Abstract
Herein, we reported on the synthesis of cpIPP, which is a new structurally-reduced analogue of cyclic ADP-ribose (cADPR), a potent Ca2+-releasing secondary messenger that was firstly isolated from sea urchin eggs extracts. To obtain cpIPP the “northern” ribose of cADPR was replaced by a pentyl chain and the pyrophosphate moiety by a phophono-phosphate anhydride. The effect of the presence of the new phosphono-phosphate bridge on the intracellular Ca2+ release induced by cpIPP was assessed in PC12 neuronal cells in comparison with the effect of the pyrophosphate bridge of the structurally related cyclic N1-butylinosine diphosphate analogue (cbIDP), which was previously synthesized in our laboratories, and with that of the linear precursor of cpIPP, which, unexpectedly, revealed to be the only one provided with Ca2+ release properties.
1. Introduction
Cyclic ADP-ribose (cADPR, 1, Figure 1) is a natural occurring metabolite of NAD+ that is capable of mobilizing Ca2+ ions from intracellular stores. cADPR was firstly isolated from sea urchin eggs extracts [1,2,3,4,5,6], but it was later established that it is also produced in many other mammalian cells, including pancreatic β-cells, T-lymphocytes, smooth and cardiac muscle cells, and cerebellar neurons, acting as a Ca2+-mobilizing agent [7,8,9].
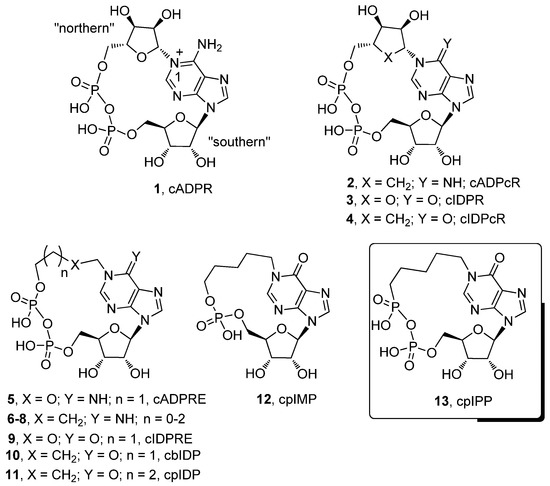
Figure 1.
Some representative cyclic ADP-ribose (cADPR) analogues.
For this activity, cADPR has been classified as a second messenger that, activating the ryanodine receptors of the sarcoplasmic reticulum, is able to mobilize the calcium ions from the intracellular stores [10]. cADPR is involved in many physiological processes that are related to the variation of the Ca2+ concentration, such as the synaptic homeostasis in neurons [11], as well as fertilization and cellular proliferation [12,13,14]. The conversion of the NAD+ into the 18-membered cyclic product (i.e., cADPR) has been attributed to the activity of the enzyme ADP-ribosyl cyclase, which was first purified from the marine mollusc Aplysia Californica [15]. In mammalian cells, the multifunctional transmembrane glycoprotein CD38 possesses this cyclase activity that converts the NAD+ into cADPR and catalyses the inverse hydrolytic reaction that produces adenosine 5’-diphosphate ribose (ADPR) [16,17,18].
In the last three decades, cADPR has been at the centre of numerous studies that were aimed at understanding its mechanism of action (the target proteins have not been yet completely identified) and at the production of potential drug candidates acting on calcium responsive physio-pathological processes. cADPR possesses a labile N1-glycosidic bond that is easily hydrolysed in aqueous solutions and in physiological conditions to the inactive ADPR [19]. To overcome this problem, most of the cADPR analogues so far prepared contain chemical modifications aimed at stabilizing the N1-ribosyl linkage and at exploring the structure activity relationship of this metabolite [20,21].
Many stable analogues of cADPR have been so far prepared by chemoenzymatic and chemical methods. The chemoenzymatic methods essentially follow a pathway that cyclizes a chemically modified NAD+ by the action of the Aplysia Californica ADP-ribosyl cyclase [15]. However, the specificity of the cyclase activity on the NAD+ analogues considerably limits the use of this strategy. On the contrary, the total chemical synthesis allows for the introduction of severe structural changes in the cADPR analogues [22].
The structural modifications so far proposed regard the northern and southern ribose, the purine base and the pyrophosphate moiety. Many of these cADPR analogues have also been biologically tested, thus allowing for the definition of some structure activity relationships (SAR) among the derivatives. For example, the substitution of the northern ribose with a carbocyclic ribose produces the stable mimic 2 (cADPcR) [23], which is more active than cADPR in the sea urchin egg homogenates and in neuronal cells, but results almost inactive in the T cell system [24]. Very recently, Shuto et al. reported on the synthesis and Ca2+ mobilizing activity of cyclic ADP-4-thioribose (cADPtR), in which the N1-ribose of cADPR is linked to 4-thioribose. cADPtR proved to be a biologically stable mimic of cADPR, as active as the latter in mobilizing Ca2+ ions in various cell systems [25].
In other cADPR analogues, the adenine base was replaced by hypoxanthine to get cIDPR (3) [26], or the corresponding N1-carbocyclic derivative cIDPcR (4) [27]. In Jurkat T cells, cIDPR showed almost the same Ca2+ mobilizing activity as cADPR. Conversely, cIDPcR resulted inactive in the same system up to 500 µM.
In other studies, the northern ribose was replaced by more flexible moieties, such as the ethoxy-alkyl or alkyl chains, both for the adenine (5–8) and inosine series (9). Compound 5 (cyclic adenosine 5’-diphosphate-ribose-ether, cADPRE) and its N1-alkyl congeners (6–8) permeate the plasma membrane and are weak agonists of cADPR in human Jurkat T cells [28,29]. The inosine analogue 9 (cIDPRE) [30] possesses an activity on intact human Jurkat-T-lymphocytes releasing Ca2+ ions from intracellular stores.
Many cADPR analogues also contain modifications on the C-8 purine position, also in combination with modifications in the “northern” ribose. For example, atoms or groups like Cl, Br, NH2, N3, CF3, SPh, and OCH3 have been introduced on the C-8 purine position [31,32,33].
In the last years, we reported on the syntheses of several cIDPR analogues, focusing our attention on derivatives containing alkyl chains in place of the “northern” (10–11) or “southern” ribose [34,35]. Among these, the N1-pentyl derivative 11 showed an interesting Ca2+ releasing activity in PC12 cells that were differentiated in neurons with the use of nerve growth factor (NGF). Furthermore, we also found a significant Ca2+ releasing activity for the derivative 12, in which the pyrophosphate moiety was replaced by a monophosphate function. In the present study, to further explore the role of the pyrophosphate group in the biological activity of cADPR analogues, we report on the synthesis and chemical and biological characterization of a new cyclic N1-pentyl inosine phosphono-phosphate anhydride analogue, cpIPP (13), in which the northern phosphate of 11 was replaced by a C-phosphonate moiety. This kind of modification is unprecedented and adds a further piece of information on the SAR of Ca2+ releasing cADPR analogues. Furthermore, the phosphono-phosphate anhydride moiety is a quite rare function in synthetic compounds, that, at the best of our knowledge, has never been explored in biologically active compounds. The Ca2+ mobilizing activities in PC12 cells of compounds 13 and of its linear precursor, as well as of the previously synthesized cyclic N1-butyl derivative 10 [35], which shares with the compound 13 the ring size, are also reported.
