The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6 β-Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
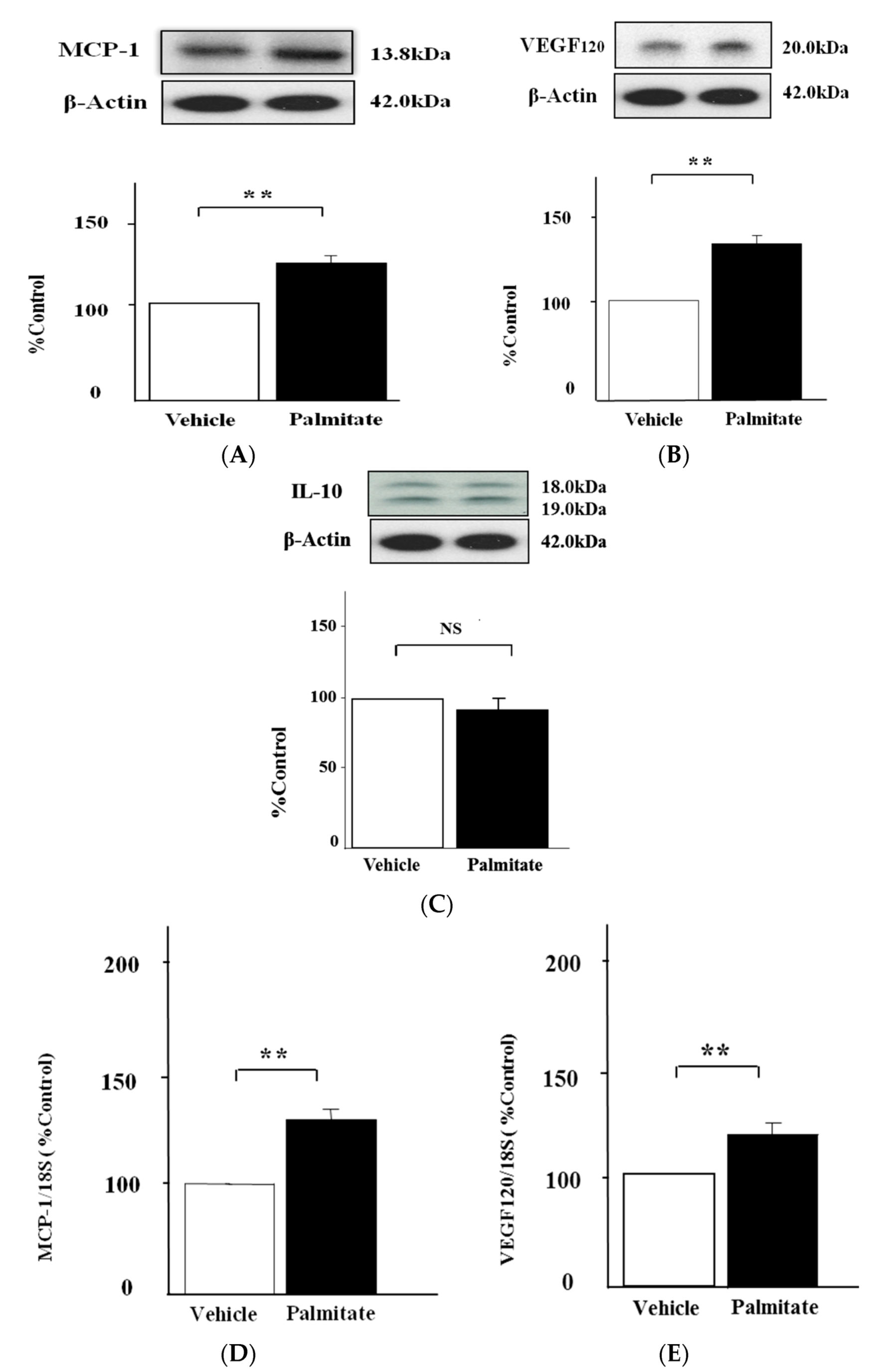
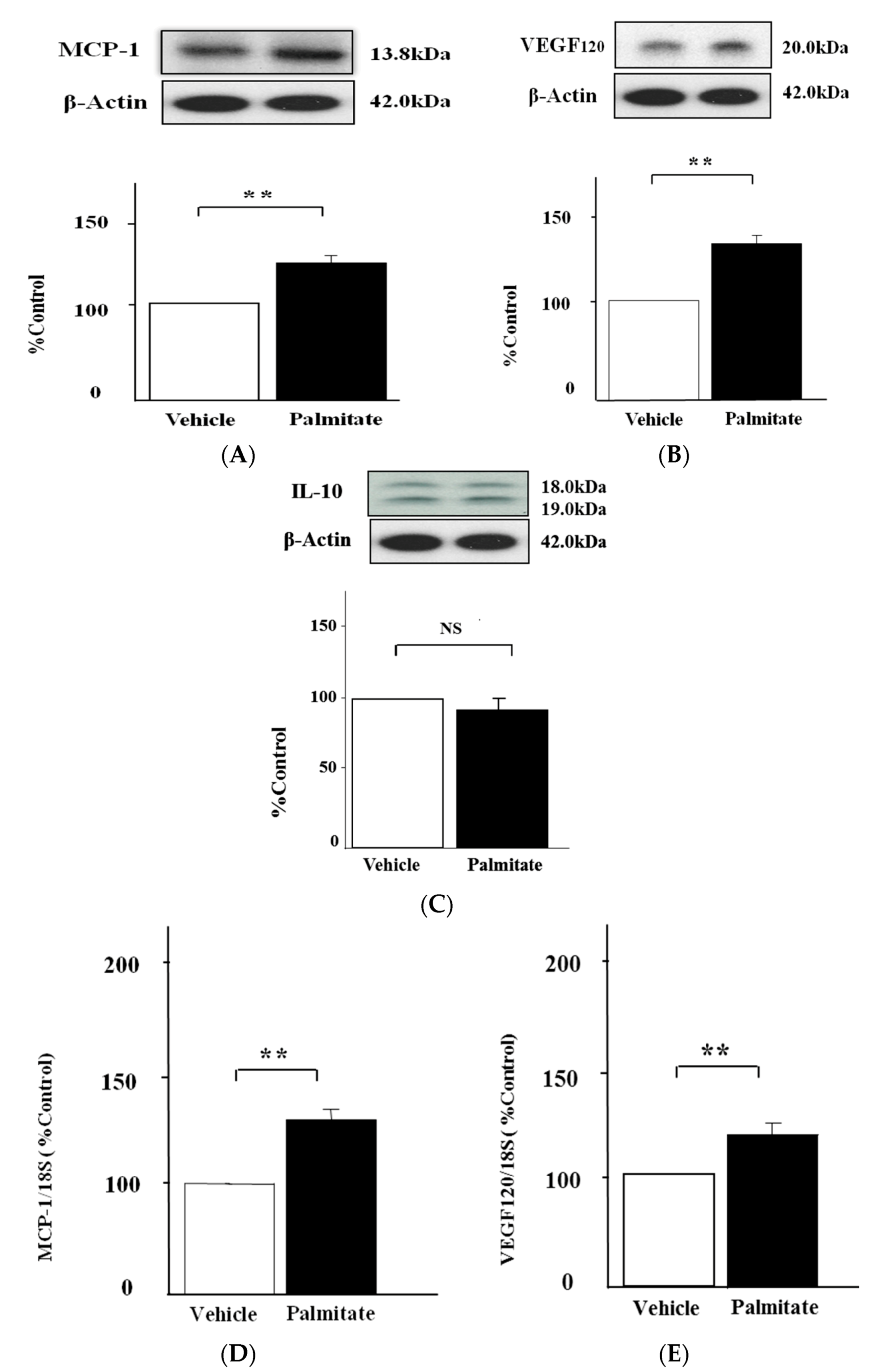
2.1. Palmitate Augments MCP-1 and VEGF120 Secretion by MIN6 Cells
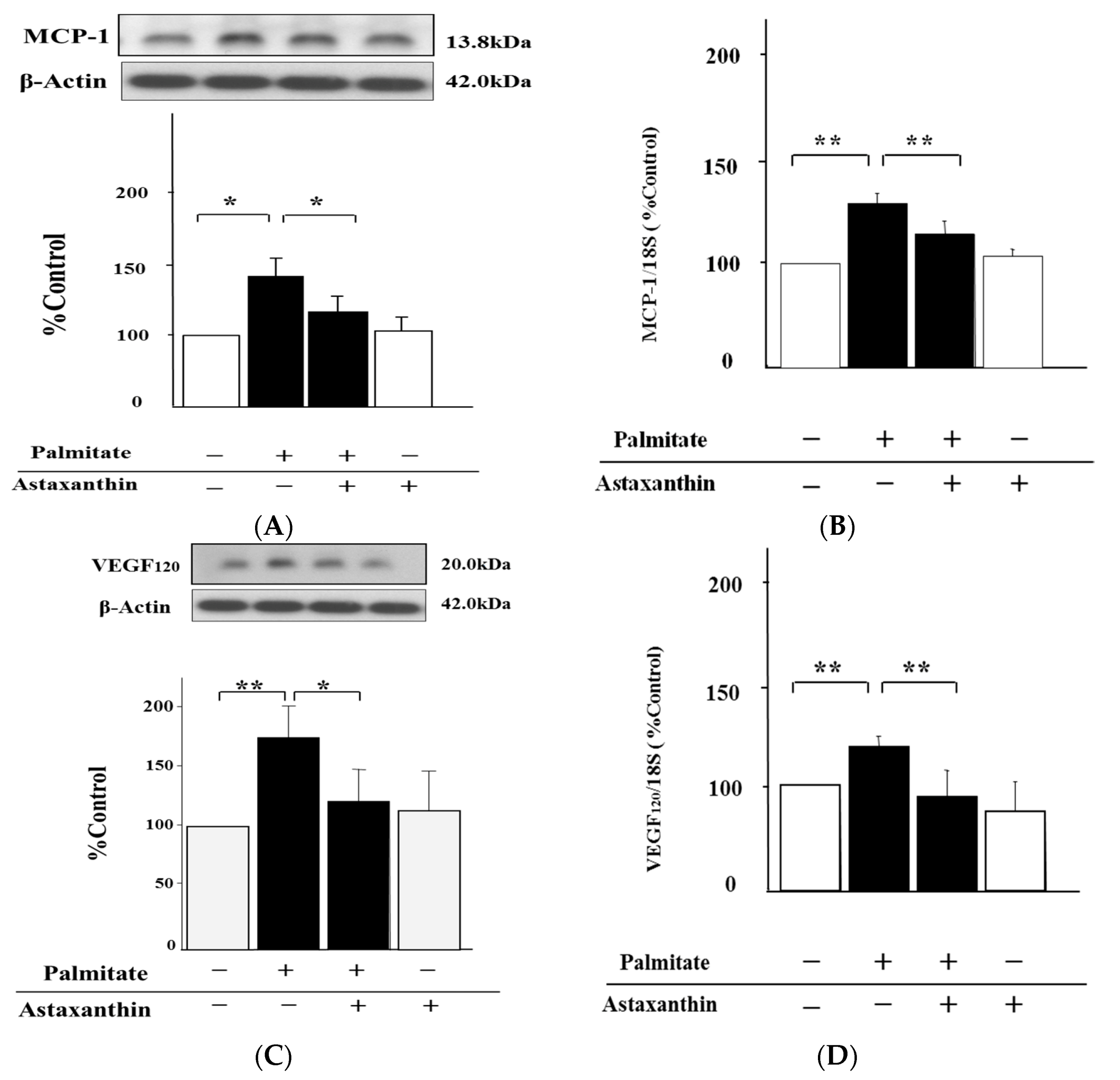
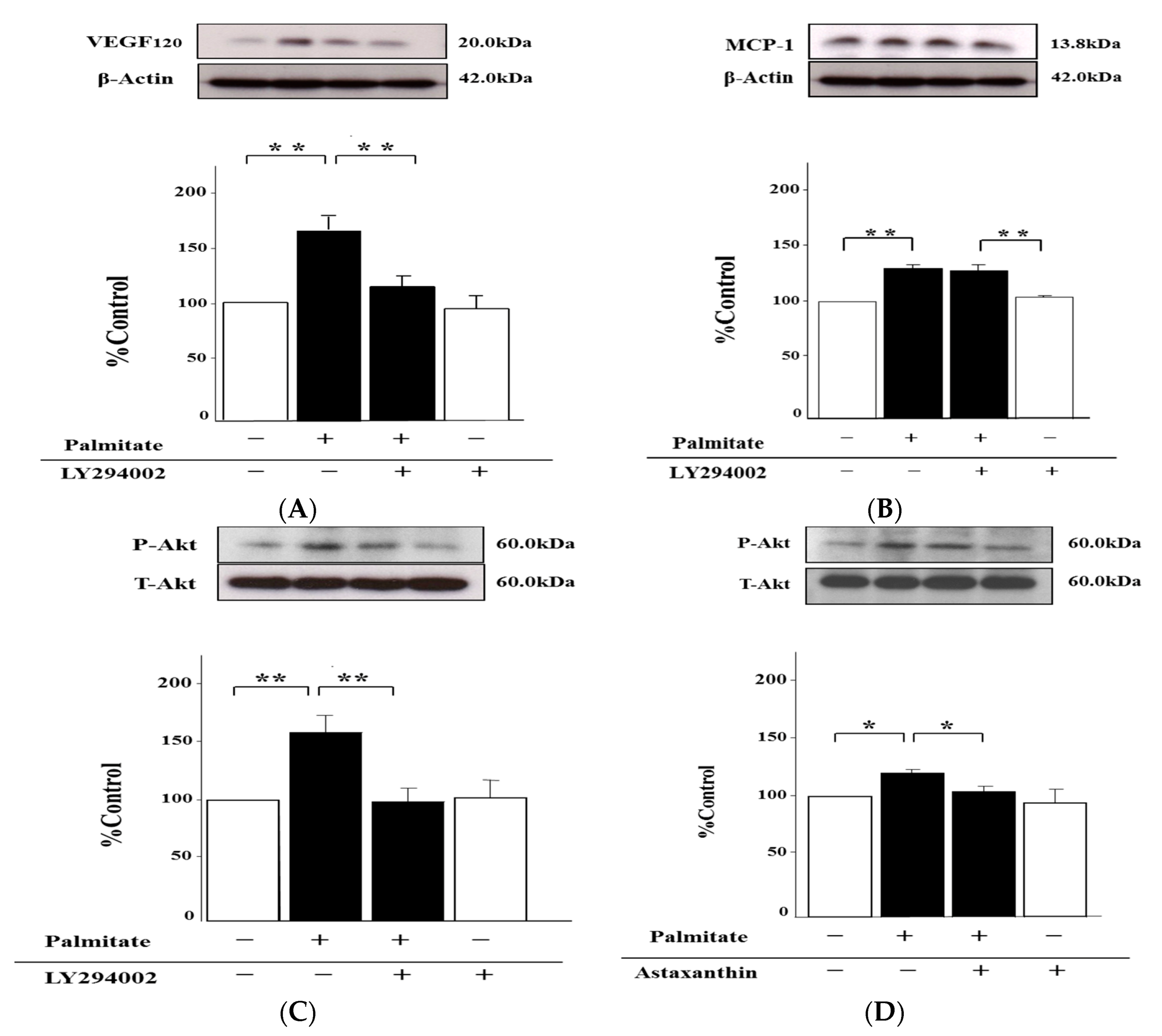
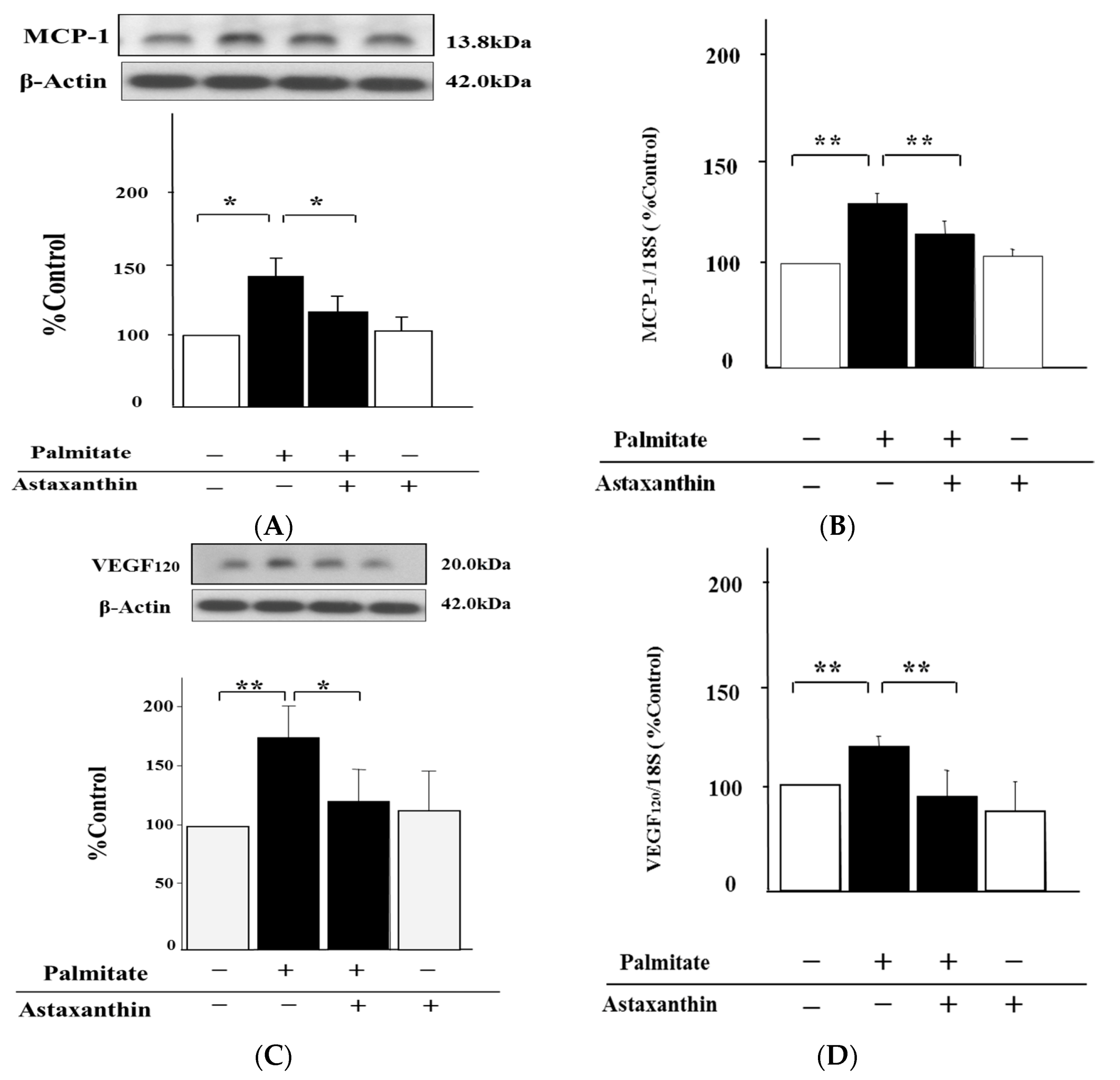
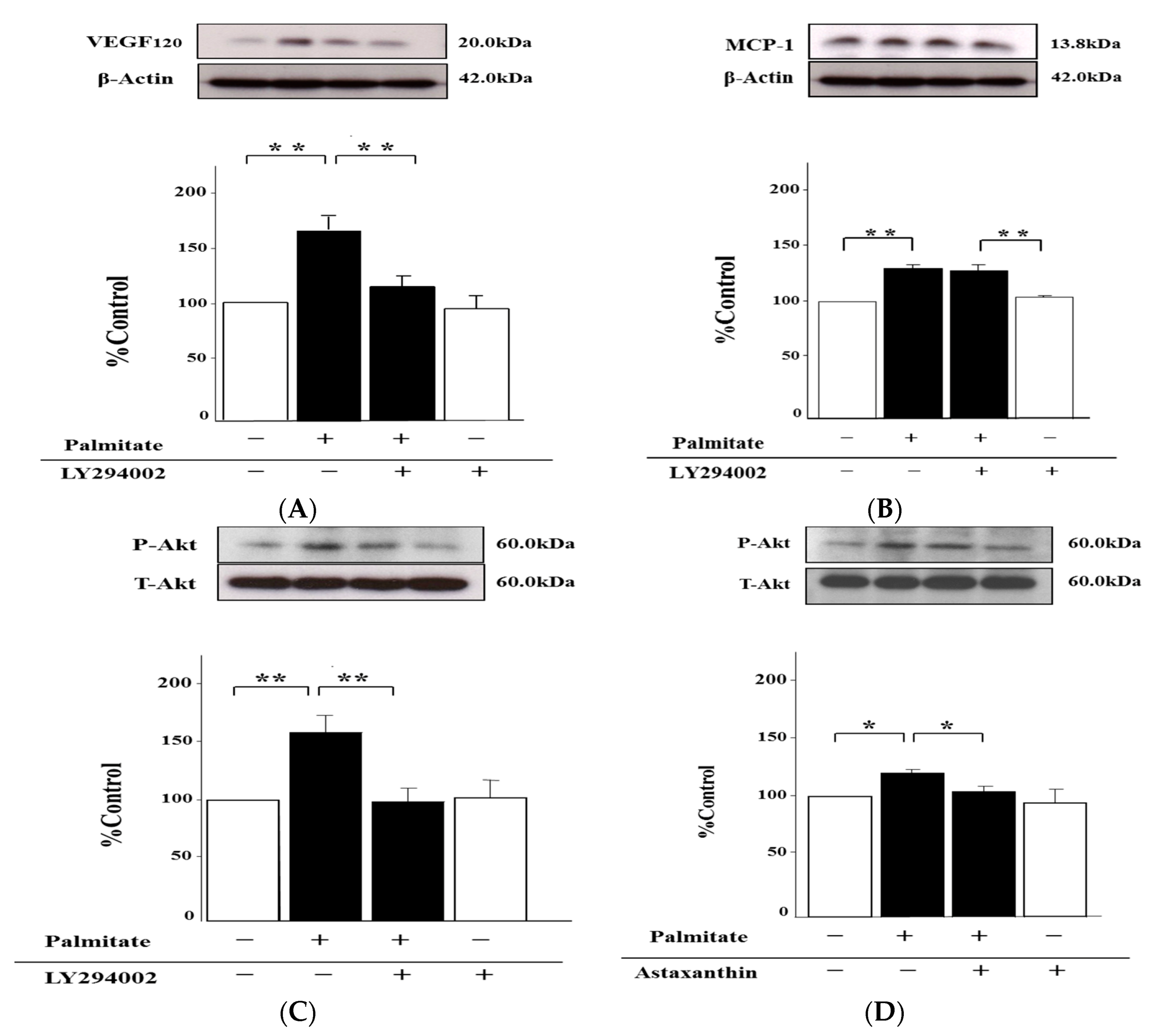
2.2. Astaxanthin Reverses Palmitate-Induced Enhancement of MCP-1 and VEGF120 Secretion
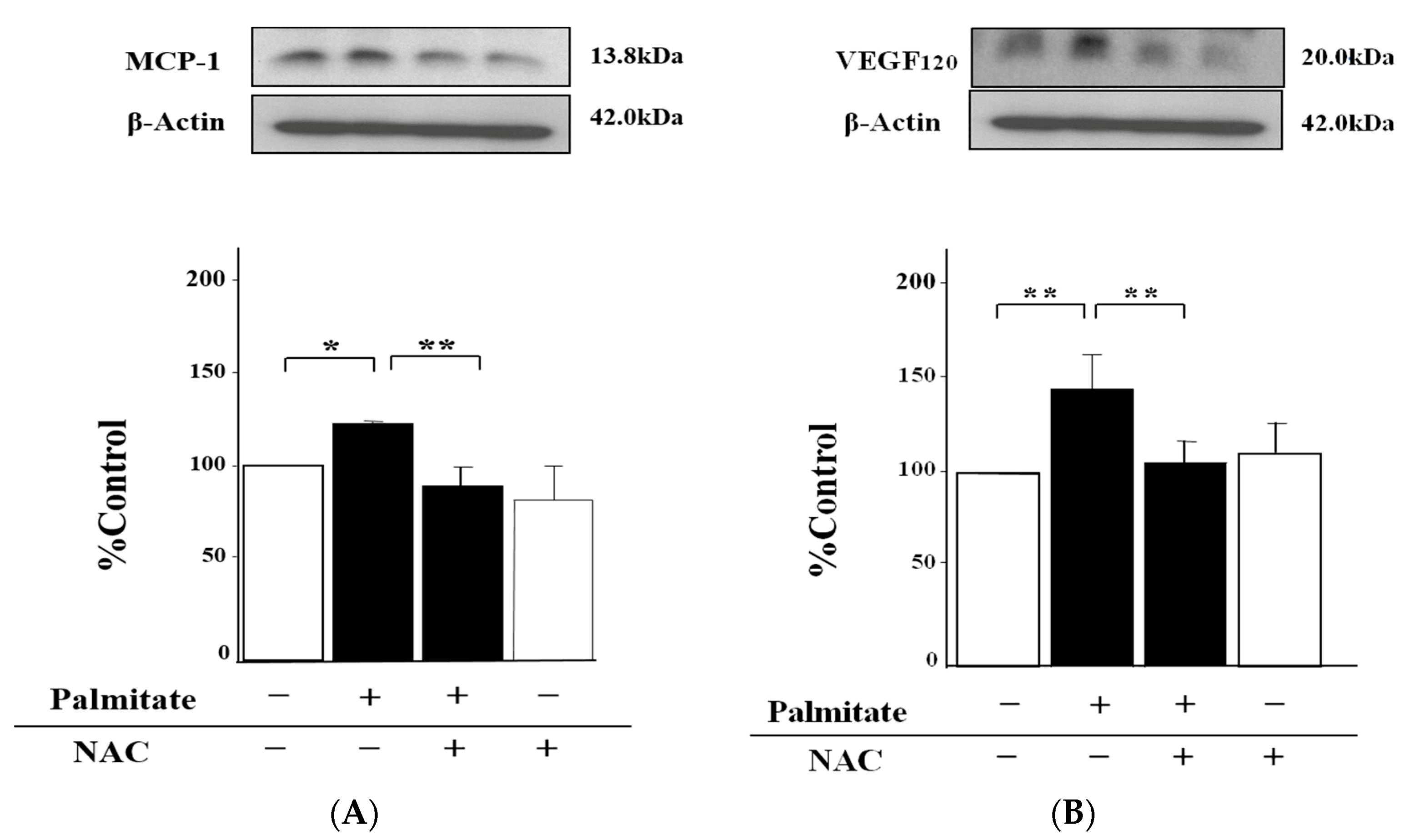
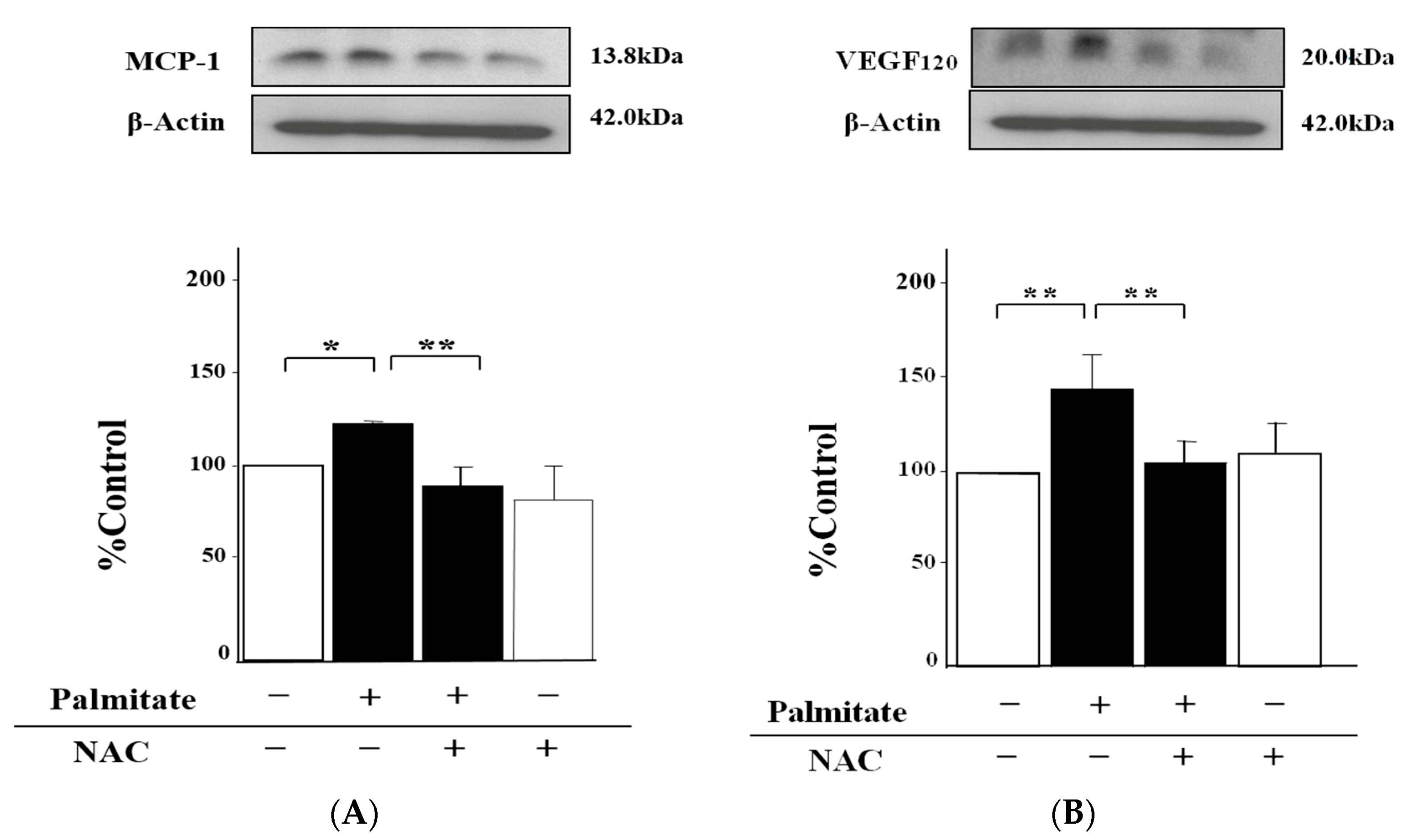
2.3. NAC, an Antioxidant Agent, Can Inhibit Palmitate-Stimulated MCP-1 and VEGF120 Secretion
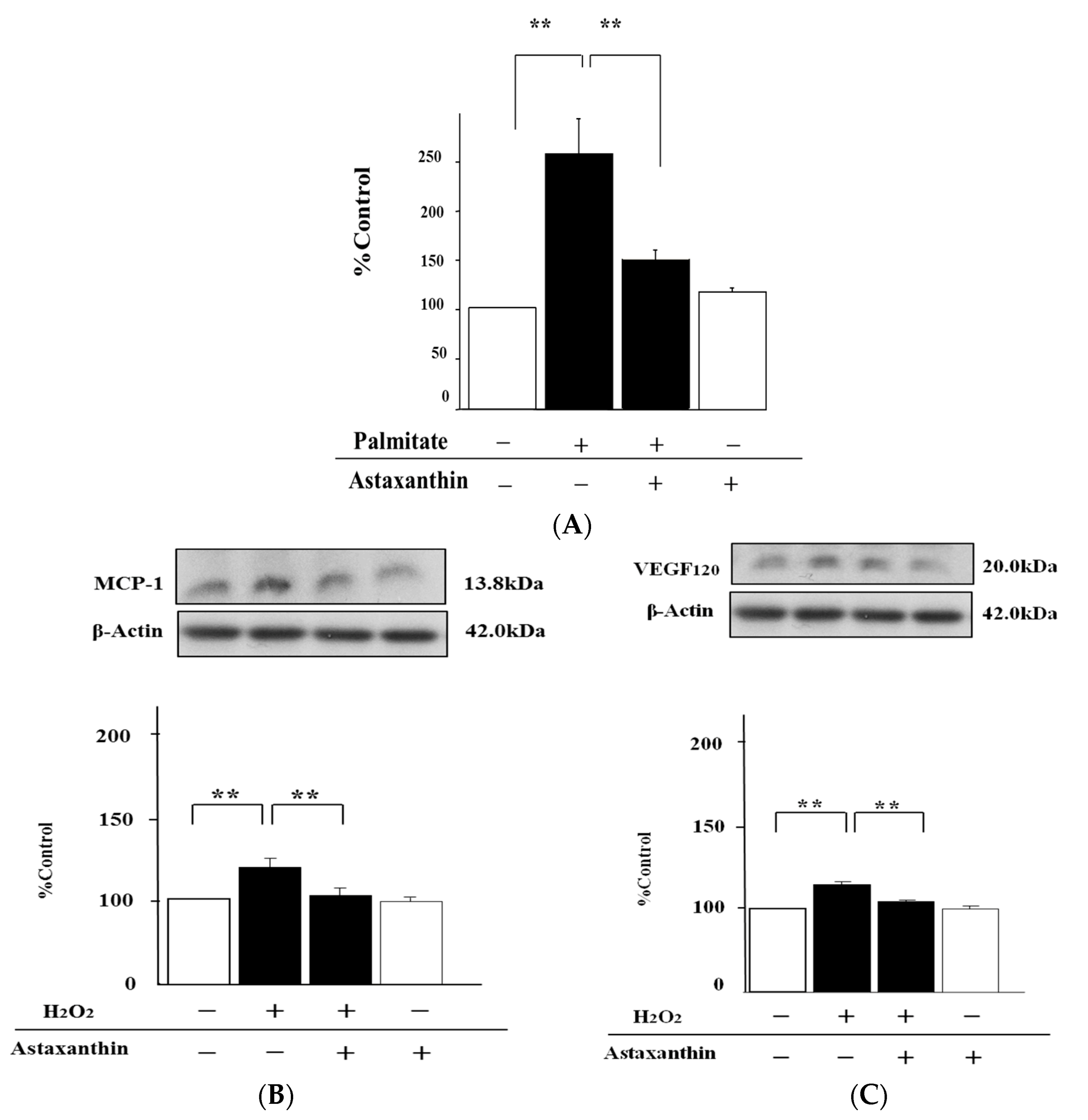
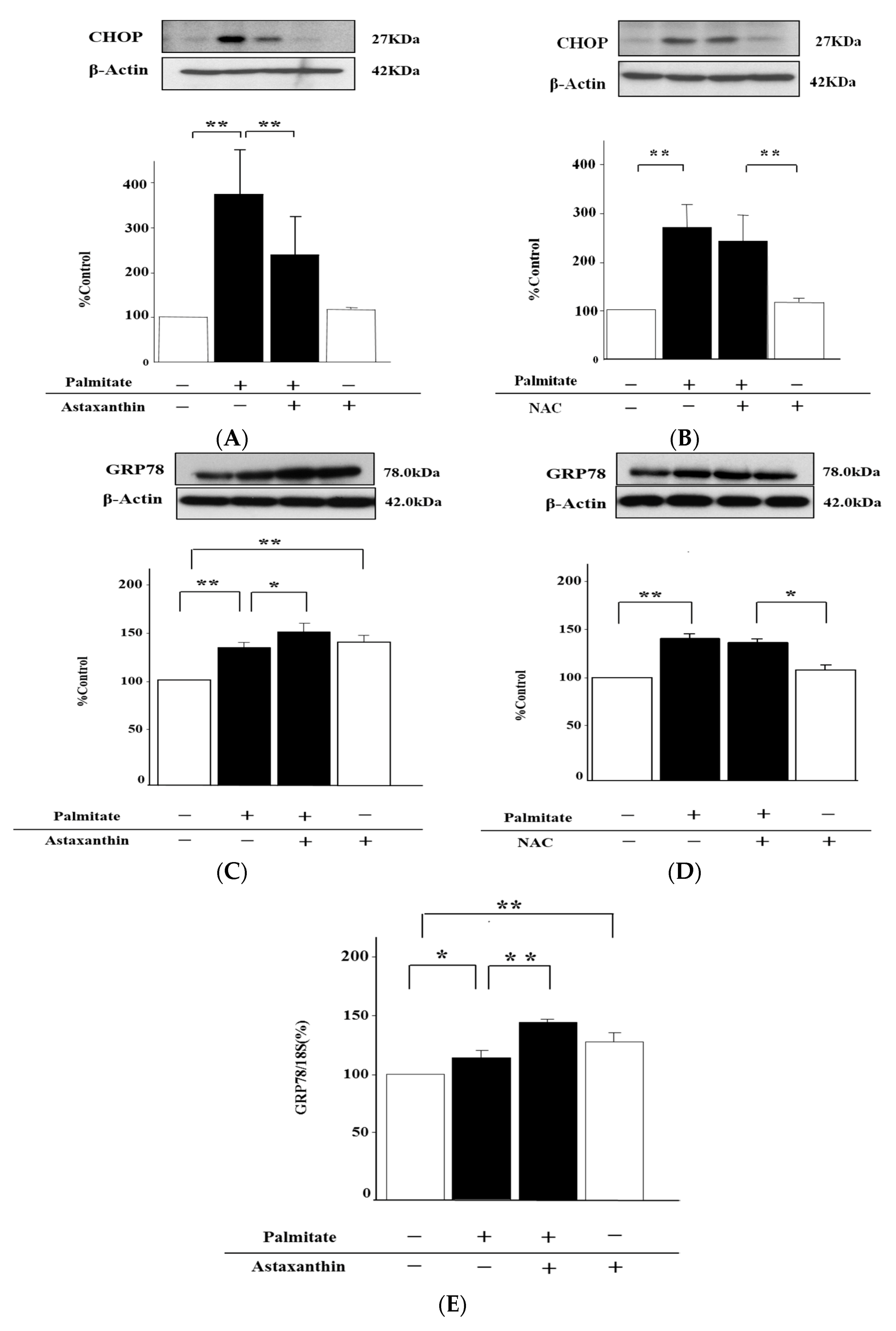
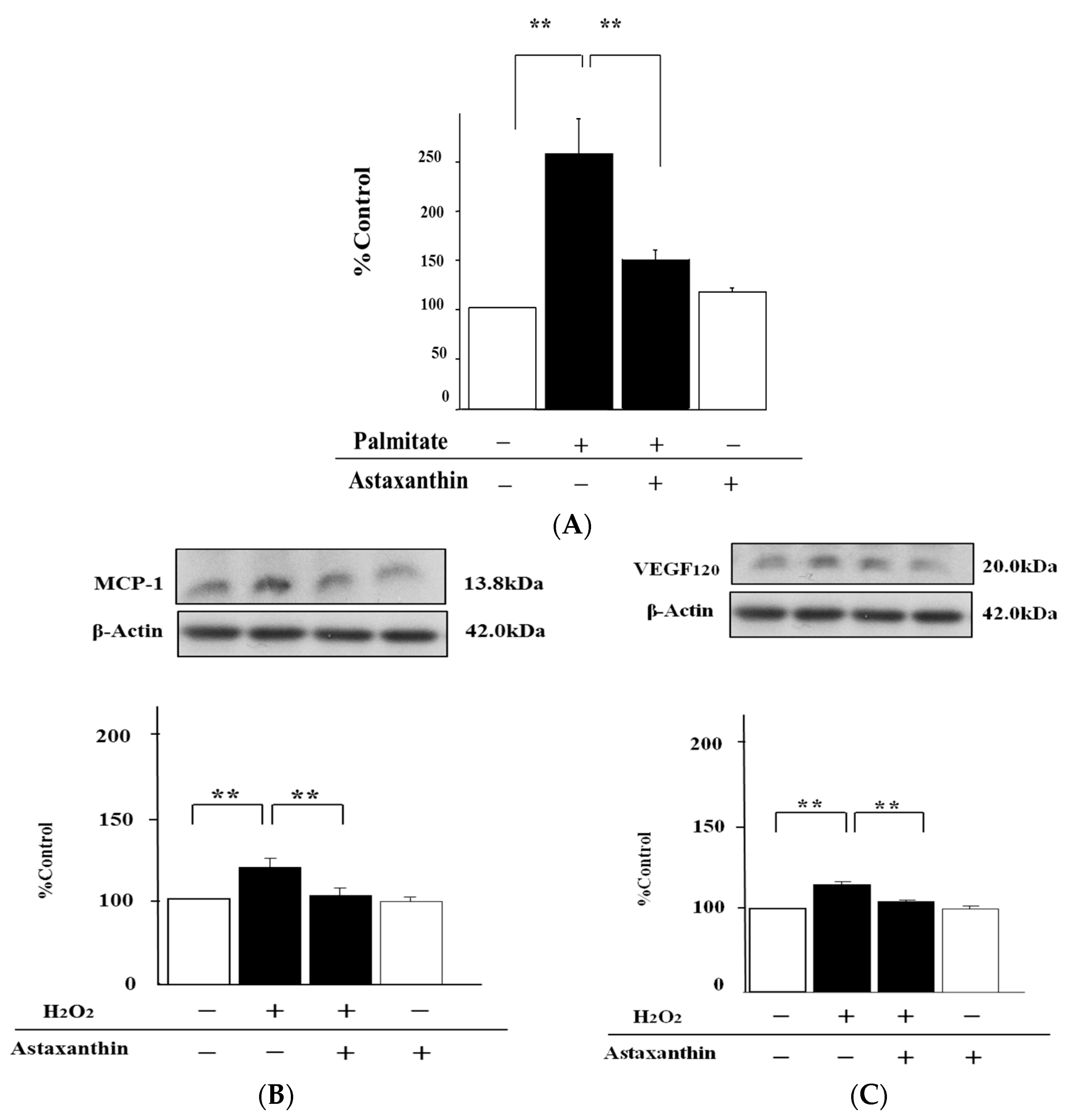
2.4. Astaxanthin Can Inhibit Oxidative Stress
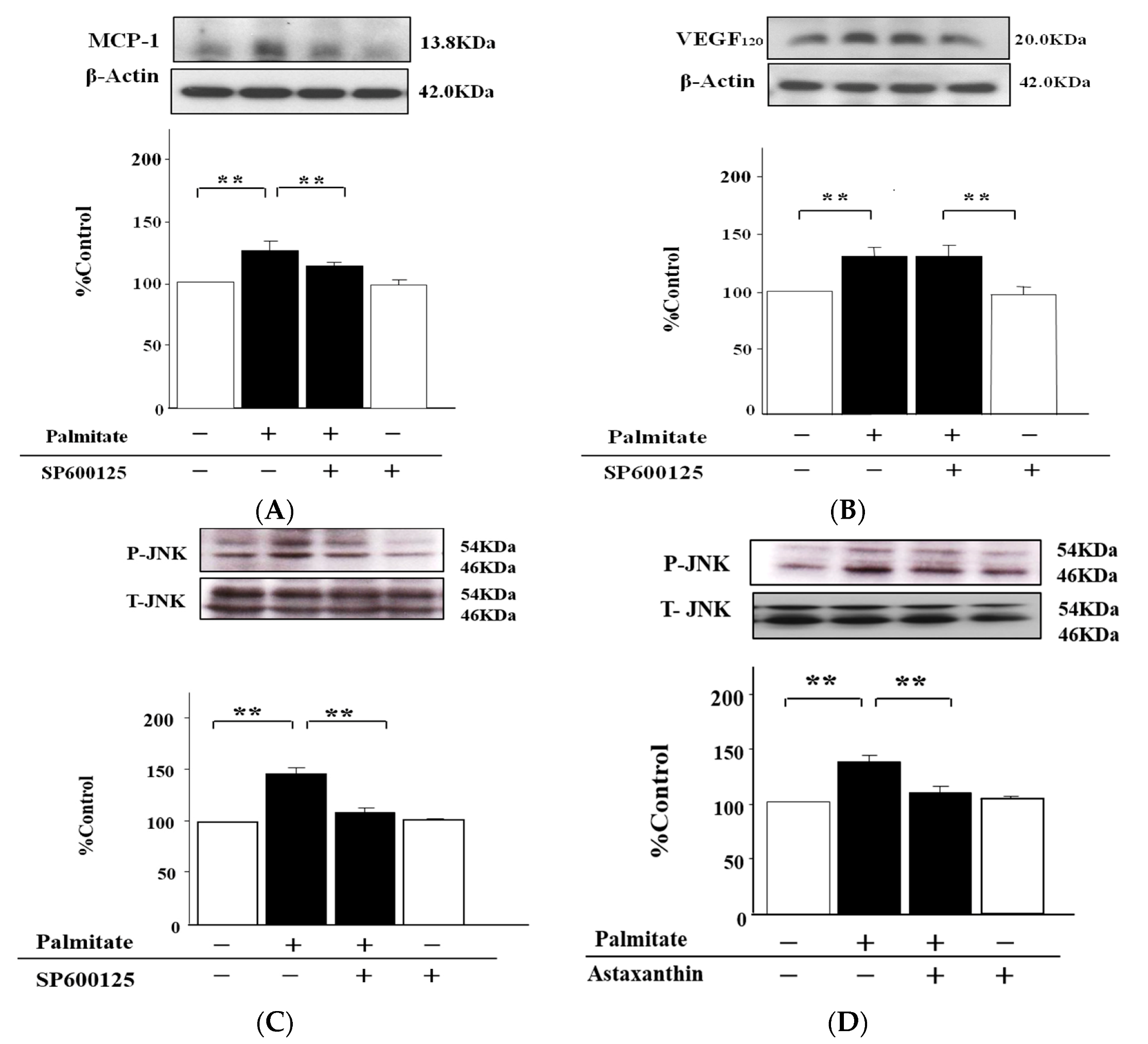
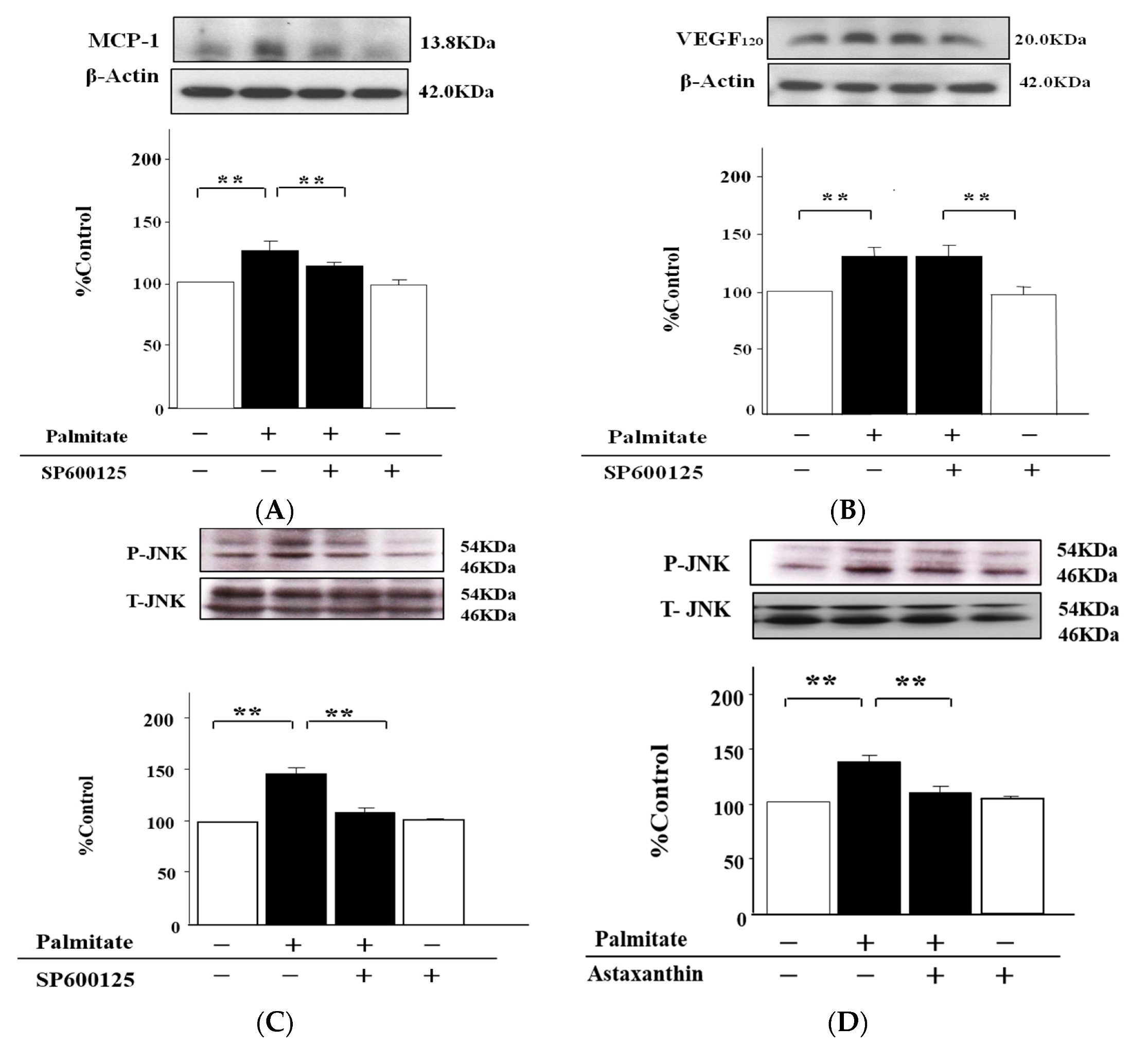
2.5. The MCP-1 Release by MIN6 Cells Treated with Palmitate Is Increased via JNK Pathways
2.6. Palmitate Upregulates VEGF120 Secretion via PI3K Pathways
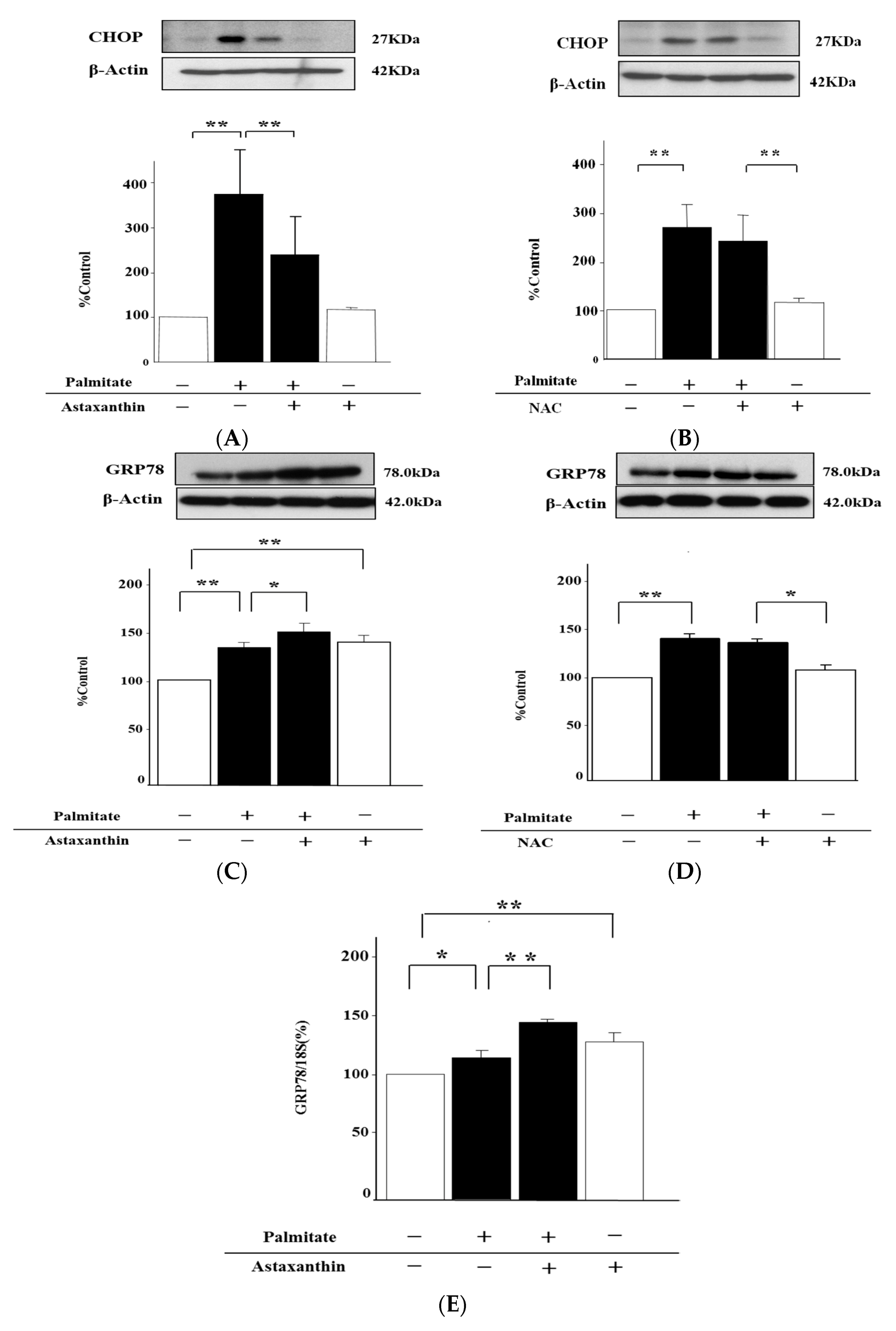
