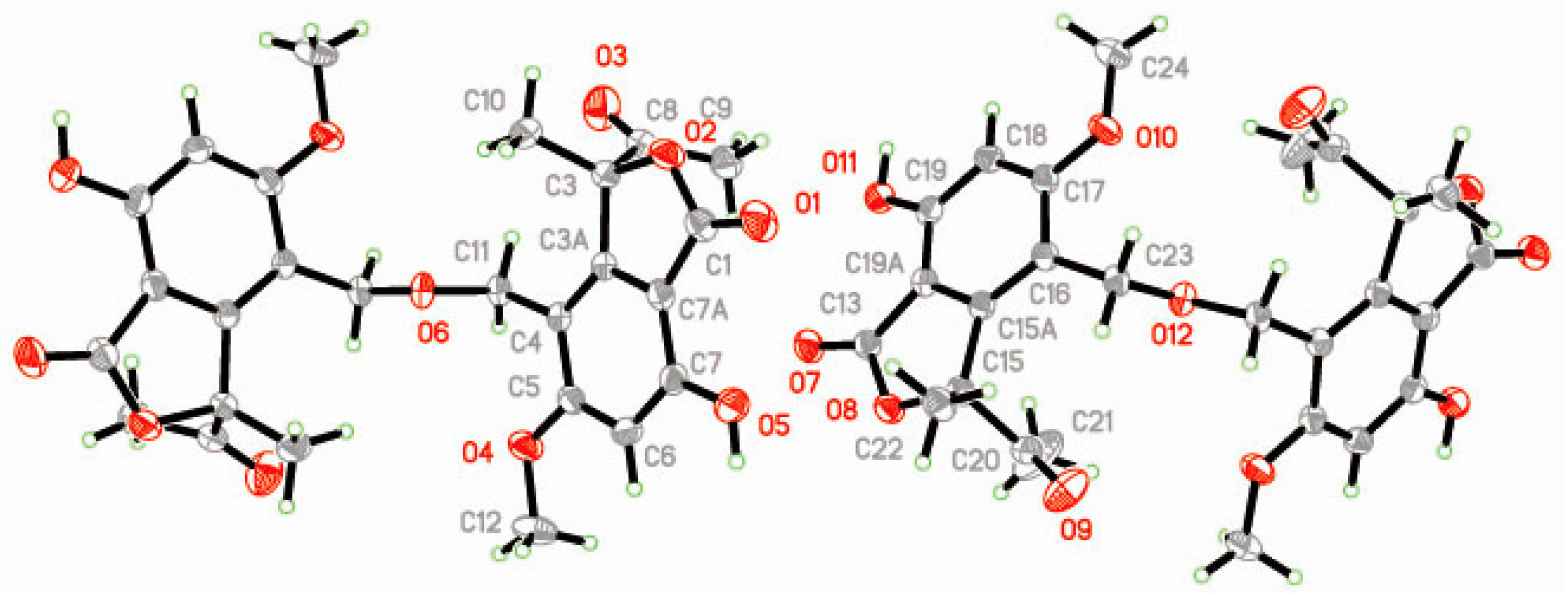
1 was supposed to be a centrosymmetric dimer skeleton of isobenzofuranone. The dimer skeleton, conjugated by an ether bond on CH
2-11 as shown in
Figure 1, was confirmed by X-ray diffraction analysis, using the anomalous scattering of Cu K
α radiation yielding a Flack parameter of 0.00 (5) (
Figure 3).
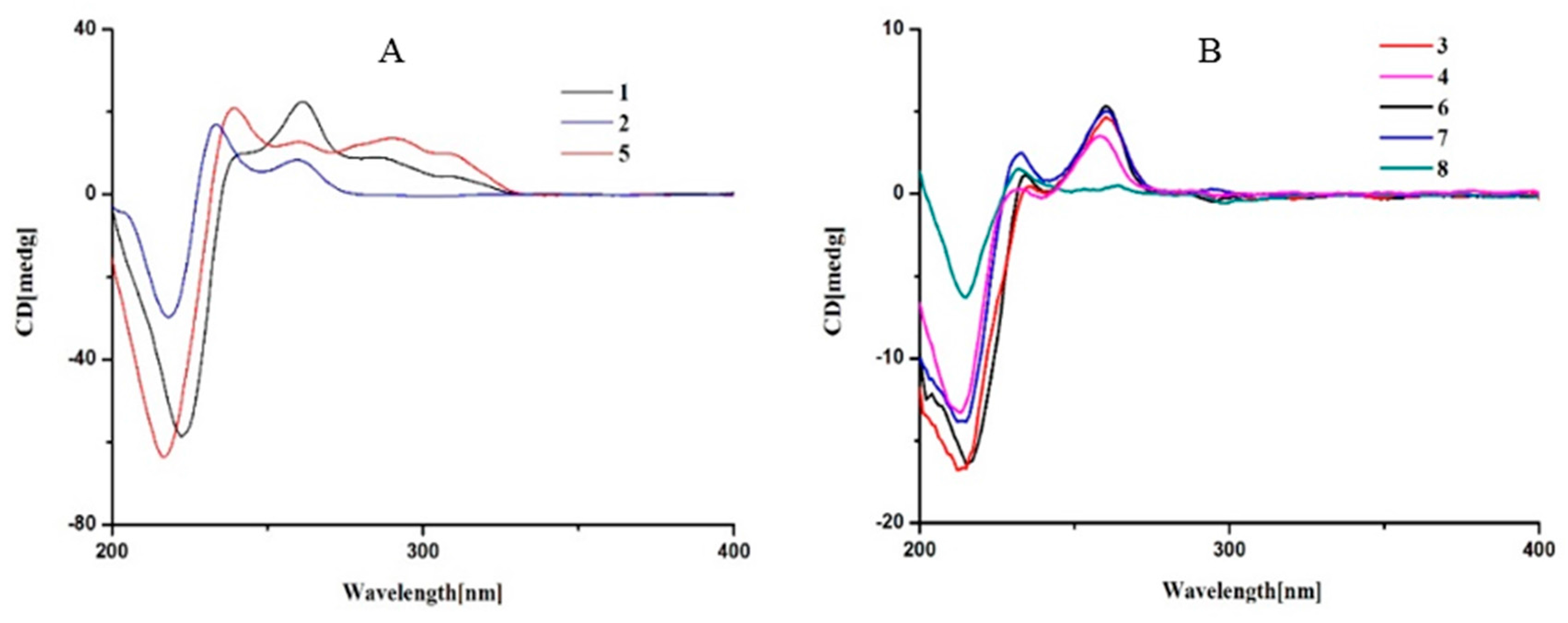
Compounds
1 and
5 showed similar optical rotation (
= +87.3° (
c 0.06, MeOH) for
1, and
= +108.4° (
c 0.15, MeOH) for
5) and similar Cotton effects in electronic circular dichroism (ECD) spectrum (
Figure 4), indicating
1 and
5 possessed the same absolute configuration of
R, which was later confirmed by the X-ray diffraction experiment (
Figure 3).