2. Results and Discussion
2.1. Chemistry
The key step for the preparation of all cADPR/cIDPR analogues is the macrocyclization reaction via pyrophosphate bond formation, which is usually performed by joining the two phosphate moieties at the end of a multistep synthesis [23]. For the construction of the 18-membered ring in 13, we have designed a convergent synthetic strategy that exploited the installation of a preformed phosphono-pentyl chain (17 or 19, Scheme 1) on the N1 position of inosine by a N-alkylation reaction. The synthesis of the phosphono-alkyl chain started from the commercially available 5-bromo-pentan-1-ol (14), which was silylated (15) [36] and then reacted with the sodium salt of di-tert-butyl phosphonate (generated in situ by reaction between NaH and di-tert-butyl phosphite), thus obtaining the intermediate 16 in very high yields. The deprotection of silyl ether in 16 with tetrabutylammonium fluoride (TBAF) allowed the quantitative recovery of the alcohol 17. The latter was converted into the iodide 19, passing through the mesylate (Ms) intermediate 18 in high yields.
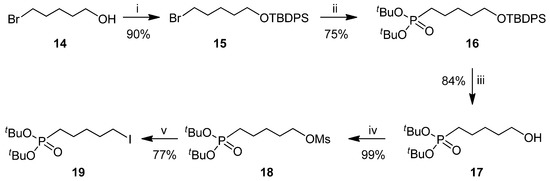
Scheme 1.
Reagents and conditions. (i) ref. [37]; (ii) NaH, HPO(OtBu)2, DMF, 0 °C, 16 h; (iii) TBAF, THF, r.t., 2 h; (iv) mesyl chloride (MsCl), TEA, CH2Cl2, 0 °C, 1 h; (v) KI, DMF, 70 °C, 5 h.
As regarded the following N1-alkylation of the hypoxanthine base, it has to be considered that the purine is an ambident nucleophile that can lead to the formation of the N1 and O6 alkylated isomers, whose ratio depends on the electrophilicity of the carbon atom that undergoes the nucleophilic attack [27,37]. At first, the alkylation of the N1 purine position was performed under the Mitsunobu conditions, [38] by reacting the alcohol 17 with the nucleoside 20 (Scheme 2) [36] in the presence of triphenylphosphine (PPh3) and diethylazodicarboxylate (DEAD) under different experimental conditions (data not shown). However, the reaction yields were low for both isomers, with the preponderance of the O6-alkylated isomer (70–80% relative to the N1-alkyl isomer). The absence of N1 regioselectivity could be a consequence of the favoured reaction between the hard alkoxy-phosphonium cation intermediate and the nucleobase O6 hard atom [39]. The two isomers were clearly distinguished by comparison of their 1H-NMR spectra. In fact, in the O6 isomer the CH2 attached to the purine O6 atom resonated as a triplet at 4.5 ppm, whereas in the N1 isomer (21) the CH2 attached to the purine N1 atom resonated as a multiplet at 4.0 ppm, according to our previous findings [40,41,42,43,44,45]. With the aim of improving the yield and the regioselectivity of the reaction toward the required N1 alkylated isomer 21, the reactivity of the soft iodide 19 was explored. At this stage, a careful selection of the alkaline conditions was crucial for the success of the reaction. Hyde, R. M. et al. [46] reported that 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was very efficient to obtain N1-allyl inosine starting from allyl bromide and inosine, but in our case, compound 21 could be isolated from a complex reaction mixture only in a 14% yield. A similar result was also observed using the tertiary amines triethylamine (TEA) and N,N-diisopropylethylamine (DIPEA). Pleasingly, the use of K2CO3 pushed the reaction to completeness with the recovery of the N1-alkylated isomer 21 in an 90% yield. This reaction represents a mild and high yielding synthetic alternative for the inosine N1-alkylation.
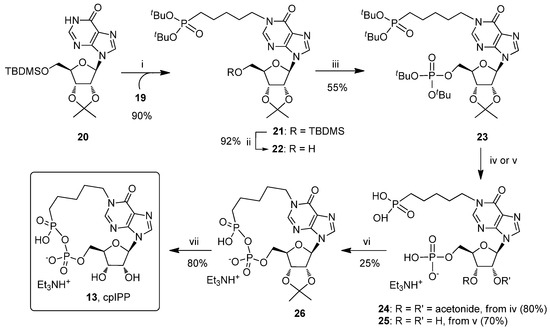
Scheme 2.
Reagents and conditions. (i) 19, K2CO3, DMF, r.t., 16 h; (ii) TBAF, THF, r.t., 1 h; (iii) (a) (tBuO)2PN(iPr)2, 1-H-tetrazole, THF, r.t., 4 h, (b) tBuOOH, THF, r.t., 1 h; (iv) 5% TFA in CH2Cl2, 0 °C, 5 h; (v) 50% TFA in H2O, r.t., 4 h; (vi) EDC, DMF, r.t., 72 h.; (vii) 60% HCO2H in H2O, r.t., 16 h.
Thereafter, the ribose tert-butyldimethylsilyl (TBDMS) group was quantitatively removed from the latter derivative with TBAF, thus obtaining the compound 22 that was phosphorylated in 55% yield by reaction with di-tert-butyl N,N-diisopropylphosphoramidite [(tBuO)2PN(iPr)2] and 1-H-tetrazole, followed by tert-butyl hydroperoxide (tBuOOH) oxidation, to give the derivative 23. Next, the protecting groups of the phosphate and phosphonate moieties in the product 23 were removed by reaction with trifluoroacetic acid (TFA) in anhydrous dichloromethane (DCM), yielding compound 24 (80% yield). This treatment preserved the ribose acetonide protecting group, which was strictly necessary for the success of the final macrocyclization reaction. Conversely, the treatment of compound 23 with aqueous TFA solution afforded the fully deprotected nucleotide 25. The compound 24, dissolved in DMF at the final concentration of 2 mM was treated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (1.2 equiv.) and the reaction allowed to stand at room temperature for 72 h. From this mixture, it was possible to isolate the cyclic compound 26 in a 25% yield. The diasterotopicity of CH2N1 protons in the 1H-NMR spectrum (resonating as two doublets of doublets of doublets at 4.45 and 4.05 ppm) and the presence of two doublets in the 1H-decoupled 31P-NMR spectrum (19.3 ppm for the phosphonate and −11.2 ppm for the phosphate) of the compound 26 were clear evidences of the 18-membered ring formation. On the contrary, for the linear precursor 24, the CH2N1 protons appeared as a triplet at 4.06 ppm in the 1H-NMR spectrum, whereas the phosphorous atoms of the phosphonate and phosphate groups resonated as two singlets at 28.2 and 1.0 ppm, respectively, in the 1H-decoupled 31P-NMR spectrum. Finally, the treatment of compound 26 with aqueous 60% HCO2H afforded the cpIPP 13 in 80% yield (7.3% overall yield starting from compound 20). In the analogue 13, the N1–C bond and the phosphono-phosphate anhydride moiety revealed to be stable to the hydrolysis, as evidenced by HPLC analyses, which indicated the absence of any degradation product after its dissolution in water at pH = 7 (data not shown).
