
Pharmacophore-Based Virtual Screening of Novel Inhibitors and Docking Analysis for CYP51A from Penicillium italicum

Abstract
:1. Introduction
2. Results
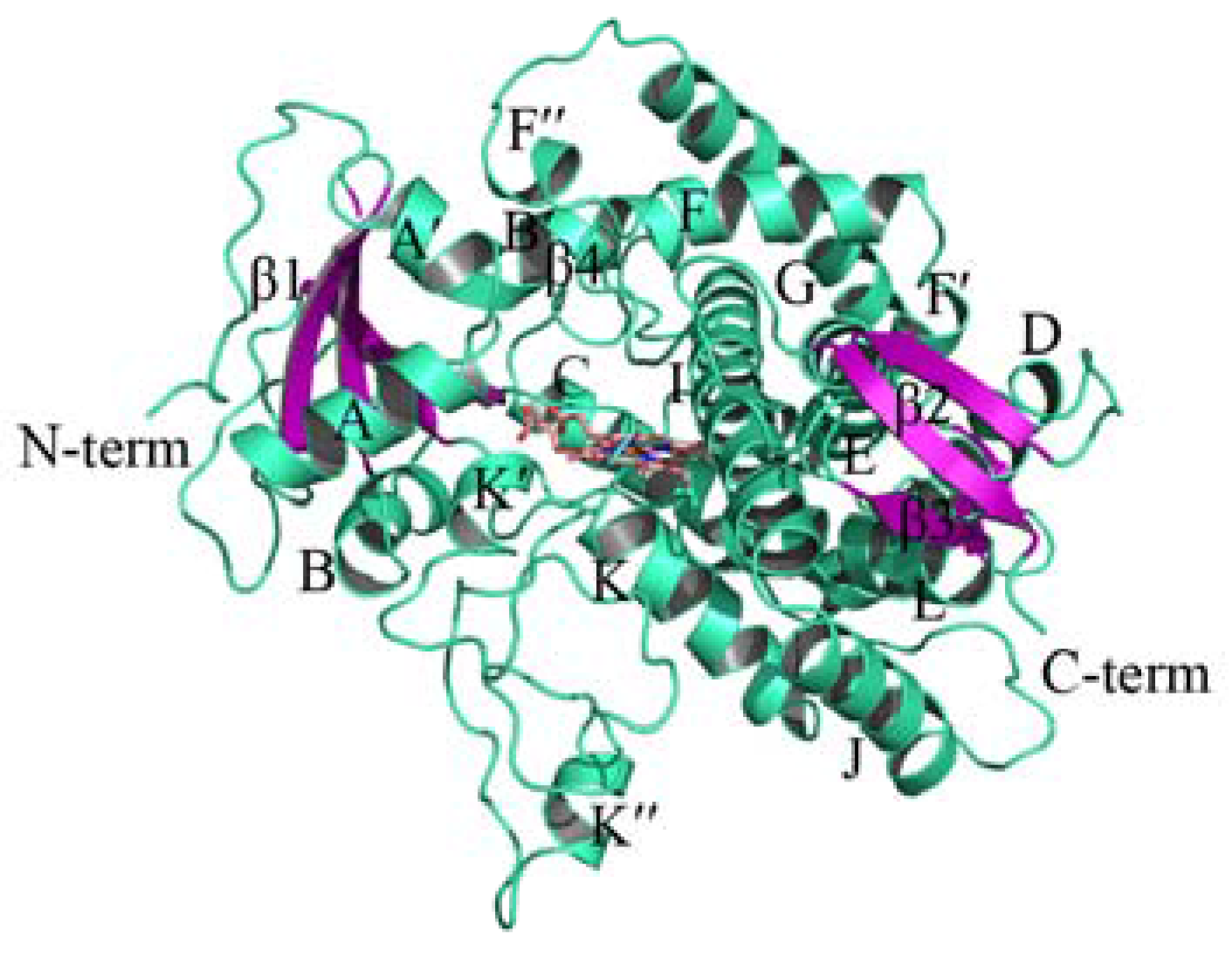
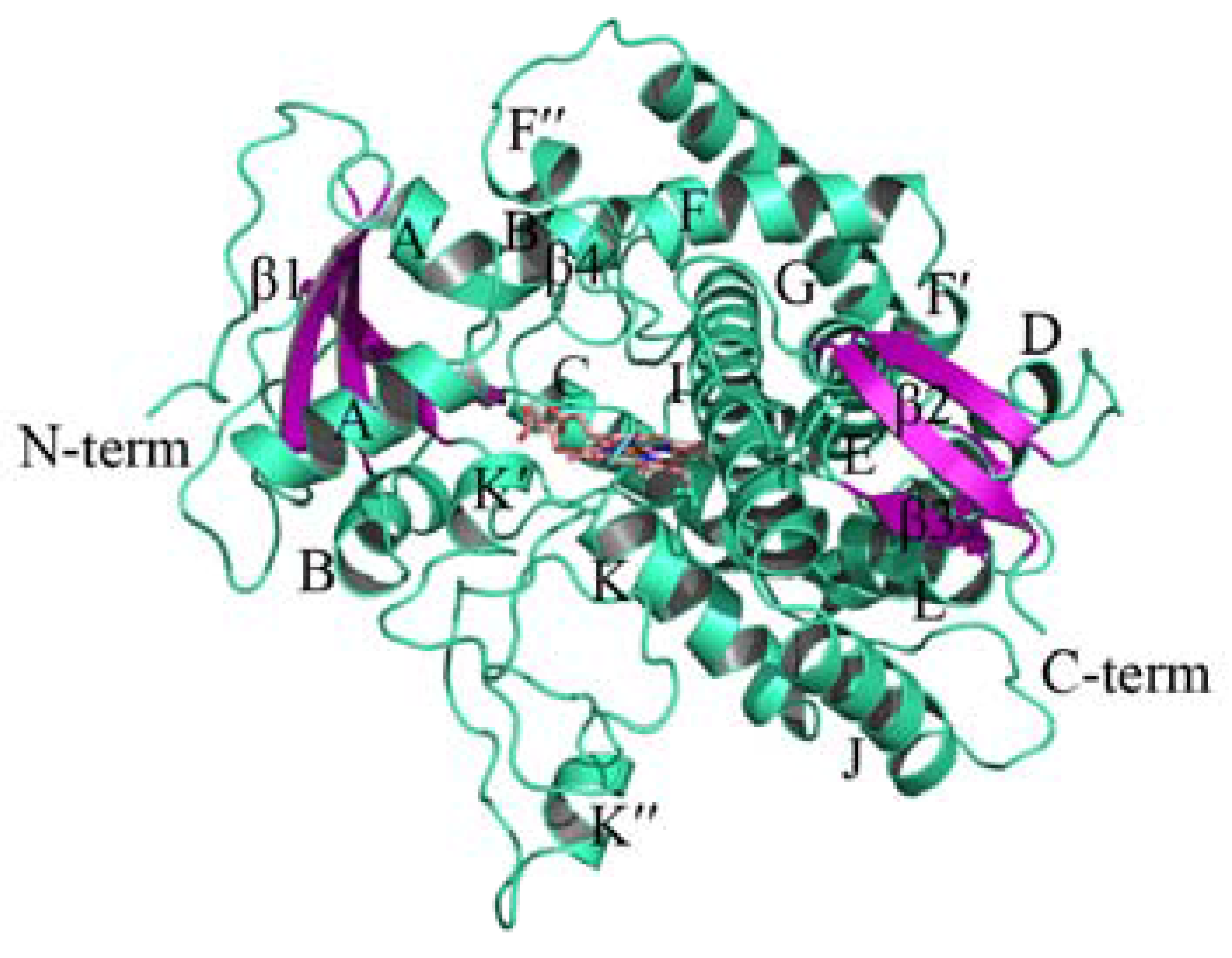
2.1. Homology Modeling of PiCYP51A
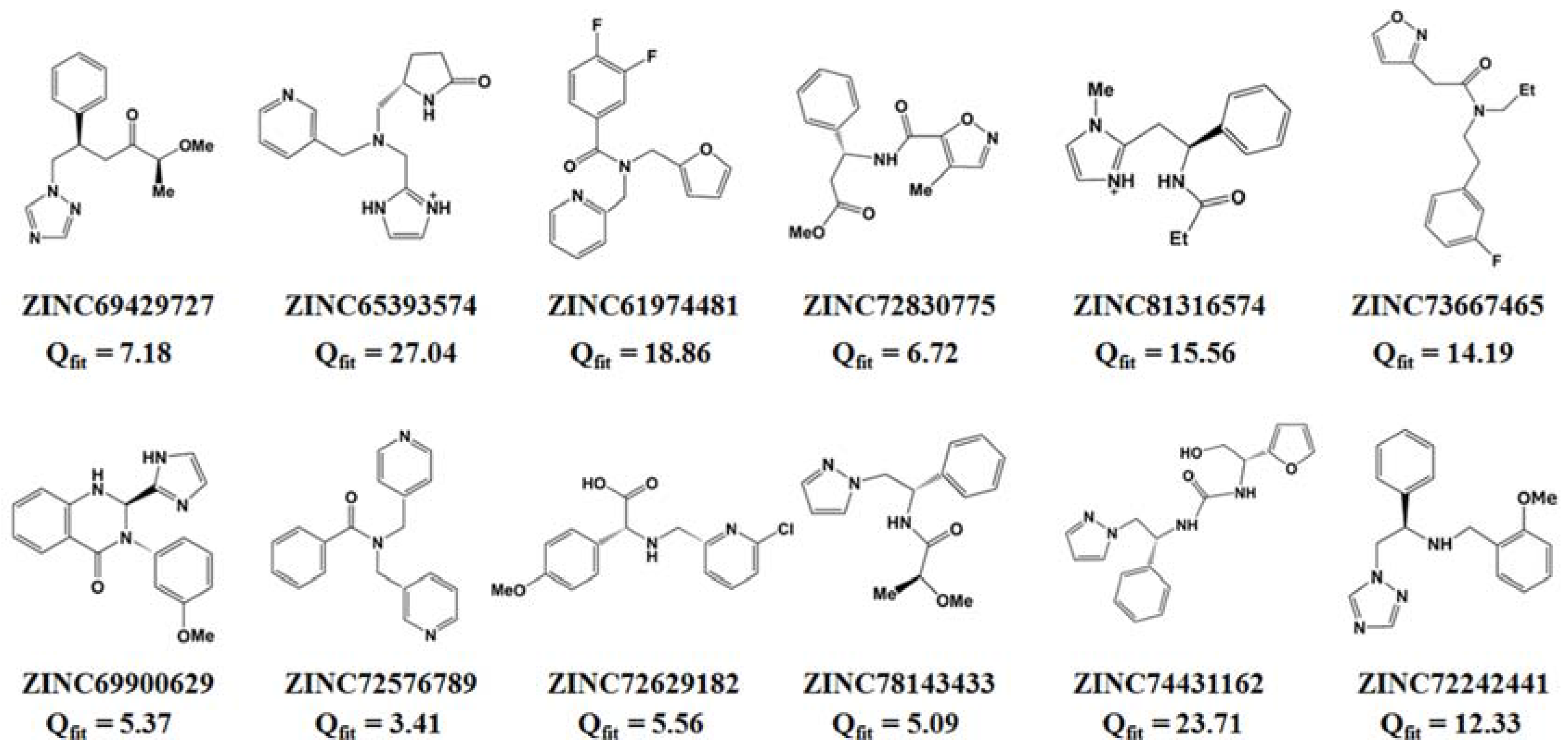
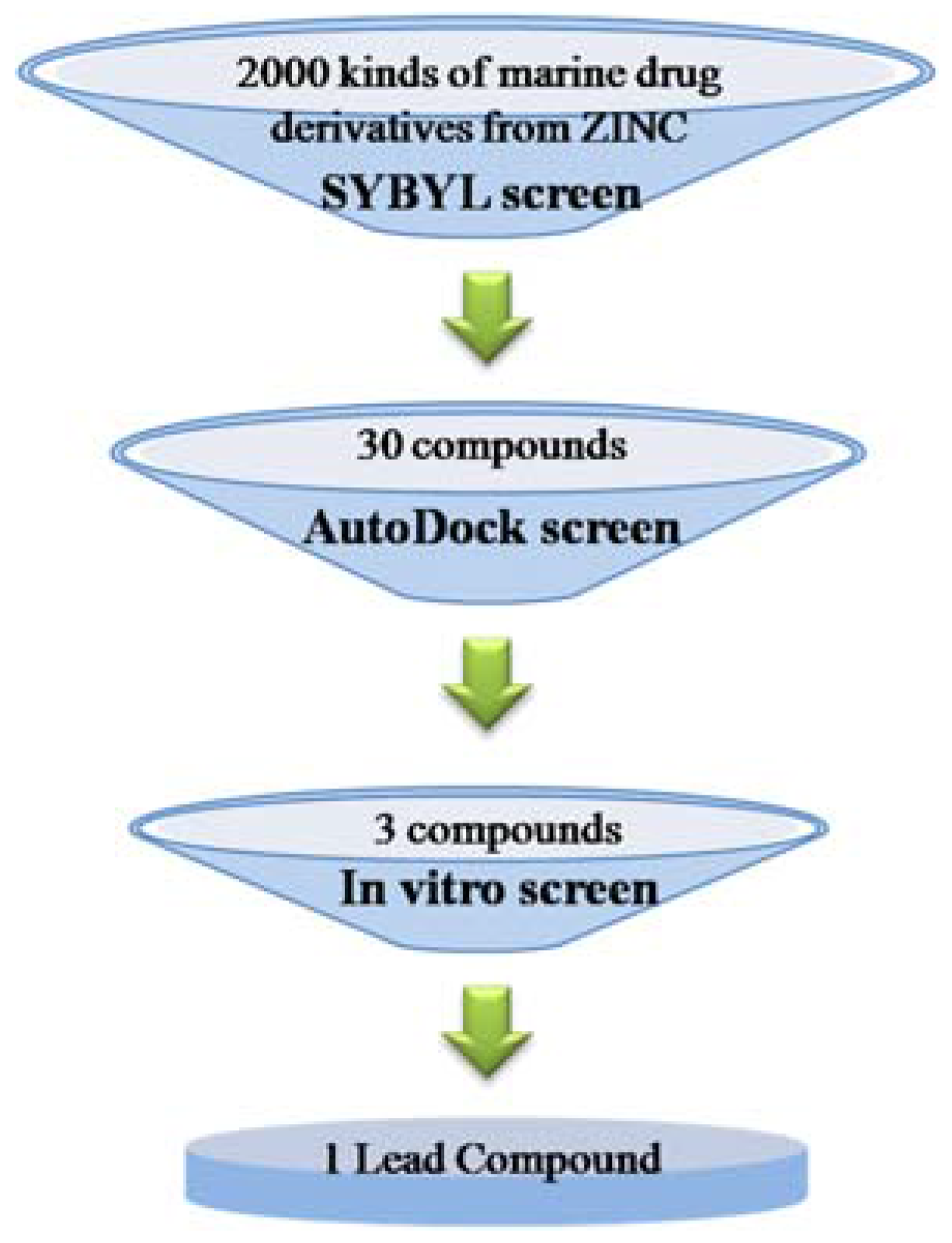
2.2. Virtual Screening
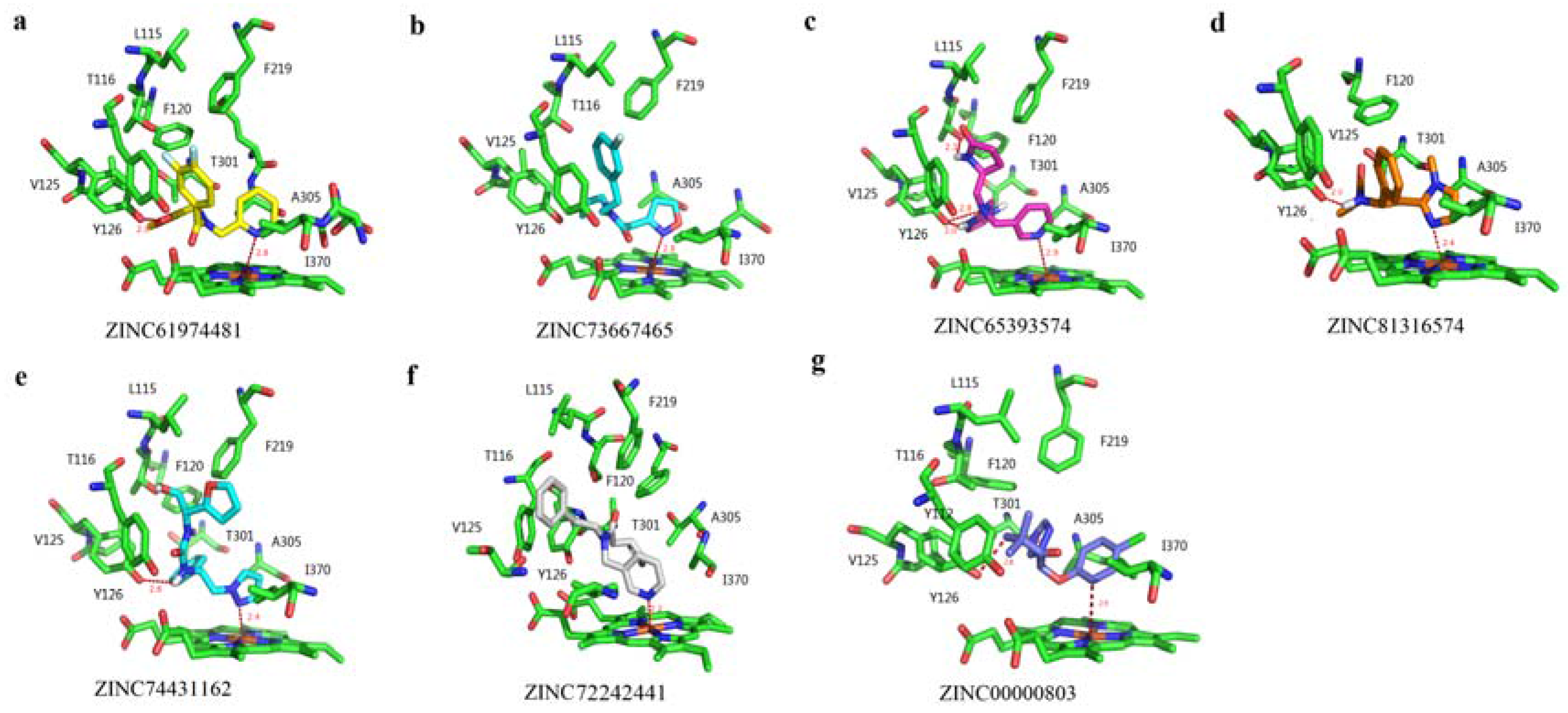
2.3. Molecular Docking with Ligands
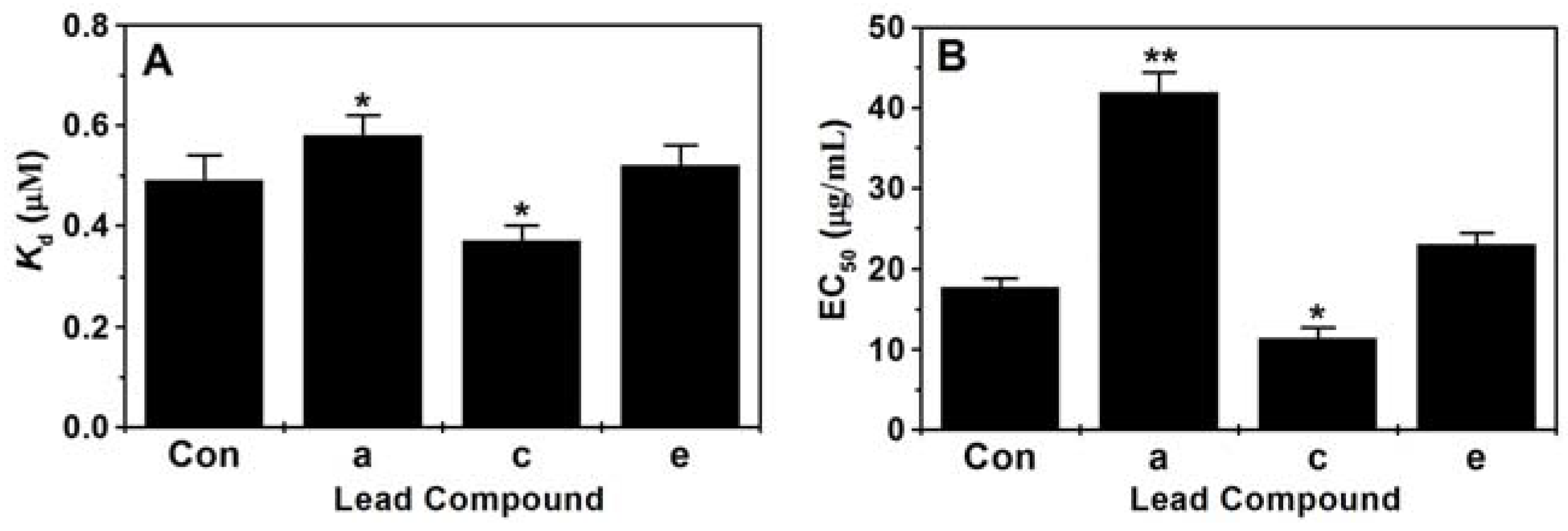
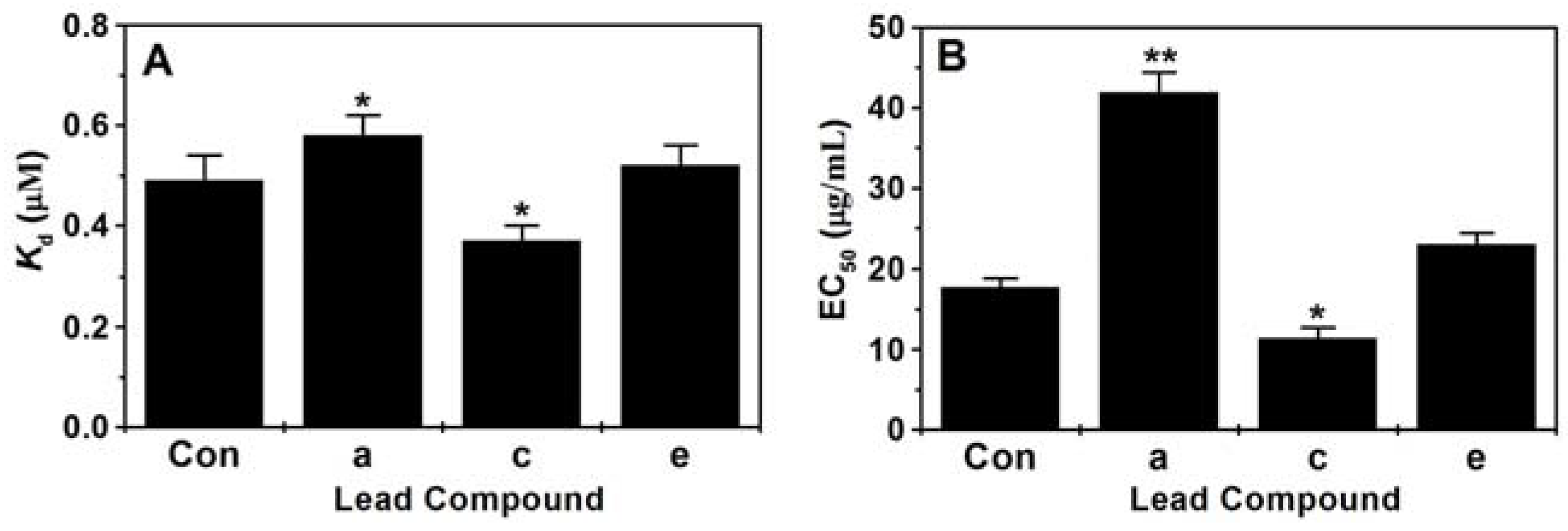
2.4. Assay of Binding Constant (Kd) and EC50
3. Discussion
4. Materials and Methods
4.1. Preparation of PiCYP51A Model
4.2. Virtual Screening
4.3. Molecular Docking
4.4. Analysis of Binding Constants (Kd) and EC50 for Fungicidal Compounds
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smilanick, J.; Mansour, M.; Margosan, D.; Gabler, F.M.; Goodwine, W. Influence of pH and NaHCO3 on effectiveness of imazalil to inhibit germination of Penicillium digitatum and to control postharvest green mold on citrus fruit. Plant Dis. 2005, 89, 640–648. [Google Scholar] [CrossRef]