Leptosphaerin K (
2) was obtained as a colorless oil. The molecular formula was deduced to be C
12H
12O
6 by the HRESIMS peak at
m/
z 251.0565 [M − H]
− (calcd. for C
12H
11O
6, 251.0556). The
1H and
13C NMR signals were assigned by combination of analyses of DEPT, HSQC and HMBC spectra. The
1H NMR spectrum (
Table 1) of
2 exhibited two primary methyls (
δH 1.78, 2.16), one methoxy (
δH 3.82) and one aromatic proton (
δH 6.38). The
13C NMR and DEPT spectra (
Table 1) revealed twelve carbons, including three methyls (one oxygenated), one aromatic methine and eight non-protonated carbons (one oxygenated, two carbonyls and five aromatics). This evidence, including UV and IR spectra (
Supplementary Materials Figures S16 and S17), revealed that there was also an isobenzofuranone unit in compound
2. Furthermore, the HMBC spectrum of
2 also showed the correlations from H-6 to C-4 and C-7a, from H
3-10 to C-3a and C-8, from H
3-11 to C-3a, C-4 and C-5, which implied that the isobenzofuranone structure of
2 was identical to that of
1 and
5. The main difference was the replacement of a methyl group (C-9) in
5 by a hydroxyl group in
2. Besides, the similar Cotton effects of
2 and
5 in their ECD spectra (
Figure 4) indicated their same absolute configuration at C-3. Thus, it was unambiguous to elucidate the structure of
2 as shown in
Figure 1.
Leptosphaerin L (
3) was obtained as colorless oil. The HRESIMS ion peak at
m/
z 237.0775 [M − H]
− (calcd. for C
12H
13O
5, 237.0763) and the NMR spectroscopy revealed a molecular formula of C
12H
14O
5. The
1H NMR spectrum (
Table 1) of
3 exhibited three primary methyls (
δH 0.85, 1.71, 2.09), one oxymethine (
δH 4.22) and one aromatic proton (
δH 6.36). The
13C NMR and DEPT spectra (
Table 1) revealed twelve carbons, assigned to three methyls, one oxymethine, one aromatic methine and seven non-protonated carbons (one oxygenated, one carbonyl and five aromatics). All these data, as well as UV and IR spectra (
Supplementary Materials Figures S24 and S25), suggested that
3 also had an isobenzofuranone moiety. The HMBC spectrum of
3 showed the correlations from H-6 to C-4 and C-7a, from H
3-9 to C-3 and C-8, from H
3-10 to C-3a and C-8, and from H
3-11 to C-3a, C-4 and C-5, which was in accordance with the structure of
6 (
Figure 2). The main difference was the replacement of a methoxy group (C-5) in
6 by a hydroxyl group in
3. Hence, the planar structure of
3 was deduced as shown in
Figure 1. Moreover, the similar optical rotation (
= −44.7° (
c 0.15, MeOH) for
3, and
= −35.4° (
c 0.68, MeOH) for
6) and almost identical curves in ECD spectrum (
Figure 4) indicated compounds
3 and
6 had the same absolute configuration (3
R, 8
S) at the position C-3 and C-8.
Leptosphaerin M (
4) was obtained as a colorless oil. Its HRESIMS ([M − H]
−, 237.0775; calcd. for C
12H
13O
5−, 237.0763) data were in agreement with the molecular formula C
12H
14O
5. The
1H NMR spectrum (
Table 1) of
4 exhibited two primary methyls (
δH 1.05, 1.59), one methoxy (
δH 3.84), one oxymethine (
δH 3.90) and two aromatic protons (
δH 6.39, 6.51). The
13C NMR and DEPT spectra (
Table 1) revealed the presence of twelve carbon resonances, involving three methyls (one oxygenated), one oxymethine, two aromatic methines and six non-protonated carbons (one oxygenated, one carbonyl and four aromatics). Analyses of the 2D NMR (HSQC and HMBC) data established an isobenzofuranone nucleus. This assignment was evident from the HMBC correlations from H-4 to C-3, C-6 and C-7a, from H-6 to C-4 and C-7a, from H
3-9 to C-3 and C-8, from H
3-10 to C-3, C-3a and C-8, and from H
3-12 to C-5 (
Figure 2). The above NMR data of
4 showed that it shared the same isobenzofuranone skeleton as
3. The main distinctions were the replacement of a hydroxyl group (OH-5) and a methyl group (C-4) in
3 by a methoxy and a proton in
4, respectively. Consequently, the planar structure of
4 was established as shown in
Figure 1. In the same way, the similar optical rotation value (
= −32.6° (
c 0.10, MeOH) for
4) and almost identical curves in ECD spectrum (
Figure 4) indicated that
4 displayed the same absolute configuration of position C-3 and C-8 (3
R, 8
S) with compounds
3 and
6.