2.2. Biology
Exogenous administration of cADPR did not produce changes in [Ca2+]i for the low ability of the metabolite to cross the membranes. To overcome this problem, the three more lipophilic derivatives 10, 13, and 25 were synthesized and tested. These analogues were perfused at the concentration of 100 nM in a Krebs-Ringer saline solution to PC12 cells previously differentiated in neurons with NGF. While the cyclic compounds 10 and 13 failed to modify [Ca2+]i, the linear precursor 25 caused a fast but low increase in [Ca2+]i (Figure 2).
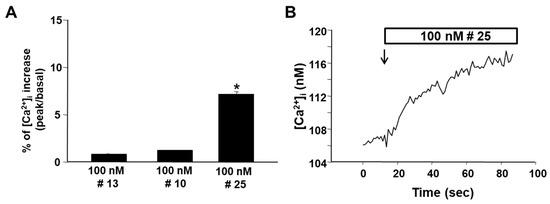
Figure 2.
Effect of compounds 10, 13 and 25 on [Ca2+]i in NGF-differentiated PC12 cells. Panel (A): quantification of [Ca2+]i increase calculated as the percentage change of plateau/basal value after the addition of compounds 10, 13 and 25. Each bar represents the mean (±S.E.M.) of the values obtained in three independent experimental sessions. For each experiment, 10 to 40 individual cells were monitored. *, p < 0.05 versus basal level. Panel (B): representative single-cell trace of the effect of compound 25 (100 nM,) on [Ca2+]i.
However, this effect was lower than that previously obtained with cyclic analogues 11 and 12 [36], likely because of a reduced ability to cross the membranes or to an inefficacious interaction with the intracellular receptor.
2.3. Conformational Analyses
Prompted by the results of biological studies, we asked whether the differences in activity that was shown by compounds 10 and 13 with respect to the analogue 11 were due to a different conformational behaviour. We therefore sampled the conformational space of compounds 10, 11, and 13 by using the macrocycle conformational sampling routine implemented in Maestro 11.2 (Schroedinger Inc.) [47]. Conformational ensembles that were obtained from the macrocycle search evidenced that all the compounds covered the same conformational space (Figure S1, Supplementary Information), albeit the phosphonate/phosphate derivative (13) clearly showed a lower number of low energy conformers. Interestingly, ring dimension does not seem to play a key role, since both pyrophosphate derivatives (10 and 11) showed a similar number of conformers.
The comparison of the lowest energy conformers of the compounds 10, 11, and 13 (Figure 3) showed a different displacement of the pyrophosphate (10, 11) or phosphonate/phosphate (13) groups with respect to the hypoxanthine ring, leading to different structural features that could play a key role in the biological activity. In particular, the southern phosphate group of 11 is generally farther from the centre of the ring when compared to compound 10 and 13, as shown by the analysis of distances d1, d2, and d3 (describing the relative position of the phosphate/phosphonate groups with respect to the hypoxanthine ring defined in Figure 4; Tables S1 and S2, Supporting Information). Nevertheless, in our opinion, the differences that were observed in conformational behaviour of compounds 11 and 13 were not so relevant to be a discriminant for the biological activity. On the other hand, the calculation of ALogP (Table S1, Supplementary Information) showed lower lipophilicity for the compounds 10 and 13. On the basis of these considerations, we hypothesise that the lack of activity characterizing compounds 10 and 13 may be due to a combination of factors, including conformational flexibility that could affect interaction with the intracellular target and a reduced ability to cross membranes.
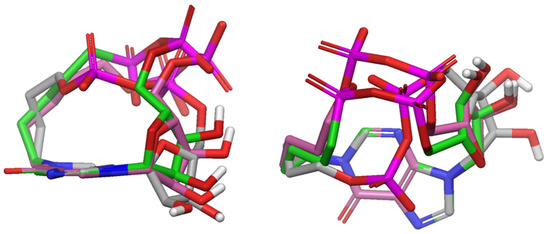
Figure 3.
Side (left) and top (right) view of the lowest energy conformers of cbIDP (10, grey), cpIDP (11, pink) and cpIPP (13, green) superimposed on the hypoxanthine ring. Non-polar hydrogens were omitted for sake of clarity. Oxygens were reported in red, nitrogens in blue, phosphates in magenta, hydrogens in white.
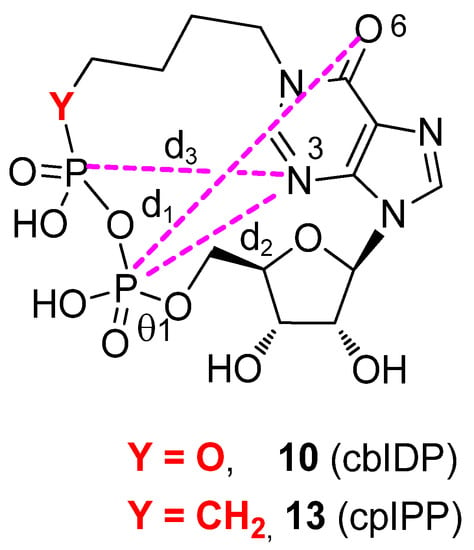
Figure 4.
Definition of the geometrical descriptors. d1: distance between O6 purine atom and P atom of the southern phosphate; d2: distance between N3 purine atom and P atom of the southern phosphate; d3: distance between N3 purine atom and P atom of the northern phosphate/phosphonate.
3. Experimental Section
3.1. General Experimental Procedures
All the reagents and solvents for the chemical syntheses were obtained from commercial sources and were used without further purification. 1H- and 13C-NMR experiments were performed using Varian Inc. NMR spectrometers (Mercury Plus 400 MHz, UNITYINOVA 500 MHz and UNITYINOVA 700 MHz, Varian Inc., Lake Forest, CA, USA) and CDCl3, CD3OD and D2O as solvents. NMR chemical shifts are reported in parts per million (δ) relative to residual solvents signals: CHCl3 7.27, CD2HOD 3.31, HOD 4.80 for 1H-NMR, and CDCl3 77.0, CD3OD 49.0 for 13C NMR. The 1H-decoupled 31P-NMR experiments were carried out on the UNITYINOVA 500 MHz instrument in CDCl3 and D2O solvents using 85% H3PO4 as external standard (0 ppm).
The 1H-NMR chemical shifts were assigned through 2D NMR experiments. High performance liquid chromatography (HPLC) was performed using a Jasco UP-2075 Plus (Jasco Europe s.r.l., Cremella (LC), Italy) pump equipped with a Jasco UV-2075 Plus UV detector (Jasco Europe s.r.l., Cremella (LC), Italy) and a Merck Purosphere Star (Merck & Co., Kenilworth, NJ, USA) 4.8 × 150 mm C-18 reversed-phase column (particle size 5 µm) eluted with a linear gradient of CH3CN in 0.1 M triethylammonium bicarbonate (TEAB) buffer (from 0 to 100% in 45 min, flow 1.3 mL/min). UV spectra were recorded on a Jasco V-530 UV spectrophotometer (Jasco Europe s.r.l., Cremella (LC), Italy). ESI-MS spectra were recorded on an AB SCIEX QTRAP 4000 LC-MS/MS system. Column chromatography was carried out on silica gel-60 (Merck, 0.063-0.200 mm). Analytical TLC analyses were performed using F254 silica gel plates (0.2 mm thick, Merck). TLC spots were detected under UV light (254 nm).