- Sharma, R.R.; Singh, D.; Singh, R. Biological control of postharvest diseases of fruits and vegetables by microbial antagonists: A review. Biol. Control 2009, 50, 205–221. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 2007, 1770, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, D.; Wei, P.; Zhang, J.; Wan, J.; Ren, Y.; Chen, Z.; Liu, D.; Yu, Z.; Feng, L. Structure-based rational screening of novel hit compounds with structural diversity for cytochrome P450 sterol 14α-demethylase from Penicillium digitatum. J. Chem. Inf. Model. 2010, 50, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Sombardier, A.; Dufour, M.C.; Blancard, D.; Corio-Costet, M.F. Sensitivity of Podosphaera aphanis isolates to DMI fungicides: Distribution and reduced cross-sensitivity. Pest Manag. Sci. 2010, 66, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, J.; Liu, J.; Yuan, Y.; Li, N.; He, M.; Qi, T.; Hui, G.; Xiong, L.; Liu, D. Novel mutations in CYP51B from Penicillium digitatum involved in prochloraz resistance. J. Microbiol. 2014, 52, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Osterhage, C.; Kaminsky, R.; König, G.M.; Wright, A.D. Ascosalipyrrolidinone A, an antimicrobial alkaloid, from the obligate marine fungus Ascochyta salicorniae. J. Org. Chem. 2000, 65, 6412–6417. [Google Scholar] [CrossRef] [PubMed]
- Poch, G.K.; Gloer, J.B. Auranticins A and B: Two new depsidones from a mangrove isolate of the fungus Preussia aurantiaca. J. Nat. Prod. 1991, 54, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, H.Y.; Li, W.S.; Zhu, X.W.; Ding, W.J.; Li, C.Y. Bioactive chaetoglobosins from the mangrove endophytic fungus Penicillium chrysogenum. Mar. Drugs 2016, 14, 172. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Brauers, G.; Ebel, R.; Wray, V.; Berg, A.; Proksch, P. Novel chromone derivatives from the fungus Aspergillus versicolor isolated from the marine sponge Xestospongia exigua. J. Nat. Prod. 2003, 66, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Lee, U.; Kang, J.S.; Hong, D.C.; Son, B.W. A new radical scavenging anthracene glycoside, asperflavin ribofuranoside, and polyketides