Compounds
7 and
8, obtained as a pair of structurally-known epimers, were elucidated as clearanol E (
7) [
19] and clearanol D (
8) [
19], by comparison of their NMR and MS data with reported literature. They were reported as a mixture with undetermined stereochemistry. In a comparison between the ECD spectra of
7 and
8, the main difference was that the positive Cotton effect at 260 nm in
7 was absent in
8 (
Figure 4), which was identical to the reported calculated CD spectra of their congeners (
R,3
1S)-7-hydroxy-3-(1-hydroxyethyl)-5-methoxy-3,4-dimethylisobenzofuran-1(3
H)-one and (
R,3
1R)-7-hydroxy-3-(1-hydroxyethyl)-5-methoxy-3,4-dimethylisobenzofuran-1(3
H)-one, respectively [
24]. Thus, the absolute configuration of clearanol E (
7) and clearanol D (
8) was determined as 3
R, 8
S and 3
R, 8
R, respectively, and that of the former was also confirmed on the basis of X-ray crystallographic experiment with a Flack parameter of 0.04(8) by using the anomalous scattering of Cu K
α radiation (
Supplementary Materials Figure S61).
Clearanol I (
9) was obtained as colorless crystals. It showed a [M + H]
+ ion peak at
m/
z 253.1077 in the positive-ion HRESIMS (calcd. for C
13H
17O
5, 253.1076), appropriate for a molecular formula of C
13H
16O
5. The
1H NMR spectrum (
Table 2) of
9 exhibited signals for two primary methyl groups (
δH 1.07, 2.07), one methoxy group (
δH 3.86), one methylene (
δH 3.90, 3.80), two methines (
δH 3.29, 4.50) and one aromatic proton (
δH 6.43). The
13C NMR and DEPT spectra (
Table 2) exhibited thirteen carbon resonances that consisted of three methyls (one oxygenated), one oxymethylene, three methines (one oxygenated and one aromatic) and six non-protonated carbons (one carbonyl and five olefinics). The above assignment suggested that
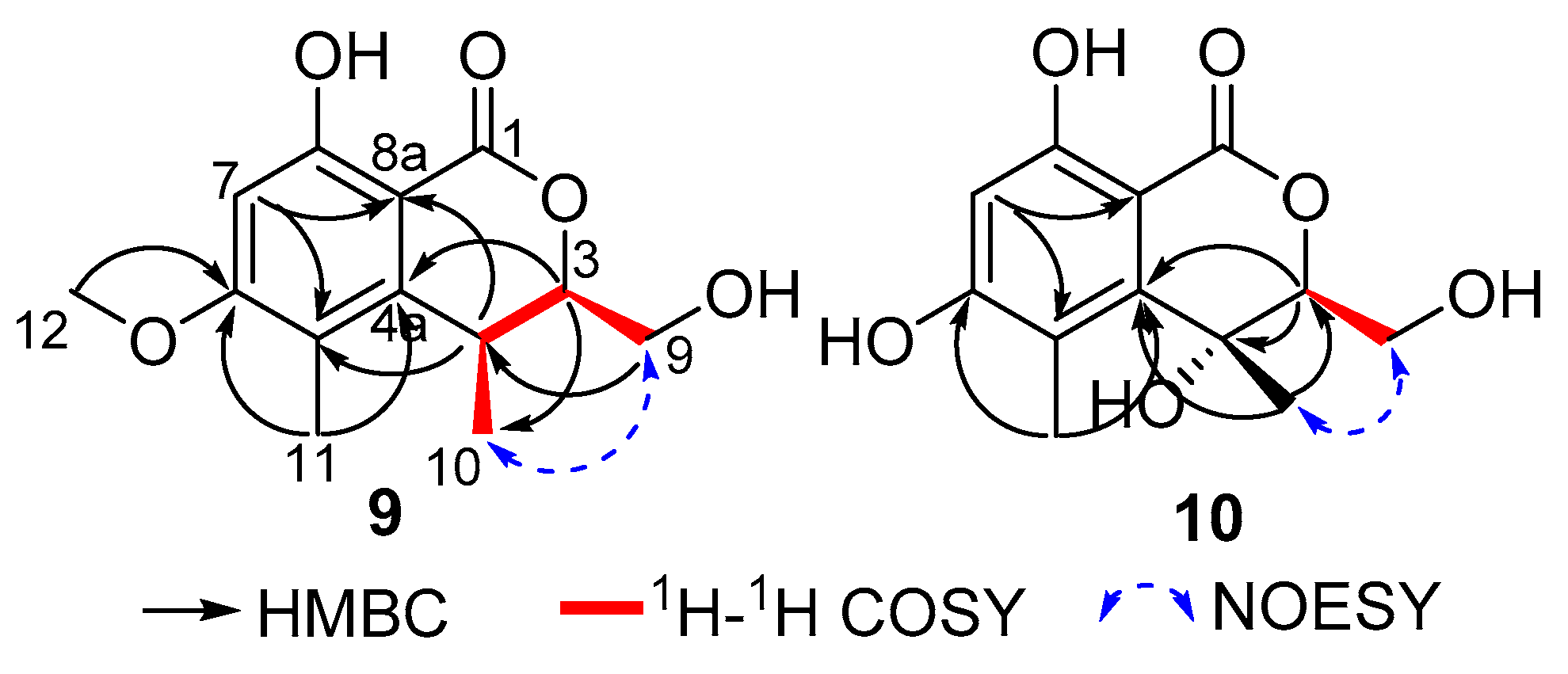
9 presented an isochromenone unit, with structural similarities to clearanol F [
25], which was further evidenced by COSY (H
2-9/H-3/H-4/H
3-10) and HMBC correlations (
Figure 5) from H-7 to C-5, C-6, C-8 and C-8a, from H-3 to C-4a, C-9 and C-10, H-4 to C-4a, C-5, C-8a and C-10, from H
2-9 to C-3 and C-4, from H
3-10 to C-3, C-4 and C-4a, in turn coupled with H
2-9. The above NMR data of
9 showed that it shared the same isochromenone skeleton as clearanol F [
25]. The main difference was the replacement of a hydroxyl group (OH-4) by a proton in
9. Therefore, the planar structure of
9, named as clearanol I, was elucidated as shown in
Figure 1.