3.2. Synthesis and Charcterization of Compounds 13, 16, 17, 19 and 21–26
Compound 16: To an ice-cooled solution of di-tert-butyl phosphite (0.8 mL, 1.3 mmol) in dry DMF (3 mL) under nitrogen atmosphere, NaH (96 mg, 4.0 mmol) was added. After 10 min, compound 15 (0.81 g, 2.0 mmol) dissolved in dry DMF (3 mL) was added via cannula. The reaction mixture was warmed to room temperature and stirred for 16 h (TLC monitoring: n-hexane/AcOEt, 8:2), quenched with CH3OH, diluted with AcOEt (50 mL), and washed with brine (50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified over a silica gel column eluted with increasing amounts of AcOEt in n-hexane (up to 70%) to afford pure 16. Oil (0.78 g, 75% yield). 1H NMR (500 MHz, CDCl3) δ 7.81–7.57 (m, 4H, arom.), 7.38 (dt, J = 13.7, 6.8 Hz, 6H, arom.), 3.65 (t, J = 6.4 Hz, 2H, CH2O), 1.70–1.36 (complex signal, 26H, 4 × CH2 and 2 × OtBu), 1.04 (s, 9H, tBu). 13C NMR (100 MHz, CDCl3) δ 135.4, 133.9, 129.4, 127.4, 81.1, 81.0, 63.6, 32.1, 30.3, 30.2 (d, J = 145.2 Hz), 30.2 (2C), 26.8 (d, J = 16.8 Hz), 23.2 (d, J = 6.0 Hz). 31P NMR (202 MHz, CDCl3) δ 25.2 (s). ESI-MS m/z 519 ([M + H]+, C29H48O4PSi, requires 519).
Compound 17: To a solution of compound 16 (0.50 g, 0.96 mmol) in dry THF (5 mL), TBAF (1.1 mL, 1.1 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 2 h (TLC monitoring: AcOEt) and then evaporated under reduced pressure. The residue was purified over a silica gel column eluted with increasing amounts of CH3OH in AcOEt (up to 1%) to afford pure 17. Oil (0.22 g, 84% yield). 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.4 Hz, 2H, CH2O), 2.58 (bs, 1H, OH), 1.70–1.34 (complex signal, 26H, 4 × CH2 and 2 × OtBu). 13C NMR (100 MHz, CDCl3) δ 81.4, 81.3, 62.4, 32.2, 30.4, 30.3, 30.1 (d, J = 145.1 Hz), 26.6 (d, J = 17.8 Hz), 23.1 (d, J = 6.2 Hz). 31P NMR (202 MHz, CDCl3) δ 25.1 (s). ESI-MS m/z 303 ([M + Na]+, C13H29NaO4P, requires 303).
Compound 19: To an iced-cooled solution of compound 17 (0.22 g, 0.80 mmol) in dry CH2Cl2 (4 mL), TEA (0.075 mL, 0.96 mmol), and then MsCl (0.22 mL, 1.6 mmol) were added. The reaction mixture was stirred at 0 °C for 1 h (TLC monitoring: AcOEt/CH3OH, 98:2), quenched with H2O, diluted with CH2Cl2 (30 mL) and washed with brine (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was dissolved in dry DMF (8 mL) and KI (0.40 g, 2.4 mmol) was added. The reaction mixture was stirred at 70 °C for 5 h (TLC monitoring: n-hexane/AcOEt, 1:1) and then cooled to room temperature. Water (30 mL) was added and the product was extracted with Et2O (3 × 30 mL). The combined organic extracts were washed with water brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a crude that was purified by silica gel column eluted with increasing amounts of AcOEt in n-hexane (up to 50%) to afford pure 19. Oil (0.24 g, 77% yield over two steps). 1H NMR (400 MHz, CDCl3) δ 3.16 (t, J = 7.0 Hz, 2H, CH2I), 1.86–1.76 (m, 2H, CH2), 1.67–1.35 (complex signal, 24H, 3 × CH2 and 2 × OtBu). 13C NMR (100 MHz, CDCl3) δ 81.3, 81.2, 62.4, 33.1, 31.3 (d, J = 16.7 Hz), 30.4, 30.3, 30.0 (d, J = 145.4 Hz), 22.4 (d, J = 6.0 Hz), 6.5. 31P NMR (202 MHz, CDCl3) δ 24.6 (s). ESI-MS m/z 413 ([M + Na]+, C13H28INaO3P, requires 413).
Compound 21: To a solution of compound 20 (0.12 g, 0.28 mmol) in dry DMF (1 mL), anhydrous K2CO3 (0.077 g, 0.56 mmol) was added. After 10 min, compound 19 (0.22 g, 0.61 mmol), dissolved in dry DMF (1 mL), was added. The reaction mixture was stirred at room temperature for 16 h (TLC monitoring: CHCl3/CH3OH, 95:5), diluted with AcOEt (20 mL) and washed with brine (20 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified over a silica gel column eluted with increasing amounts of CH3OH in CHCl3 (up to 1%) to afford pure 21. Oil (0.17 g, 90% yield). 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H, 8-H), 7.91 (s, 1H, 2-H), 6.04 (bs, 1H, 1′-H), 5.05-4.99 (s, 1H, 2′-H), 4.86-4.80 (s, 1H, 3′-H), 4.34 (bs, 1H, 4′-H), 4.07-3.88 (m, 2H, CH2N), 3.79 (dd, J = 3.4, 11.5 Hz, 1H, 5′-Ha), 3.75 (dd, J = 3.7, 11.5 Hz, 1H, 5′-Hb), 1.78–1.65 (m, 2H, CH2), 1.60–1.46 (complex signal, 7H, 2 × CH2 and CH3), 1.42–1.36 (complex signal, 20H, CH2 and 2 × OtBu), 1.32 (s, 3H, CH3), 0.77 (s, 9H, SitBu), −0.04 (s, 3H, CH3), −0.05 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 156.3, 147.1, 146.8, 138.2, 124.8, 114.1, 91.2, 87.0, 85.3, 81.3, 81.2, 81.1, 63.4, 46.7, 30.4, 30.3, 30.0 (d, J = 145.6 Hz), 29.4, 27.3 (d, J = 17.0 Hz), 27.2, 25.8, 25.3, 23.1 (d, J = 5.9 Hz), −5.4, −5.6. 31P NMR (202 MHz, CDCl3) δ 24.4 (s). ESI-MS m/z 707 ([M + Na]+, C32H57N4NaO8PSi, requires 707).