from a marine isolate for the fungus microsporum. Chem. Pharm. Bull. 2006, 54, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.G.; Zhang, H.; Mochizuki, M.; Adachi, K.; Shizuri, Y.; Lee, W.J.; Kim, S.K. Novel antifungal diketopiperazine from marine fungus. ChemInform 2003, 34, 102–106. [Google Scholar] [CrossRef]
- Neto, A.C.; Maraschin, M.; Piero, R.M. Antifungal activity of salicylic acid against Penicillium expansum and its possible mechanisms of action. Int. J. Food Microbiol. 2015, 215, 64–70. [Google Scholar] [CrossRef] [PubMed]
- El-Megharbel, S.M.; Adam, A.M.; Megahed, A.S.; Refat, M.S. Synthesis and molecular structure of moxifloxacin drug with metal ions as a model drug against some kinds of bacteria and fungi. Russ. J. Gen. Chem. 2015, 85, 2366–2373. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Paliwal, S.K.; Sharma, M.; Mittal, A.; Sharma, S. In silico and in vitro screening to identify structurally diverse non-azole CYP51 inhibitors as potent antifungal agent. J. Mol. Graph. Model. 2016, 63, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Abagyan, R.A. Protein flexibility in ligand docking and virtual screening to protein kinases. J. Mol. Biol. 2004, 337, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Elshaier, Y.A.; Barakat, A.; Al-Qahtany, B.M.; Al-Majid, A.M.; Al-Agamy, M.H. Synthesis of pyrazole-thiobarbituric acid derivatives: Antimicrobial activity and docking studies. Molecules 2016, 21, 1337. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, M.M.; Salim, N.; Himmat, M.; Ahmed, A.; Saeed, F. Quantum probability ranking principle for ligand-based virtual screening. J. Comput. Aided Mol. Des. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.Q.; Zhang, W.N.; Zhang, M.; Song, Y.L.; Ji, H.T.; Zhu, J.; Yao, J.Z.; Yu, J.X.; Yang, S.; Zhou, Y.J. Homology modeling of lanosterol 14α-demethylase of Candida albicans and Aspergillus fumigatus and insights into the enzyme-substrate interactions. J. Biomol. Struct. Dyn. 2004, 22, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Miao, Z.; Ji, H.; Yao, J.; Wang, W.; Che, X.; Dong, G.; Lü, J.; Guo, W.; Zhang, W. Three-dimensional model of lanosterol 14α-demethylase from Cryptococcus neoformans: Active-site characterization and insights into azole binding. Antimicrob. Agents Chemother. 