In the NOESY experiment of
9 (
Figure 5), the correlation between H
2-9 and H
3-10 suggested that CH
2-9 and CH
3-10 are positioned on the same face. Besides, compound
9 (
= +18.2° (
c 0.19, MeOH)) and clearanol F (
= +18° (
c 0.05, CHCl
3)) shared perfectly identical optical rotation value. Furthermore, in analyzing the ECD spectroscopic data,
9 and clearanol F showed almost identical curves (Cotton effect was negative at 224, 250 nm (
9) and 228, 261 nm (clearanol F), and positive at 237, 270 nm (
9) and 242, 273 nm (clearanol F), respectively). These data indicated
9 and clearanol F possibly possessed similar stereochemistry, which was further determined as 3
R, 4
S on the basis of X-ray crystallographic study with a Flack parameter of −0.03 (5) by using the anomalous scattering of Cu K
α radiation (
Supplementary Materials Figure S62).
Clearanol J (
10) was obtained as colorless crystals and had the molecular formula C
12H
14O
6, as evidenced by HRESIMS ([M − H]
−, 253.0721, calcd. for C
12H
13O
6−, 253.0712) and the NMR data (
Table 2). The
1H NMR spectrum (
Table 2) of
10 exhibited two primary methyls (
δH 1.37, 2.29), one methylene (
δH 3.85, 4.06), one methine (
δH 4.26) and one aromatic proton (
δH 6.28). The analysis of the
13C NMR and DEPT spectra (
Table 2) revealed twelve carbons, including two methyls, one oxymethylene, two methines (one oxygenated and one aromatic) and seven non-protonated carbons (one oxygenated, one carbonyl and five aromatics). These data suggested that
10 presented an isochromenone moiety, which was similar to that of
9 and clearanol F [
25], with the replacement of CH
3O-6 in clearanol F by a hydroxyl group in
10, and this was further confirmed by COSY and HMBC correlations (
Figure 5).
In the NOESY spectra of
10 and
9, there were same correlations between H
2-9 and H
3-10 (
Figure 5), suggesting that CH
2-9 and CH
3-10 are positioned on the same face, both in
10 and
9. Meanwhile, the perfectly identical optical rotation value (
= +17.4° (
c 0.10, MeOH)) and similar ECD spectroscopic data (Cotton effect was negative at 230, 258 nm, and positive at 242, 276 nm) of
10 with those of
9 and clearanol F, illustrated
10 possessed the same absolute configuration of 3
R, 4
R with
9 and clearanol F.
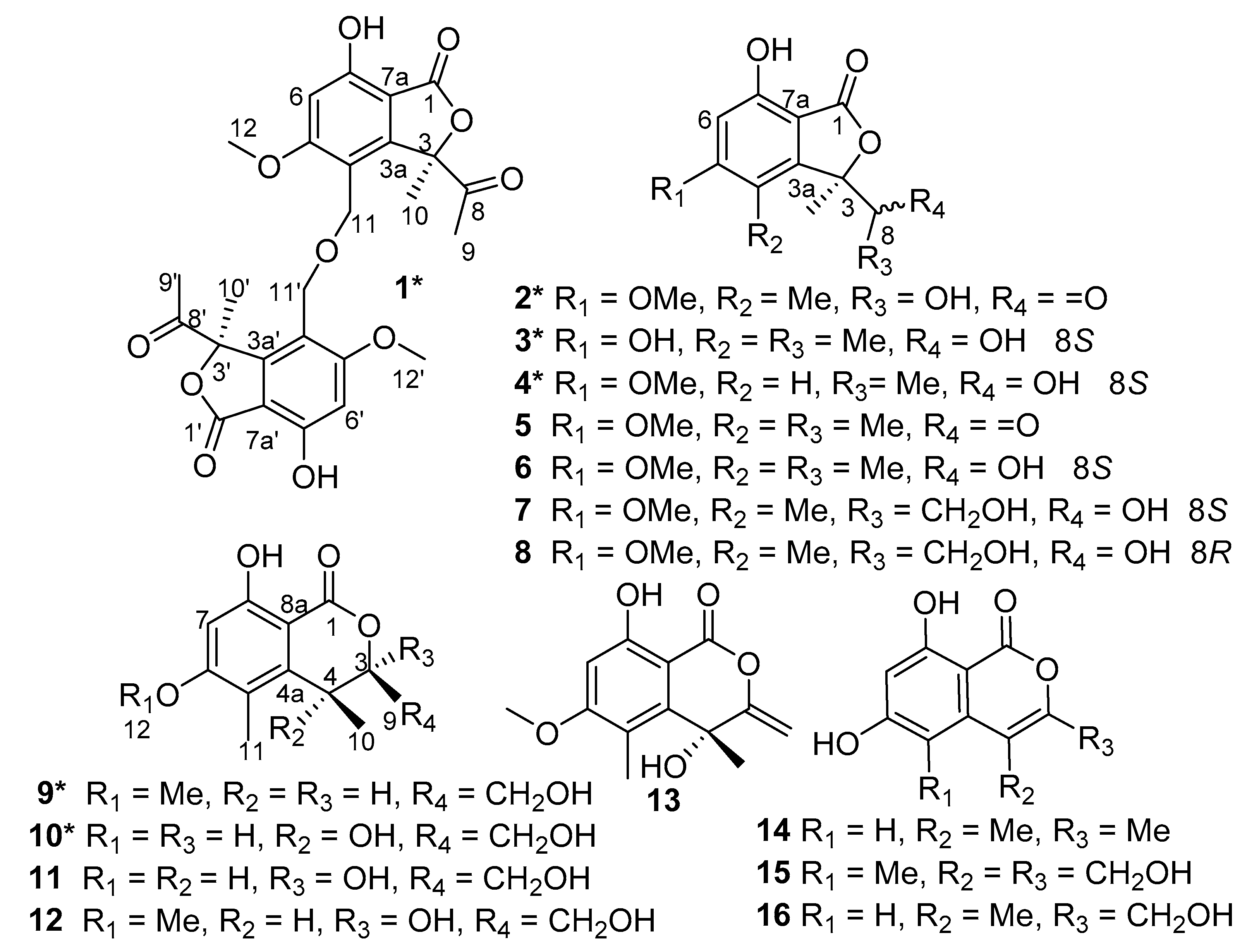
Other eight known isobenzofuranones or isochromenones were elucidated by comparison of their NMR and MS data with reported literature, determined to be (
R)-3-acetyl-7-hydroxy-5-methoxy-3,4-dimethylisobenzofuran-1(3
H)-one (
5) [
6], (3
R,3
1S)-7-hydroxy-3-(1-hydroxyethyl)-5-methoxy-3,4-dimethylisobenzofuran-1(3
H)-one (
6) [
6], dothideomynone A (
11) [
20], 3,8-dihydroxy-3-hydroxymethyl-6-methoxy-4,5-dimethylisochromen-l-one (