Compound 22: To a solution of compound 21 (0.15 g, 0.22 mmol) in dry THF (5 mL), TBAF (0.3 mL, 0.26 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 1 h (TLC monitoring: CHCl3/CH3OH, 95:5) and then evaporated under reduced pressure [48,49]. The residue was purified over a silica gel column eluted with increasing amounts of CH3OH in CHCl3 (up to 2%) to afford pure 22. Oil (0.13 g, 92% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (bs, 1H, 8-H), 8.01 (s, 1H, 2-H), 5.92 (d, J = 3.9 Hz, 1H, 1′-H), 5.15–5.08 (m, 1H, 2′-H), 5.07-5.02 (m, 1H, 3′-H), 4.52 (bs, 1H, 4′-H), 4.11–4.00 (m, 2H, CH2N), 3.95 (d, J = 12.3 Hz, 1H, 5′-Ha), 3.79 (d, J = 12.4 Hz, 1H, 5′-Hb), 3.19 (bs, 1H, OH), 1.85–1.73 (m, 2H, CH2), 1.65–1.55 (complex signal, 7H, 2 × CH2 and CH3), 1.51–1.40 (complex signal, 20H, CH2 and 2 × OtBu), 1.37 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 155.7, 147.5, 146.1, 139.7, 125.2, 114.2, 93.8, 86.5, 84.1, 81.6, 81.4, 63.0, 47.1, 30.4, 30.0 (d, J = 146.0 Hz), 29.4, 27.4, 27.3 (d, J = 16.3 Hz), 25.8, 25.2, 23.1 (d, J = 5.5 Hz). 31P NMR (202 MHz, CDCl3) δ 24.4 (s). ESI-MS m/z 593 ([M + Na]+, C26H43N4NaO8P, requires 593).
Compound 23: To a solution of compound 22 (0.11 g, 0.19 mmol), in dry THF (5 mL), 1-H-tetrazole (0.094 g, 1.3 mmol), and then iPr2NP(tBuO)2 (0.84 mL, 2.6 mmol) were added under nitrogen atmosphere [50,51]. The reaction mixture was stirred at room temperature for 4 h (TLC monitoring: CHCl3/CH3OH, 95:5) and then tBuOOH (0.35 mL of a solution 5.5 M in decane, 1.9 mmol) was added at room temperature. After 1h (TLC monitoring: CHCl3/CH3OH, 95:5) the reaction mixture was evaporated under reduced pressure, diluted with AcOEt (30 mL), and washed with brine (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was purified over a silica gel column eluted with increasing amounts of CH3OH in CHCl3 (up to 5%) to afford pure 23. Oil (76 mg, 55% over two steps). 1H NMR (400 MHz, CDCl3) δ 8.01 (bs, 1H, 8-H), 7.98 (s, 1H, 2-H), 6.10 (bs, 1H, 1′-H), 5.22–5.17 (m, 1H, 2′-H), 5.04–4.99 (m, 1H, 3′-H), 4.47 (bs, 1H, 4′-H), 4.18–4.11 (m, 2H, CH2N), 4.09–3.99 (m, 2H, 5′-Ha,b), 1.83–1.73 (m, 2H, CH2), 1.64–1.58 (complex signal, 7H, 2 × CH2 and CH3), 1.49–1.45 (complex signal, 29H, CH2 and 3 × OtBu), 1.42 (s, 9H, OtBu), 1.37 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ 156.3, 147.3, 146.9, 138.6, 124.5, 114.6, 90.7, 84.9, 84.6, 83.1, 83.0, 81.5, 81.4, 81.3, 66.0, 46.9, 30.4, 30.0 (d, J = 145.4 Hz), 29.8, 29.5, 27.4 (d, J = 17.1 Hz), 27.2, 25.3, 23.1 (d, J = 5.5 Hz). 31P NMR (202 MHz, CDCl3) δ 24.4 (s), –8.7 (s). ESI-MS m/z 785 ([M + Na]+, C34H60N4NaO11P2, requires 785).
Compound 24: To an ice-cooled solution of compound 23 (50 mg, 0.065 mmol) in dry CH2Cl2 (0.95 mL) TFA (0.050 mL) was added. The reaction mixture was stirred at 0 °C for 5 h (TLC monitoring: iPrOH/NH3(aq)/H2O, 6:3:1), warmed to room temperature and evaporated under reduced pressure. The residue was dissolved in TEAB 0.1 M buffer, passed over a polyvinylidene difluoride (PVDF) 0.45 μm filter and purified by HPLC (see General experimental procedures). The fractions containing the triethylammonium salt of compound 24 (tR = 21.3 min) were collected and lyophilized. Amorphous white solid (33 mg, 80% yield). 1H NMR (400 MHz, D2O) δ 8.40 (bs, 1H, 8-H), 8.39 (s, 1H, 2-H), 6.30 (d, J = 3.1 Hz, 1H, 1′-H), 5.43 (dd, J = 6.1, 3.1 Hz, 1H, 2′-H), 5.21 (dd, J = 6.1, 2.2 Hz, 1H, 3′-H), 4.70-4.65 (m, 1H, 4′-H), 4.21-4.10 (m, 2H, 5′-Ha,b), 4.06 (t, J = 4.5 Hz, 2H, CH2N), 3.21 (q, J = 7.3 Hz, 6H, 3 × CH2, triethylammonium), 1.88–1.78 (m, 2H, CH2), 1.67 (s, 3H, CH3), 1.65–1.54 (complex signal, 4H, 2 × CH2), 1.50–1.41 (complex signal, 5H, CH2 and CH3), 1.29 (t, J = 7.3 Hz, 9H, 3 × CH3, triethylammonium). 13C NMR (175 MHz, D2O) δ 158.1, 148.8, 147.5, 140.1, 123.5, 114.8, 90.5, 85.2 (d, J = 8.8 Hz), 84.1, 81.4, 64.7 (J = 4.1 Hz), 47.2, 46.6, 28.2, 27.6 (d, J = 144.3), 27.0 (d, J = 17.5 Hz), 26.0, 24.1, 22.6 (d, J = 4.2 Hz), 8.1. 31P NMR (202 MHz, D2O) δ 28.2 (s), 1.0 (s). ESI-MS m/z 537 ([M − H]−, C18H27N4O11P2, requires 537).
Compound 25: To a solution of compound 23 (10 mg, 0.013 mmol) in H2O (0.20 mL) TFA (0.20 mL) was added. The reaction mixture was stirred at room temperature for 4 h (TLC monitoring: iPrOH/NH3(aq)/H2O, 6:3:1) and then evaporated under reduced pressure. The residue was dissolved in TEAB 0.1 M buffer, passed over a PVDF 0.45 μm filter and then purified by HPLC (see General experimental procedures). The fractions containing the triethylammonium salt of compound 25 (tR = 19.1 min) were collected and lyophilized. Amorphous white solid (4.5 mg, 70% yield). 1H NMR (500 MHz, D2O) δ 8.47 (bs, 1H, 8-H), 8.42 (s, 1H, 2-H), 6.15 (d, J = 5.6 Hz, 1H, 1′-H), 4.80 (1H, 2′-H, covered by residual solvent signal), 4.53–4.50 (m, 1H, 3′-H), 4.42–4.38 (m 1H, 4′-H), 4.20–4.10 (complex signal, 4H, 5′-Ha,b and CH2N), 3.22 (q, J = 7.3 Hz, 6H, 3 × CH2, triethylammonium), 1.87–1.79 (m, 2H, CH2), 1.66-1.54 (complex signal, 4H, 2 × CH2), 1.50–1.42 (m, 2H, CH2), 1.30 (t, J = 7.3 Hz, 9H, 3 × CH3, triethylammonium) 13C NMR (100 MHz, D2O) δ 158.1, 148.9, 147.9, 139.8, 123.4, 87.2, 84.1 (d, J = 8.5 Hz), 74.4, 70.3, 64.2 (d, J = 3.7 Hz), 47.3, 46.6, 28.2. 27.6 (d, J = 143.7 Hz), 27.0 (d, J = 17.1 Hz) 22.6 (d, J = 3.3 Hz), 8.1. 31P NMR (202 MHz, D2O) δ 27.4 (s), 1.7 (s). ESI-MS m/z 497 ([M − H]−, C15H23N4O11P2, requires 497).