2009, 53, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, D.; Zhang, Q.; Zhang, S.; Wan, J.; Xiao, W. Expression and homology modeling of sterol 14α-demethylase from Penicillium digitatum. FEMS Microbiol. Lett. 2007, 277, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, J.; Cao, S.; Han, R.; Yuan, Y.; Yang, J.; Yan, Y.; Liu, D. Homology modeling, molecular docking and spectra assay studies of sterol 14α-demethylase from Penicillium digitatum. Biotechnol. Lett. 2011, 33, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, T.M.; Kumar, R.M.; Agrawal, A.; Dubey, G.P.; Ilango, K. Comparative inhibitory potential of selected dietary bioactive polyphenols, phytosterols on CYP3A4 and CYP2D6 with fluorometric high-throughput screening. J. Food Sci. Technol. 2015, 52, 4537–4543. [Google Scholar] [CrossRef] [PubMed]
- Su, B.H.; Tu, Y.S.; Lin, C.; Shao, C.Y.; Lin, O.A.; Tseng, Y.J. Rule-based prediction models of cytochrome P450 inhibition. J. Chem. Inf. Model. 2015, 55, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010, 38, 545–549. [Google Scholar] [CrossRef] [PubMed]
- SYBYL7.3. Tripos Inc.: St. Louis, MO, USA, 2003. Available online: http://www.tripos.com (accessed on 21 January 2014).
- Li, W.J.; Li, Q.; Liu, D.L.; Ding, M.W. Synthesis, fungicidal activity, and sterol 14α-demethylase binding interaction of 2-azolyl-3,4-dihydroquinazolines on Penicillium digitatum. J. Agric. Food Chem. 2013, 61, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.J.; Eckert, J.W. Sensitivity of Penicillium digitatum and P. italicum to postharvest citrus fungicides in California. Phytopathology 1999, 89, 716–721. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Compound | Binding Energy (kcal·mol−1) |
|---|---|---|
| ZINC61974481 | a | −7.01 |
| ZINC73667465 | b | −6.91 |
| ZINC65393574 | c | −7.96 |
| ZINC81316574 | d | −6.95 |
| ZINC74431162 | e | −7.23 |
| ZINC72242441 | f | −6.44 |