2.7. Astaxanthin Can Eliminate ER Stress via the Enhancement of GRP78 Expression
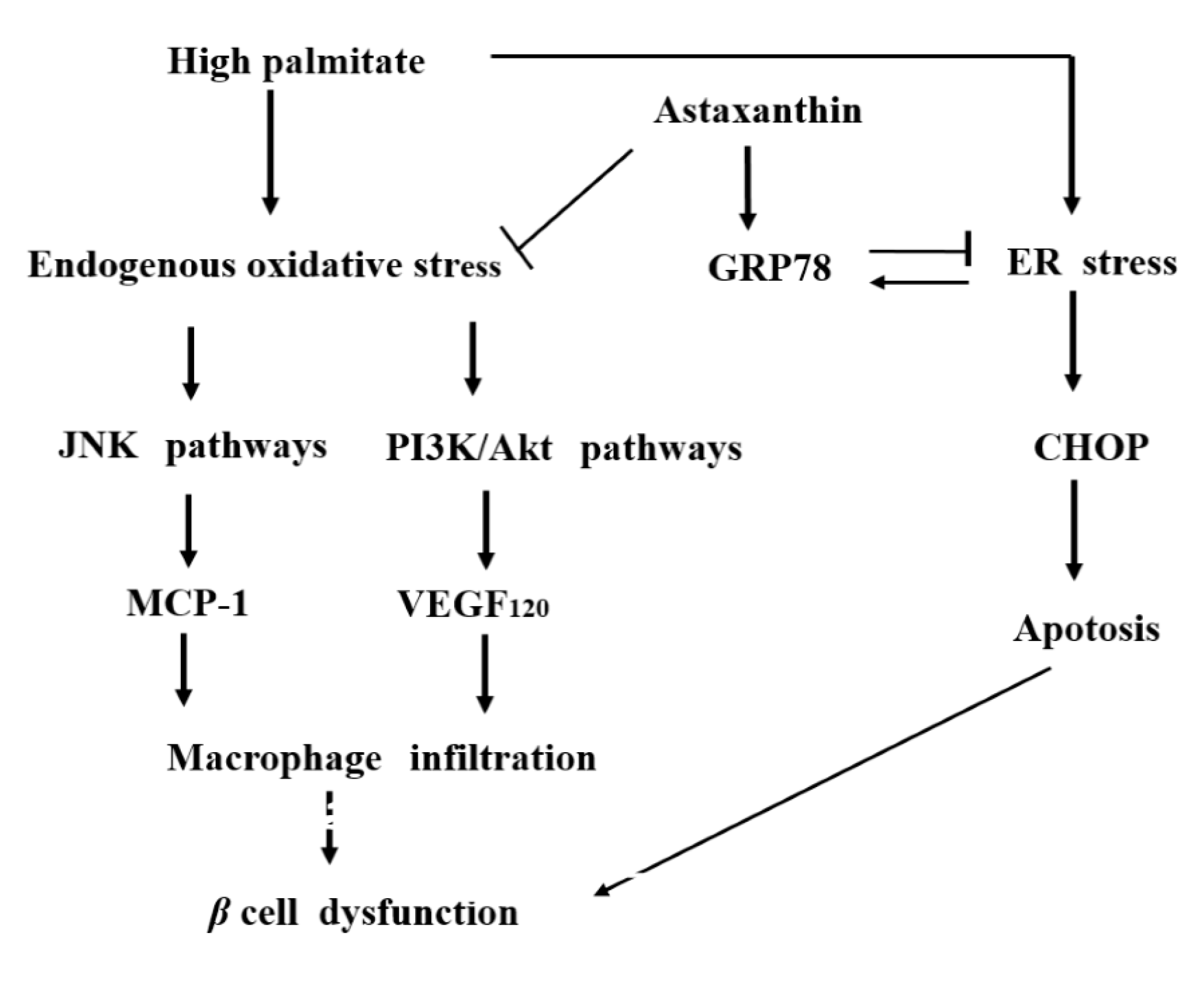
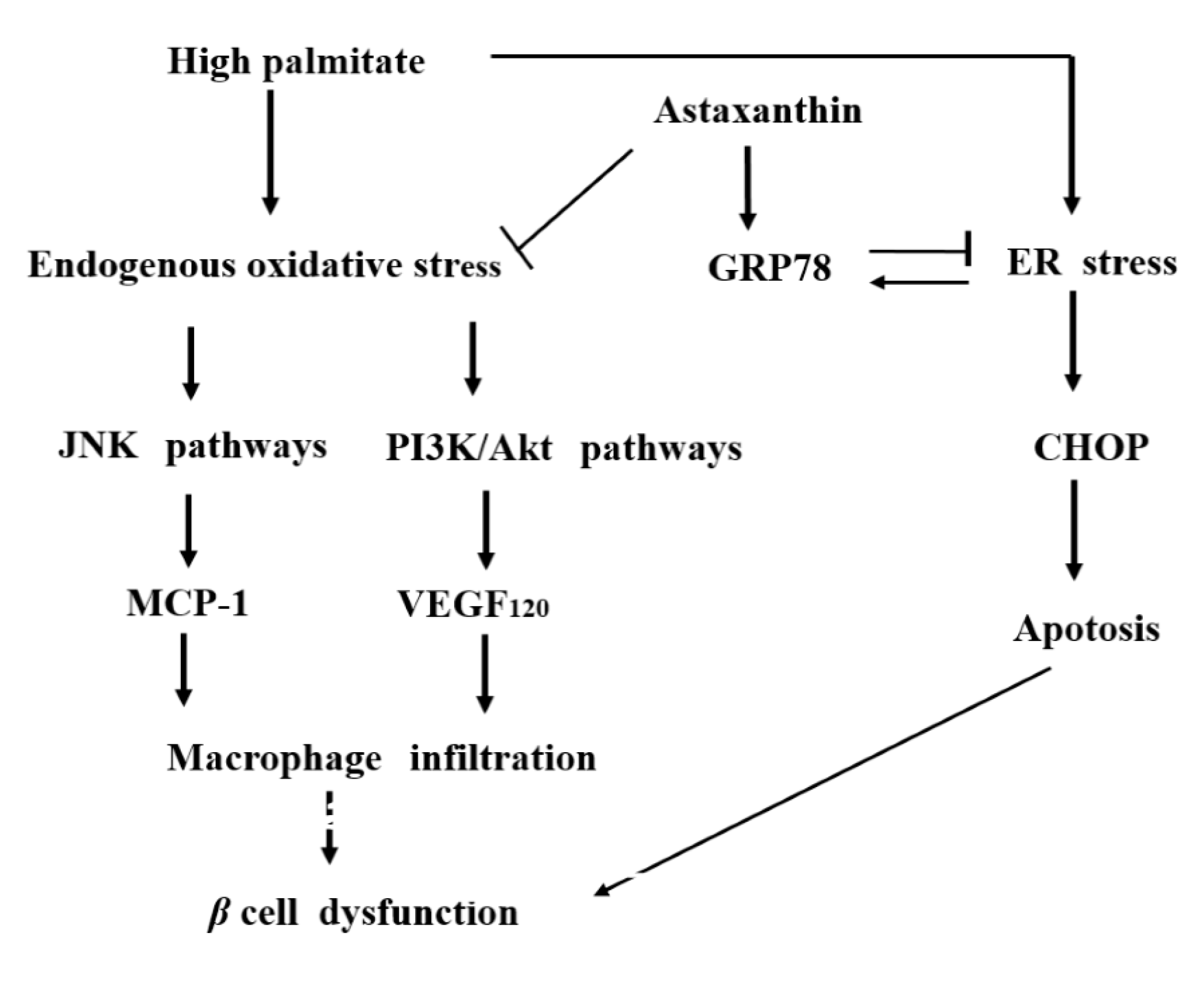
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Preparation of MIN6 Cells
4.3. Treatment of MIN6 Cells
4.4. Immunoblotting
4.5. Real-Time Quantitative PCR
4.6. Quantstudio 3D dPCR
4.7. Quantification of Hydroperoxides
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications-a review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Naito, Y.; Takanami, Y.; Ishii, T.; Kawai, Y.; Akagiri, S.; Kato, Y.; Osawa, T.; Yoshikawa, T. Astaxanthin improves muscle lipid metabolism in exercise via inhibitory effect of oxidative CPT I modification. Biochem. Biophys. Res. Commun. 2008, 366, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Koyama, T.; Takahashi, J.; Yazawa, K. Effects of astaxanthin supplementation on exercise-induced fatigue in mice. Biol. Pharm. Bull. 2006, 29, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Tani, M.; Uto-Kondo, H.; Iizuka, M.; Saita, E.; Sone, H.; Kurata, H.; Kondo, K. Astaxanthin suppresses scavenger receptor expression and matrix metalloproteinase activity