Compound 26: To a solution of compound 24 (20 mg, 0.031 mmol) in dry DMF (15 mL) EDC (0.012 g, 0.062 mmol) was added. The reaction mixture was stirred at room temperature for 72 h and evaporated under reduced pressure. The residue was dissolved in TEAB 0.1 M buffer, passed over a PVDF 0.45 μm filter and purified by HPLC (see General experimental procedures). The fractions containing the triethylammonium salt of compound 26 (tR = 23.8 min) were collected and lyophilized. Amorphous white solid (4.8 mg, 25% yield). 1H NMR (500 MHz, D2O) δ 8.49 (s, 1H, 8-H), 8.25 (s, 1H, 2-H), 6.37 (bs, 1H, 1′-H), 5.84 (d, J = 6.0 Hz, 1H, 2′-H), 5.40 (dd, J = 6.0, 2.2 Hz, 1H, 3′-H), 4.65–4.60 (m, 1H, 4′-H), 4.45 (ddd, J = 13.4, 5.6, 3.9 Hz, 1H, CHaN), 4.05 (ddd, J = 13.2, 8.9, 3.6 Hz, 1H, CHbN), 3.98–3.87 (m, 2H, 5′-Ha,b), 3.22 (q, J = 7.3 Hz, 6H, 3 × CH2, triethylammonium), 2.00–1.86 (m, 2H, CH2), 1.66 (s, 3H, CH3), 1.61–1.51 (m, 2H, CH2), 1.50 (s, 3H, CH3), 1.45-1.32 (m, 4H, 2 × CH2), 1.30 (t, J = 7.3 Hz, 9H, 3 × CH3, triethylammonium). 13C NMR (175 MHz, D2O) δ 158.3, 148.4, 147.2, 141.7, 124.0, 114.0, 91.1, 87.2 (d, J = 10 Hz), 83.6, 81.5, 65.2 (d, J = 5.0 Hz), 48.0, 46.5, 28.2 (d, J = 140.1 Hz), 27.2 (d, J = 17.5 Hz), 26.3, 25.6, 22.7, 8.1. 31P NMR (202 MHz, D2O) δ 19.3 (d, 2JP,P = 27.8 Hz), –11.4 (d, 2JP,P = 27.2 Hz). ESI-MS m/z 519 ([M − H]−, C18H25N4O10P2, requires 519).
Compound 13 (cpIPP): A solution of compound 16 (3.0 mg, 0.0053 mmol) in aqueous 60% HCO2H (0.5 mL) was stirred at room temperature for 16 h and then evaporated under reduced pressure. The crude was dissolved in TEAB 0.1 M buffer, passed over a PVDF 0.45 μm filter and purified by HPLC (see General experimental procedures). The fractions containing the triethylammonium salt of compound 13 (tR = 19.8 min) were collected and lyophilized. Amorphous white solid (2.0 mg, 80% yield). 1H NMR (700 MHz, D2O) δ 8.47 (s, 1H, 8-H), 8.21 (s, 1H, 2-H), 6.06 (d, J = 3.5 Hz, 1H, 1′-H), 5.36 (dd, J = 3.6, 5.0 Hz, 1H, 2′-H), 4.80 (1H, 3′-H, covered by residual solvent signal), 4.34–4.28 (complex signal, 2H, 4′-H and CHaN), 4.18–4.13 (m, 1H, CHbN), 4.12–4.08 (m, 1H, 5′-Ha), 4.07–4.03 (m, 1H, 5′-Hb), 3.20 (q, J = 7.3 Hz, 6H, 3 × CH2, triethylammonium),1.95–1.86 (m, 2H, CH2CH2N), 1.60–1.52 (m, 1H, CHaCH2CH2N), 1.42–1.30 (m, 2H, PCH2), 1.29–1.22 (complex signal, 10H, 3 × CH3 triethylammonium and CHbCH2CH2N), 1.16–1.06 (m, 2H, PCH2CH2). 13C NMR (175 MHz, D2O) δ 158.2, 148.2, 147.6, 141.5, 123.9, 89.6, 83.4, 72.1, 69.6, 64.0, 47.6, 46.5, 27.8, 26.9, 25.9, 22.2, 8.1. 31P NMR (202 MHz, D2O) δ 19.3 (d, 2JP,P = 26.4 Hz), −11.2 (d, 2JP,P = 26.8 Hz). ESI-MS m/z 479 ([M − H]−, C15H21N4O10P2, requires 479).
3.3. Cell Cultures and Intracellular [Ca2+]i Measurement for Compounds 10, 13 and 25
PC12 cells, which grown on plastic dishes in RPMI medium (composed of 10% horse serum, 5% FBS, 100 UI/mL penicillin, and 100 μg/mL streptomycin) were differentiated in neurons with NGF (50 ng/mL; 7 days). Cells were cultured in the atmosphere of 5% CO2. Culture medium was changed every 2 days. Intracellular Ca2+ concentration ([Ca2+]i) was measured by single cell computer-assisted video-imaging [52]. Briefly, differentiated PC12 cells cultured on glass coverslips coated with poly-L-lysine (5 μg/mL) (Sigma, St. Louis, MO, USA) were loaded with 10 µM Fura-2AM for 1 h at 22 °C in Krebs-Ringer saline solution containing the following: 5.5 mM KCl, 160 mM NaCl, 1.2 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose, and 10 mM HEPES-NaOH, pH 7.4. At the end of the loading period, the coverslips were placed in a perfusion chamber (Medical System, Greenvale, NY, USA), mounted on a Zeiss Axiovert 200 microscope (Carl Zeiss, Oberkochen, Germany) equipped with a FLUAR 40× oil objective lens. The experiments employed a digital imaging system that was composed of a MicroMax 512BFT cooled CCD camera (Princeton Instruments, Trenton, NJ, USA), LAMBDA 10-2 filter wheeler (Sutter Instruments, Novato, CA, USA), and Meta-Morph/MetaFluor Imaging System software (Universal Imaging, West Chester, PA, USA). After loading, cells were illuminated alternately at 340 and 380 nm by a Xenon lamp. The emitted light was passed through a 512 nm barrier filter. Fura-2AM fluorescence intensity was measured every 3s. Forty to sixty-five individual cells were selected and monitored simultaneously from each cover slip. Results are presented as the cytosolic Ca2+ concentration. Calibrations used the relation of Grynkiewicz et al. [53] assuming that the KD for Fura-2AM was 224 nM.