12) [
21], (
R)-4,8-dihydroxy-6-methoxy-4,5-dimethyl-3-methyleneisochromen-1-one (
13) [
6], 6,8-dihydroxy-3,4-dimethylisocoumarin (
14) [
22], acremonone F (
15) [
23], acremonone G (
16) [
23], whose absolute configurations were assigned by comparison of optical rotation data and ECD with the reported data.
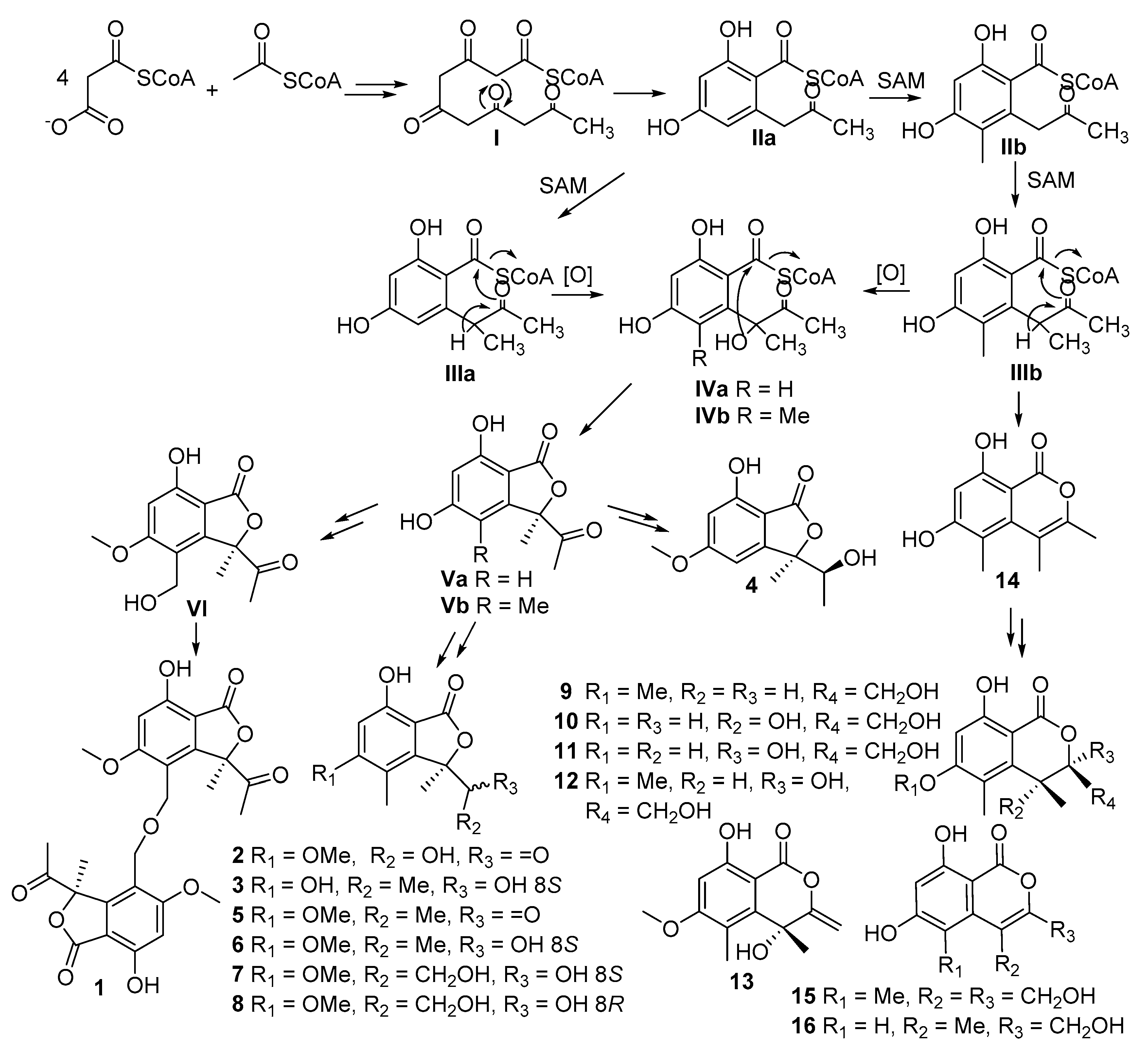
Fungal polyketides with structural diversity derived from the polymerization of short-chain carboxylic acids units are mainly synthesized by iterative type I polyketide synthases (IPKSs) via a series of decarboxylative condensations of malonyl-CoA extender units and
β-keto modification [
26,
27,
28]. Isobenzofuranones and isochromenones, suggested to share the same biogenetic pathway, could be hypothesized as derivatives from the pentaketide intermediate
I. In brief, other intermediates (
IIa–
VI) originated from intermediate
I via several steps as shown in
Scheme 1 [
29,
30,
31]. Compound
14 was obtained as an intermediate derived from intermediate
IIIb by cyclization and dehydration, which then yielded a series of isochromenones (
9–
13,
15–
16) after several reaction steps such as methylation, reduction, oxidation, etc. Methylation and reduction of the ketone carbonyl of intermediate
Va offered compound
4. Besides, a succession of isobenzofuranones (
2–
3,
5–
8) was produced by derivatization of intermediate
Vb. Interestingly, methylation and oxidation of the phenolic ring in intermediate
Vb gave one important intermediate
VI, which afterward formed a dimer, leptosphaerin J (
1), via intermolecular condensation of alcoholic hydroxyls. To the best of our knowledge, leptosphaerin J (
1) was isolated as a novel, rare, naturally-occurring isobenzofuranone dimer.
All of the obtained compounds (
1–
16) were evaluated for their cytotoxic and antiviral activities, according to previously reported methods [
18,
32]. The results showed that none of them displayed obvious cytotoxicity against three cancer cell lines, K562, MCF-7 and SGC7901 (IC
50 > 50 μM). Meanwhile, all of the isobenzofuranones (
1–
8) and isochromenones (
9–
16) were inactive (IC
50 > 100 μM) against H3N2, EV71 and HIV viruses.
Natural isobenzofuranones and isochromenones have been mostly reported from fungal sources, with modest or weak antimicrobial activities [
6,
19,
23,
25,
29,
33,
34,
35], and weak or no cytotoxicities [
24,
30,
36]. Nevertheless, in those reported isochromenones, (
R)-4,6,8-trihydroxy-4,5-dimethyl-3-methyleneisochromen-1-one and (
R)-4,8-dihydroxy-6-methoxy-4,5-dimethyl-3-methyleneisochromen-1-one (
13) were reported with antifungal activity against
Cochliobolus miyabeanus with IC
50 values of 10 and 0.5 μM [
6], respectively. Additionally,