in macrophages. Eur. J. Nutr. 2010, 49, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hellsten, A.; Jacobsson, L.S.; Blomqvist, H.M.; Olsson, A.G.; Yuan, X.M. Alpha-tocopherol and astaxanthin decrease macrophage infiltration, apoptosis and vulnerability in atheroma of hyperlipidaemic rabbits. J. Mol. Cell. Cardiol. 2004, 37, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ohgami, K.; Shiratori, K.; Jin, X.H.; Ilieva, I.; Koyama, Y.; Yazawa, K.; Yoshida, K.; Kase, S.; Ohno, S. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-kappaB signaling pathway. Exp. Eye Res. 2006, 82, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Inokuchi, Y.; Shimazawa, M.; Otsubo, K.; Ishibashi, T.; Hara, H. Astaxanthin, a dietary carotenoid, protects retinal cells against oxidative stress in vitro and in mice in vivo. J. Pharm. Pharmacol. 2008, 60, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.S.; Cho, H.H.; Cho, S.; Lee, S.R.; Shin, M.H.; Chung, J.H. Supplementating with dietary astaxanthin combined with collagen hydrolysate improves facial elasticity and decreases matrix metalloproteinase-1 and -12 expression: A comparative study with placebo. J. Med. Food 2014, 17, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox Rep. 2002, 7, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, E.; Bhuvaneswari, S.; Anuradha, C.V. An intervention study in obese mice with astaxanthin, a marine carotenoid-effects on insulin signaling and pro-inflammatory cytokines. Food Funct. 2012, 3, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Bhuvaneswari, S.; Anuradha, C.V. Astaxanthin prevents loss of insulin signaling and improves glucose metabolism in liver of insulin resistant mice. Can. J. Physiol. Pharmacol. 2012, 90, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, M.; Nishida, Y.; Ishibashi, H.; Wada, T.; Fujisaka, S.; Takikawa, A.; Urakaze, M.; Sasaoka, T.; Usui, I.; Tobe, K. Impact of divergent effects of astaxanthin on insulin signaling in L6 cells. Endocrinology 2013, 154, 2600–2612. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Murakami, K.; Bujo, H.; Unoki, H.; Jiang, M.; Nakayama, T.; Saito, Y. Matrix metalloproteinase-3 enhances the free fatty acids-induced VEGF expression in adipocytes through toll-like receptor 2. Exp. Biol. Med. (Maywood) 2008, 233, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa-Hoshimoto, S.; Takahashi, K.; Bujo, H.; Hashimoto, N.; Yagui, K.; Saito, Y. Roles of degree of fat deposition and its localization on VEGF expression in adipocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, e1128–e1136. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Miyokawa-Gorin, K.; Handa, K.; Kitahara, A.; Moriya, R.; Onuma, H.; Sumitani, Y.; Tanaka, T.; Katsuta, H.; Nishida, S.; et al. Endogenous oxidative stress, but not ER stress, induces hypoxia-independent VEGF120 release through PI3K-dependent pathways in 3T3-L1 adipocytes. Obesity 2013, 21, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Hosoya, Y.; Yamashita, H.; Fujita, H.; Ohsugi, M.; Tobe, K.; Kadowaki, T.; Nagai, R.; et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 2007, 56, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, L.; Ubhayasekera, S.J.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-induced impairments of β-cell function are linked with generation of specific ceramide species via acylation of sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.; Jensen, V.H.; Jastroch, M.; Affourtit, C. Palmitate-induced impairment of glucose-stimulated insulin secretion precedes mitochondrial dysfunction in mouse pancreatic islets. Biochem. J. 2016, 473, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamaguchi, S.; Shimoyama, T.; Seki, H.; Miyokawa, K.; Katsuta, H.; Tanaka, T.; Yoshimoto, K.; Ohno, H.; Nagamatsu, S.; et al. JNK- and IkappaB-dependent pathways regulate MCP-1 but not adiponectin release from artificially hypertrophied 3T3-L1 adipocytes preloaded with palmitate in vitro. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, A.; Takahashi, K.; Moriya, R.; Onuma, H.; Handa, K.; Sumitani, Y.; Tanaka, T.; Katsuta, H.; Nishida, S.; Sakurai, T.; et al. Ghrelin augments the expressions and secretions of proinflammatory adipokines, VEGF120 and MCP-1, in differentiated 3T3-L1 adipocytes. J. Cell. Physiol. 2015, 230, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Bhuvaneswari, S.; Yogalakshmi, B.; Sreeja, S.; Anuradha, C.V. Astaxanthin reduces hepatic endoplasmic reticulum stress and nuclear factor-κB-mediated inflammation in high fructose and high fat diet-fed mice. Cell Stress Chaperones 