3.4. Conformational Search
Calculations were performed using the Macromodel Conformational sampling from Schrödinger [47] using the OPLS 2005 force field and a generalized Born continuum solvent model (electrostatic treatment of water solvent). 10000 Large Scale Low Mode (LLMOD) steps were performed and normal modes were recalculated every time a new conformation was generated. Conformers with RMSD ≤ 0.75 Å were deemed redundant and removed. Only conformations within 10 kcal of the observed global minimum were retained.
4. Conclusions
cADPR is a ubiquitous second messenger responsible for Ca2+ mobilization from intracellular stores. Its instability in neutral aqueous conditions and its inability to cross the blood-brain barrier pushed chemists to develop synthetic or chemo-enzymatic strategies to obtain more stable and less polar analogues to better understand its physiological roles. In this frame, we have previously synthesized the cADPR analogue 11, in which the “northern” ribose is replaced by a pentyl chain and the corresponding structurally reduced derivative 12 having a phosphodiester moiety in the place of the pyrophosphate one. When considering the encouraging biological results that were obtained by analogues 11 and 12 on PC12 neuronal cells and to better explore the exact role of the pyrophosphate in the Ca2+ releasing activity, herein we have reported on the synthesis of a more lipophilic and not-hydrolysable cADPR analogue 13 (cpIPP), containing the not so far explored phosphono-phosphate anhydride moiety. In accordance with our recent results, the final cyclization step was performed at the end of the synthesis by joining the fully deprotected C-phosphonate and phosphate functionalities. This reaction, even if low yielding, allowed for a rapid recovery of the cADPR derivative 13 for the evaluation of its biological activity. Preliminary experiments on PC12 neuronal cells have shown that the cyclic compounds 10 and 13 failed to modify the [Ca2+]i, whereas the linear compound 25 caused a fast but low increase in [Ca2+]i if compared to the efficacy of the previously described cyclic analogues 11 and 12.
From these results, we could conclude that the pyrophosphate and the pentyl chain that is installed on the inosine N1 position are key elements for the Ca2+ releasing activity in PC12 neuronal cells of the cIDPR analogues having alkyl chains in the place of the “northern” ribose. Furthermore, the activity shown by the linear analogue 25, in which the phosphonate and the phosphate moieties are not joined, paves the way for new studies that are aimed at the identification of the minimal structural unit required for the biological activity of linear analogues of IDPR.
Supplementary Materials
The following are available online at http://www.mdpi.com/1660-3397/16/3/89/s1, S3–S12: copies of 1H-NMR and 31P-NMR spectra of compounds 13, 16, 17, 19 and 21–26; S13–S21: copies of 13C-NMR spectra of compounds 16, 17, 19 and 21–26; S22: Table S1; S22: Figure S1; S23: Table S2.
Acknowledgments
The authors are grateful to Luisa Cuorvo and Davide Cardella for their technical assistance. B.C. gratefully acknowledge the support of NVIDIA Corporation with the donation of the Tesla K40 GPU used for this research.
Author Contributions
S.D., G.O., G.P. and L.M. conceived and designed the experiments; S.D. performed the synthetic experiments; N.B. and V.C. performed the spectroscopic experiments and analyzed the data; A.S., T.P., I.P. and A.P. performed the biological experiments and analyzed the data; B.C. performed the computational experiments and analyzed the data; S.D., G.O. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [PubMed]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Li, P.L. Vascular physiology of a Ca2+ mobilizing second messenger - Cyclic ADP-ribose. J. Cell. Mol. Med. 2006, 10, 407–422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, H.C. Physiological Functions of Cyclic ADP-Ribose and NAADP as Calcium Messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Liu, H.-X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H. Calcium mobilizing second messengers derived from NAD. Biochim. Biophys. Acta 2015, 1854, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Hashii, M.; Yokoyama, S.; Hoshi, N.; Asai, K.; Kato, T. Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J. Neurochem. 2001, 76, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and NAADP: Fraternal twin messengers for calcium signaling. Sci. China Life Sci. 2011, 54, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Harde, M.; Empson, R.; Potter, B.V.; Galione, A.; Stanton, P.K. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc. Natl. Acad. Sci. USA 1999, 96, 4061–4066. [Google Scholar] [CrossRef] [PubMed]
- Guse, A. Biochemistry, Biology, and Pharmacology of Cyclic Adenosine Diphosphoribose (cADPR). Curr. Med. Chem. 2004, 11, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Chini, E.N.; Nagamune, K.; Wetzel, D.M.; Sibley, L.D. Evidence that the cADPR signalling pathway controls calcium-mediated microneme secretion in Toxoplasma gondii. Biochem. J. 2005, 389, 269–277. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okamoto, H.; Takasawa, S.; Sugawara, A. The CD38-Cyclic ADP-Ribose System in Mammals: Historical Background, Pathophysiology and Perspective. Messenger 2014, 3, 27–34. [Google Scholar] [CrossRef]
- Zhang, F.J.; Gu, Q.M.; Sih, C.J. Bioorganic chemistry of cyclic ADP-ribose (cADPR). Bioorg. Med. Chem. 1999, 7, 653–664. [Google Scholar] [CrossRef]
- Cakir-Kiefer, C.; Muller-Steffner, H.; Schuber, F. Unifying mechanism for Aplysia ADP-ribosyl cyclase and CD38/NAD(+) glycohydrolases. Biochem. J. 2000, 349, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Kirchberger, T.; Zhang, B.; Thomas, M.P.; Weber, K.; Guse, A.H.; Potter, B.V.L. Aberrant cyclization affords a C-6 modified cyclic adenosine 5′-diphosphoribose analogue with biological activity in Jurkat T cells. J. Med. Chem. 2012, 55, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Walseth, T.F.; Lee, H.C. Synthesis and characterization of antagonists of cyclic-ADP-ribose-induced Ca2+ release. BBA Mol. Cell Res. 1993, 1178, 235–242. [Google Scholar] [CrossRef]
- Guse, A.H.; Cakir-Kiefer, C.; Fukuoka, M.; Shuto, S.; Weber, K.; Bailey, V.C.; Matsuda, A.; Mayr, G.W.; Oppenheimer, N.; Schuber, F.; et al. Novel hydrolysis-resistant analogues of cyclic ADP-ribose: Modification of the “northern” ribose and calcium release activity. Biochemistry 2002, 41, 6744–6751. [Google Scholar] [CrossRef] [PubMed]
- Bailey, V.C.; Fortt, S.M.; Summerhill, R.J.; Galione, A.; Potter, B.V.L. Cyclic aristeromycin diphosphate ribose: A potent and poorly hydrolysable Ca2+ -mobilising mimic of cyclic adenosine diphosphate ribose. FEBS Lett. 1996, 379, 227–230. [Google Scholar] [CrossRef]
- Shuto, S.; Matsuda, A. Chemistry of Cyclic ADP-Ribose and Its Analogs. Curr. Med. Chem. 2004, 11, 827–845. [Google Scholar] [CrossRef] [PubMed]