| ZINC00000803 | Triazolone | −7.15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Han, R.; Cao, Q.; Yu, J.; Mao, J.; Zhang, T.; Wang, S.; Niu, Y.; Liu, D. Pharmacophore-Based Virtual Screening of Novel Inhibitors and Docking Analysis for CYP51A from Penicillium italicum. Mar. Drugs 2017, 15, 107. https://doi.org/10.3390/md15040107
Yuan Y, Han R, Cao Q, Yu J, Mao J, Zhang T, Wang S, Niu Y, Liu D. Pharmacophore-Based Virtual Screening of Novel Inhibitors and Docking Analysis for CYP51A from Penicillium italicum. Marine Drugs. 2017; 15(4):107. https://doi.org/10.3390/md15040107
Chicago/Turabian StyleYuan, Yongze, Rui Han, Qianwen Cao, Jinhui Yu, Jiali Mao, Tingfu Zhang, Shengqiang Wang, Yuhui Niu, and Deli Liu. 2017. "Pharmacophore-Based Virtual Screening of Novel Inhibitors and Docking Analysis for CYP51A from Penicillium italicum" Marine Drugs 15, no. 4: 107. https://doi.org/10.3390/md15040107
APA StyleYuan, Y., Han, R., Cao, Q., Yu, J., Mao, J., Zhang, T., Wang, S., Niu, Y., & Liu, D. (2017). Pharmacophore-Based Virtual Screening of Novel Inhibitors and Docking Analysis for CYP51A from Penicillium italicum. Marine Drugs, 15(4), 107. https://doi.org/10.3390/md15040107