2014, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, E.; Artemenko, K.; Manukyan, L.; Bergquist, J.; Bergsten, P. Oleate protects beta-cells from the toxic effect of palmitate by activating pro-survival pathways of the ER stress response. Biochim. Biophys. Acta 2016, 1861, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Bikopoulos, G.; Wheeler, M.B.; Rozakis-Adcock, M.; Volchuk, A. Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Ladrière, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes. Metab. 2010, 12, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Hao, L.; Li, S.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.; Zi, T.; Chu, X.; Na, L.; et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Teodoro-Morrison, T.; Schuiki, I.; Zhang, L.; Belsham, D.D.; Volchuk, A. GRP78 overproduction in pancreatic beta cells protects against high-fat-diet-induced diabetes in mice. Diabetologia 2013, 56, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Yoon, K.H. Glucolipotoxicity in Pancreatic β-Cells. Diabetes Metab. J. 2011, 35, 444–450. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitahara, A.; Takahashi, K.; Morita, N.; Murashima, T.; Onuma, H.; Sumitani, Y.; Tanaka, T.; Kondo, T.; Hosaka, T.; Ishida, H. The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6 β-Cells. Mar. Drugs 2017, 15, 185. https://doi.org/10.3390/md15060185
Kitahara A, Takahashi K, Morita N, Murashima T, Onuma H, Sumitani Y, Tanaka T, Kondo T, Hosaka T, Ishida H. The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6 β-Cells. Marine Drugs. 2017; 15(6):185. https://doi.org/10.3390/md15060185
Chicago/Turabian StyleKitahara, Atsuko, Kazuto Takahashi, Naru Morita, Toshitaka Murashima, Hirohisa Onuma, Yoshikazu Sumitani, Toshiaki Tanaka, Takuma Kondo, Toshio Hosaka, and Hitoshi Ishida. 2017. "The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6 β-Cells" Marine Drugs 15, no. 6: 185. https://doi.org/10.3390/md15060185
APA StyleKitahara, A., Takahashi, K., Morita, N., Murashima, T., Onuma, H., Sumitani, Y., Tanaka, T., Kondo, T., Hosaka, T., & Ishida, H. (2017). The Novel Mechanisms Concerning the Inhibitions of Palmitate-Induced Proinflammatory Factor Releases and Endogenous Cellular Stress with Astaxanthin on MIN6 β-Cells. Marine Drugs, 15(6), 185. https://doi.org/10.3390/md15060185