- Shuto, S.; Fukuoka, M.; Manikowsky, A.; Ueno, Y.; Nakano, T.; Kuroda, R.; Kuroda, H.; Matsuda, A. Total synthesis of cyclic ADP-carbocyclic-ribose, a stable mimic of Ca2+-mobilizing second messenger cyclic ADP-ribose. J. Am. Chem. Soc. 2001, 123, 8750–8759. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure-activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Sakaguchi, N.; Kudoh, T.; Takano, S.; Uehara, M.; Murayama, T.; Sakurai, T.; Hashii, M.; Higashida, H.; Weber, K.; et al. Design and Synthesis of Cyclic ADP-4-Thioribose as a Stable Equivalent of Cyclic ADP-Ribose, a Calcium Ion-Mobilizing Second Messenger. Angew. Chem. Int. Ed. 2013, 52, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.K.; Guse, A.H.; Potter, B.V.L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5′-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [Google Scholar] [CrossRef] [PubMed]
- Shuto, S.; Shirato, M.; Sumita, Y.; Ueno, Y.; Matsuda, A. Nucleosides and Nucleotides. 173. Synthesis of Cyclic IDP-carbocyclic-ribose, a Stable Mimic of Cyclic ADP-ribose. Significant Facilitation of the Intramolecular Condensation Reaction of N-1-(Carbocyclic-ribosyl)inosine 5′,6′-Diphosphate Derivatives by an. J. Org. Chem. 1998, 63, 1986–1994. [Google Scholar] [CrossRef]
- Zhang, L.; Guse, A.H. Cyclic ADP-Ribose Analogues with Minimal Structure: Synthesis and Calcium-Release Activity. In Drug Discovery Research; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 186–202. ISBN 9780470131862. [Google Scholar]
- Xu, J.; Yang, Z.; Dammermann, W.; Zhang, L.; Guse, A.H.; Zhang, L.H. Synthesis and agonist activity of cyclic ADP-ribose analogues with substitution of the northern ribose by ether or alkane chains. J. Med. Chem. 2006, 49, 5501–5512. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yang, Z.; Zhang, L.; Kunerth, S.; Fliegert, R.; Weber, K.; Guse, A.H.; Zhang, L. Synthesis and biological evaluation of novel membrane-permeant cyclic ADP-ribose mimics: N1-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylinosine 5′,5″-cyclicpyrophosphate (cIDPRE) and 8-substituted derivatives. J. Med. Chem. 2004, 47, 5674–5682. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, M.; Liu, J.; Zhang, K.; Yang, Z.; Zhang, L.; Zhang, L. Concise syntheses of trifluoromethylated cyclic and acyclic analogues of cADPR. Molecules 2010, 15, 8689–8701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wagner, G.K.; Weber, K.; Garnham, C.; Morgan, A.J.; Galione, A.; Guse, A.H.; Potter, B.V.L. 2′-deoxy cyclic adenosine 5′-diphosphate ribose derivatives: Importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Kirchberger, T.; Huang, X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. Trifluoromethylated cyclic-ADP-ribose mimic: Synthesis of 8-trifluoromethyl-N(1)-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylino sine-5′,5″-cyclic pyrophosphate (8-CF(3)-cIDPRE) and its calcium release activity in T cells. Org. Biomol. Chem. 2010, 8, 4705–4715. [Google Scholar] [CrossRef] [PubMed]
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a New N1-Pentyl Analogue of Cyclic Inosine Diphosphate Ribose (cIDPR) as a Stable Potential Mimic of Cyclic ADP Ribose (cADPR). Eur. J. Org. Chem. 2002, 2002, 4234–4238. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Piccialli, G.; Mayol, L. A solid-phase approach to the synthesis of N-1-alkyl analogues of cyclic inosine-diphosphate-ribose (cIDPR). Tetrahedron 2010, 66, 1931–1936. [Google Scholar] [CrossRef]
- Chang, S.H.; Han, J.L.; Tseng, S.Y.; Lee, H.Y.; Lin, C.W.; Lin, Y.C.; Jeng, W.Y.; Wang, A.H.J.; Wu, C.Y.; Wong, C.H. Glycan array on aluminum oxide-coated glass slides through phosphonate chemistry. J. Am. Chem. Soc. 2010, 132, 13371–13380. [Google Scholar] [CrossRef] [PubMed]
- De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Piccialli, G.; Varra, M. Synthetic studies on the glycosylation of the base residues of inosine and uridine. J. Chem. Soc. Perkin Trans. 1 1999, 3489–3493. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza Dörwald, F. Side Reactions in Organic Synthesis: A Guide to Successful Synthesis Design; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2005; ISBN 3527310215. [Google Scholar]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Piccialli, G.; Luciano, M. Facile solid-phase synthesis of AICAR 5-monophosphate (ZMP) and its 4-N-Alkyl derivatives. Eur. J. Org. Chem. 2010, 1517–1524. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D’Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. A facile synthesis of 5′-Fluoro-5′-deoxyacadesine (5′-F-AICAR): A novel non-phosphorylable AICAR Analogue. Molecules 2012, 17, 13036–13044. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Synthesis of new acadesine (AICA-riboside) analogues having acyclic D-ribityl or 4-hydroxybutyl chains in place of the ribose. Molecules 2013, 18, 9420–9431. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, S.; Oliviero, G.; Amato, J.; Borbone, N.; Cerullo, V.; Hemminki, A.; Piccialli, V.; Zaccaria, S.; Mayol, L.; Piccialli, G. Synthesis and biological evaluation of unprecedented ring-expanded nucleosides (RENs) containing the imidazo[4,5-d][1,2,6]oxadiazepine ring system. Chem. Commun. 2012, 48, 9310–9312. [Google Scholar] [CrossRef] [PubMed]
- Hyde, R.M.; Broom, A.D.; Buckheit, R.W. Antiviral amphipathic oligo- and polyribonucleotides: Analogue development and biological studies. J. Med. Chem. 2003, 46, 1878–1885. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Schrodinger, Inc. Release 11.3. 2017. Available online: https://www.schrodinger.com/products/macrocycles.
- Oliviero, G.; Borbone, N.; Amato, J.; D’Errico, S.; Galeone, A.; Piccialli, G.; Varra, M.; Mayol, L. Synthesis of quadruplex-forming tetra-end-linked oligonucleotides: Effects of the linker size on quadruplex topology and stability. Biopolymers 2009, 91, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Oliviero, G.; Roviello, G.N.; Bucci, E.M.; Piccialli, G. G-quadruplex-forming oligonucleotide conjugated to magnetic nanoparticles: Synthesis, characterization, and enzymatic stability assays. Bioconjug. Chem. 2012, 23, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Piccialli, V.; Borbone, N.; Oliviero, G. Ruthenium-catalyzed oxidative cyclization of 1,7-dienes. A novel diasteroselective synthesis of 2,7-disubstituted trans-oxepane diols. Tetrahedron Lett. 2007, 48, 5131–5135. [Google Scholar] [CrossRef]
- Piccialli, V.; D’Errico, S.; Borbone, N.; Oliviero, G.; Centore, R.; Zaccaria, S. A general synthesis of bis-α-acyloxy-1,4- and -1,5-diketones through catalytic oxidative opening of acylated THF and THP diols. Eur. J. Org. Chem. 2013, 2, 1781–1789. [Google Scholar] [CrossRef]
- Secondo, A.; Staiano, R.I.; Scorziello, A.; Sirabella, R.; Boscia, F.; Adornetto, A.; Valsecchi, V.; Molinaro, P.; Canzoniero, L.M.T.; Di Renzo, G.; et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium 2007, 42, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).