13 and clearanol C showed weak inhibitory activity against
Candida albicans biofilm formation with MIC (minimum inhibitory concentration) values of 86 ± 3 and 101 ± 3 μM, respectively [
19]. However, the three bioactive isochromenones mentioned above share the presence of an exocyclic olefinic group anchored at C-3, which probably plays a role in improving the antimicrobial activities.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured with a Perkin Elmer MPC 500 (Waltham, MA, USA) polarimeter. UV spectra were measured on a Shimadzu UV-2600 PC spectrometer (Shimadzu, Beijing, China). IR spectra were measured on an IR Affinity-1 spectrometer (Shimadzu, Beijing, China). Circular dichroism spectra were measured with a Chirascan circular dichroism spectrometer (Applied Photophysics, Ltd., Leatherhead, UK). 1D and 2D NMR spectra were recorded on a Bruker Avance 500 MHz and 700 MHZ NMR (Bruker, Fällanden, Switzerland) spectrometer using TMS as an internal standard, and chemical shifts were recorded as δ-values. HRESIMS were recorded on a Bruker miXis TOF-QII mass spectrometer (Bruker, Fällanden, Switzerland). X-ray diffraction intensity data were collected on an Agilent Xcalibur Nova single-crystal diffractometer (Santa Clara, CA, USA) using Cu Kα radiation. TLC and column chromatography (CC) were performed on plates precoated with silica gel GF254 (10–40 μm) and over silica gel (200–300 mesh) (Qingdao Marine Chemical Factory, Qingdao, China), and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden), respectively. Spots were detected on TLC (Qingdao Marine Chemical Ltd., Qingdao, China) under 254 nm UV light or by heating after spraying with 5% H2SO4 in EtOH. All solvents used were of analytical grade (Tianjin Fuyu Chemical and Industry Factory, Tianjin, China). Semi-preparative HPLC was performed using an ODS column (YMC-pack ODS-A, YMC Co. Ltd., Kyoto, Japan, 10 × 250 mm, 5 μm, 2.5 mL/min).
3.2. Fungal Material
The fungal strain SCSIO 41005 was isolated from deep-sea sediments, which were collected from the Indian Ocean (Lat: 7°9.43716667′ N, Long: 89°4.4266667′ E) at a depth of 3614 m in 2013. The isolates were stored on Muller Hinton broth (MB) agar (malt extract 15 g, sea salt 15 g and agar 15 g) slants at 4 °C, and a voucher specimen was deposited in the CAS Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China. Fungal identification was performed by analysis of its ITS region of the rDNA as described in the
Supplementary Materials. The resulting sequence data, which were most similar (99%) to the sequence of
Leptosphaeria sp. EIODSF012 (accession no. KJ173535), have been deposited in GenBank (accession no. KY290887). Thus, the strain was identified as
Leptosphaeria sp. SCSIO 41005.
3.3. Fermentation, Extraction and Isolation
The strain Leptosphaeria sp. SCSIO 41005 was cultured on MB-agar plates at 25 °C for 7 days. The seed medium (malt extract 15 g and sea salt 2.5 g in 1.0 L tap distilled water, pH 7.4–7.8) was inoculated with strain SCSIO 41005 and incubated at 25 °C for 3 days on a rotating shaker (180 rpm). Then, large-scale fermentation of fungal isolate SCSIO 41005 was incubated for 90 days at room temperature in 1 L × 48 conical flasks with solid rice medium (each flask contained 200 g rice, 3 g sea salt and 200 mL naturally sourced water). The whole fermented cultures were harvested and extracted with acetone and then filtered through cheesecloth to separate them into filtrate and mycelia. The filtrate was concentrated under vacuum to remove the organic solvents and then extracted three times with EtOAc, while the mycelia were also extracted three times with EtOAc. Both EtOAc solutions were combined and concentrated under vacuum to yield a brown-colored EtOAc extract (180 g).
3.4. Purification
The crude EtOAc extract was subjected to silica gel column chromatography eluted with CH2Cl2/MeOH in a gradient eluent (v/v, 100:0, 98:2, 97:3, 95:5, 90:10, 80:20, 50:50) to obtain six fractions (fractions 1–6) based on TLC properties. Fraction 2 was purified by further Sephadex LH-20 (CH2Cl2/MeOH, 1:1) and then by semi-preparative reversed-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeOH/H2O 45:55) to yield 5 (19.3 mg) and 13 (15.9 mg). Fraction 3 was purified by further Sephadex LH-20 (CH2Cl2/MeOH, 1:1) and then by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeOH/H2O 39:61) to yield 1 (12.0 mg). Fraction 4 was applied to Sephadex LH-20 (CH2Cl2/MeOH, 1:1) to yield three subfractions. Fraction 4.2 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeOH/H2O 57:43) to yield 6 (508 mg) and 9 (4 mg). Fraction 4.3 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeOH/H2O 57:43) to yield 14 (3.0 mg). Fraction 5 was chromatographed on a silica gel column with a CH2Cl2/MeOH gradient system (from 1:0 to 0:1) to afford three subfractions. Fraction 5.1 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeCN/H2O 15:85) to yield 4 (22.0 mg). Fraction 5.2 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeCN/H2O 23:77) to yield 8 (41.0 mg), 12 (329 mg) and 16 (5.0 mg). Fraction 5.3 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeCN/H2O 22:78) to yield 10 (10.2 mg), 11 (104 mg) and 15 (2.8 mg). Fraction 6 was chromatographed on a silica gel column with a CH2Cl2/MeOH gradient system (from 1:0 to 0:1) to afford three subfractions. Fraction 6.1 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeCN/H2O 21:79) to yield 2 (4.0 mg). Fraction 6.2 was purified by semi-preparative reverse-phase HPLC (2 mL/min, detector UV λmax 220 nm, MeOH/H2O 26:74) to yield 3 (59.1 mg) and 7 (1.12 g).
3.5. Spectral Data
Leptosphaerin J (
1). Colorless crystals;
= +87.3° (
c 0.06, MeOH); UV (MeOH) λ
max (log
ε) 299 (2.79), 260 (3.19), 222 (3.63) nm; CD (4.61 × 10
−4 M, MeOH) λ
max (Δ
ε) 261 (+14.79), 222 (−38.61) nm; IR (film) ν
max 3400, 2937, 1708, 1562, 1217, 1051 cm
−1; HRESIMS (positive) [M + Na]
+ m/
z 537.1361, calcd. for C
26H
26O
11Na
+.
1H and
13C NMR spectrum data are seen in
Table 1.
Leptosphaerin K (
2). Colorless oil;
= +21.0° (
c 0.10, MeOH); UV (MeOH) λ
max (log
ε) 300 (3.51), 259 (3.81), 220 (4.20) nm; CD (5.67 × 10
−4 M, MeOH) λ
max (Δ
ε) 259 (+4.50), 234 (+9.00), 221 (−14.33) nm; IR (film) ν
max 3448, 2972, 1714, 1645, 1205, 1028 cm
−1; HRESIMS (negative) [M − H]
− m/
z 251.0565, calcd. for C
12H
11O
6−, 251.0556; [2M − H]
− m/
z 503.1204, calcd. for C
24H
23O
12−, 503.1190.
1H and
13C NMR spectrum data are seen in
Table 1.
Leptosphaerin L (
3). Colorless oil;
= −44.7° (
c 0.15, MeOH); UV (MeOH) λ
max (log
ε) 299 (3.76), 259 (4.02), 220 (4.28) nm; CD (9.15 × 10
−4 M, MeOH) λ
max (Δ
ε) 260 (+1.53), 212 (−5.56) nm; IR (film) ν
max 3302, 2981, 1708, 1562, 1244, 1055 cm
−1; HRESIMS (negative) [M − H]
− m/
z 237.0775, calcd. for C
12H
13O
5−, 237.0763; [2M − H]
− m/
z 475.1605, calcd. for C
24H
27O
10−, 475.1604.
1H and
13C NMR spectrum data are seen in
Table 1.
Leptosphaerin M (
4). Colorless oil;
= −32.6° (
c 0.10, MeOH); UV (MeOH) λ
max (log
ε) 291 (3.58), 257 (4.06), 220 (4.24) nm; CD (6.30 × 10
−4 M, MeOH) λ
max (Δ
ε) 258 (+1.69), 213 (−6.41) nm; IR (film) ν
max 3358, 2943, 1726, 1610, 1209, 1020 cm
−1; HRESIMS (negative) [M − H]
− m/
z 237.0775, calcd. for C
12H
13O
5−, 237.0763; [M + Cl]
− m/
z 273.0530, calcd. for C
12H
14O
5Cl
−, 273.0530; [2M − H]
− m/
z 475.1605, calcd. for C
24H
27O
10−, 475.1604.
1H and
13C NMR spectrum data are seen in
Table 1.
Clearanol I (
9). Colorless crystals;
= +18.2° (
c 0.19, MeOH); UV (MeOH) λ
max (log
ε) 308 (3.86), 265 (4.11), 229 (4.10) nm; CD (1.10 × 10
−3 M, MeOH) λ
max (Δ
ε) 309 (−0.94), 270 (+4.71), 250 (+0.60), 237 (+4.56), 224 (−2.85) nm; IR (film) ν
max 3419, 2924, 1651, 1616, 1244, 1165 cm
−1; HRESIMS (positive) [M + Na]
+ m/
z 275.0890, calcd. for C
13H
16O
5Na
+, 275.0895; [M + H]
+ m/
z 253.1077; calcd. for C
13H
17O
5+, 253.1076.
1H and
13C NMR spectrum data are seen in
Table 2.
Clearanol J (
10). Colorless crystals;
= + 17.4° (
c 0.10, MeOH); UV (MeOH) λ
max (log
ε) 314 (3.74), 271 (3.92), 229 (3.97), 216 (4.20) nm; CD (7.71 × 10
−4 M, MeOH) λ
max (Δ
ε) 276 (+0.07), 258 (−0.12), 242 (+0.29), 230 (−0.62), 211 (+1.64) nm; IR (film) ν
max 3334, 2937, 1645, 1367, 1253, 1014 cm
−1; HRESIMS (negative) [M − H]
− m/
z 253.0721, calcd. for C
12H
13O
6−, 253.0712; [M + Cl]
− m/
z 289.0486, calcd. for C
12H
14O
6Cl
−, 289.0479.
1H and
13C NMR spectrum data are seen in
Table 2.
3.6. X-ray Crystal Structure Analysis
Crystallographic data for compounds leptosphaerin J (
1), clearanol E (
7) and clearanol I (
9) were collected on an Agilent Xcalibur Nova single-crystal diffractometer using Cu K
α radiation. The structures of these compounds were solved by direct methods (SHELXS97), expanded using difference Fourier technniques, and refined by full-matrix least-squares calculation. The non-hydrogen atoms were refined anisotropically, and hydrogen atoms were fixed at calculated positions. Crystallographic data for the structures of leptosphaerin J (
1), clearanol E (
7) and clearanol I (
9) have been deposited in the Cambridge Crystallographic Data Centre database (deposition number CCDC 1539529, CCDC 1539538, CCDC 1539537). Copies of the data can be obtained free of charge from the CCDC at
www.ccdc.cam.ac.uk.Crystal data for 1: C26H26O11, M = 514.47, Orthorhombic, a = 23.08124(16) Å, b = 10.49438(8) Å, c = 10.03167(10) Å, a = 90°, b = 90°, g = 90°, V = 2429.90 (3) Å3, T = 150(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.938 mm−1, 23,207 reflections measured, 4841 independent reflections (Rint = 0.0236). The final R1 values were 0.0289 (I > 2σ(I)). The final wR (F2) values were 0.0297 (all data). The goodness of fit on F2 was 1.031. Flack parameter = 0.00 (5).
Crystal data for 7: C13H16O6, M = 268.26, Orthorhombic, a = 12.50776(10) Å, b = 13.73220(10) Å, c = 14.93010(10) Å, a = 90°, b = 90°, g = 90°, V = 2564.38 (3) Å3, T = 150(2) K, space group P212121, Z = 8, μ(Cu Kα) = 0.937 mm−1, 19,111 reflections measured, 5109 independent reflections (Rint = 0.0366). The final R1 values were 0.0312 (I > 2σ (I)). The final wR (F2) values were 0.0321 (all data). The goodness of fit on F2 was 1.052. Flack parameter = 0.04 (8).
Crystal data for 9: C13H16O5, M = 252.26, Orthorhombic, a = 4.91205(6) Å, b = 13.99641(14) Å, c = 17.18834(19) Å, a = 90°, b = 90°, g = 90°, V= 1181.72 (2) Å3, T= 150(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.914 mm−1, 8470 reflections measured, 2216 independent reflections (Rint = 0.0181). The final R1 values were 0.0256 (I > 2σ (I)). The final wR (F2) values were 0.0259 (all data). The goodness of fit on F2 was 1.059. Flack parameter = −0.03 (5).
3.7. Cytotoxic Bioassay
All isolated compounds were evaluated against the three human tumor cell lines (K562, MCF-7 and SGC7901) guided by previously reported methods [
17,
18]. Taxol was used as the positive control with IC
50 values of 3.67, 7.56 and 3.41 nM against K562, MCF-7 and SGC7901, respectively.
3.8. Antiviral Assay against H3N2, EV71 and HIV Viruses
The antiviral assay against H3N2 was evaluated by the cytopathic effect (CPE) inhibition assay, on Madin–Darby canine kidney (MDCK) cells with CCK-8 assay, while EV71 virus was measured by the ability to inhibit the CPE induced by EV71 virus on Vero cells with a CCK-8 assay, according to previously reported methods [
18]. The 50% inhibitory concentration (IC
50) of the testing compound was calculated using the GraphPad Prism software. Oseltamivir was used as the positive control in an anti-H3N2 assay with IC
50 values of 36.8 nM, and ribavirin was used as the positive control in an anti-EV71 assay with an IC
50 value of 0.60 μM. In addition, all isolated compounds were evaluated for their inhibitory activities against infection with HIV-1 SF162 in TZM-bl cells, according to previously reported methods [
32]. A nucleoside reverse transcriptase inhibitor, abacavir, was used as the positive control with an IC
50 value of 0.8 μM.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}