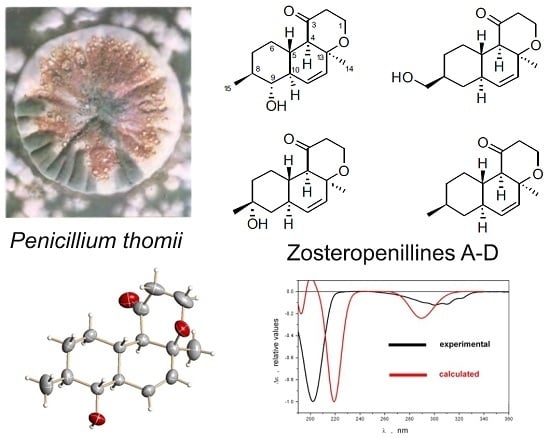
Zosteropenillines: Polyketides from the Marine-Derived Fungus Penicillium thomii

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
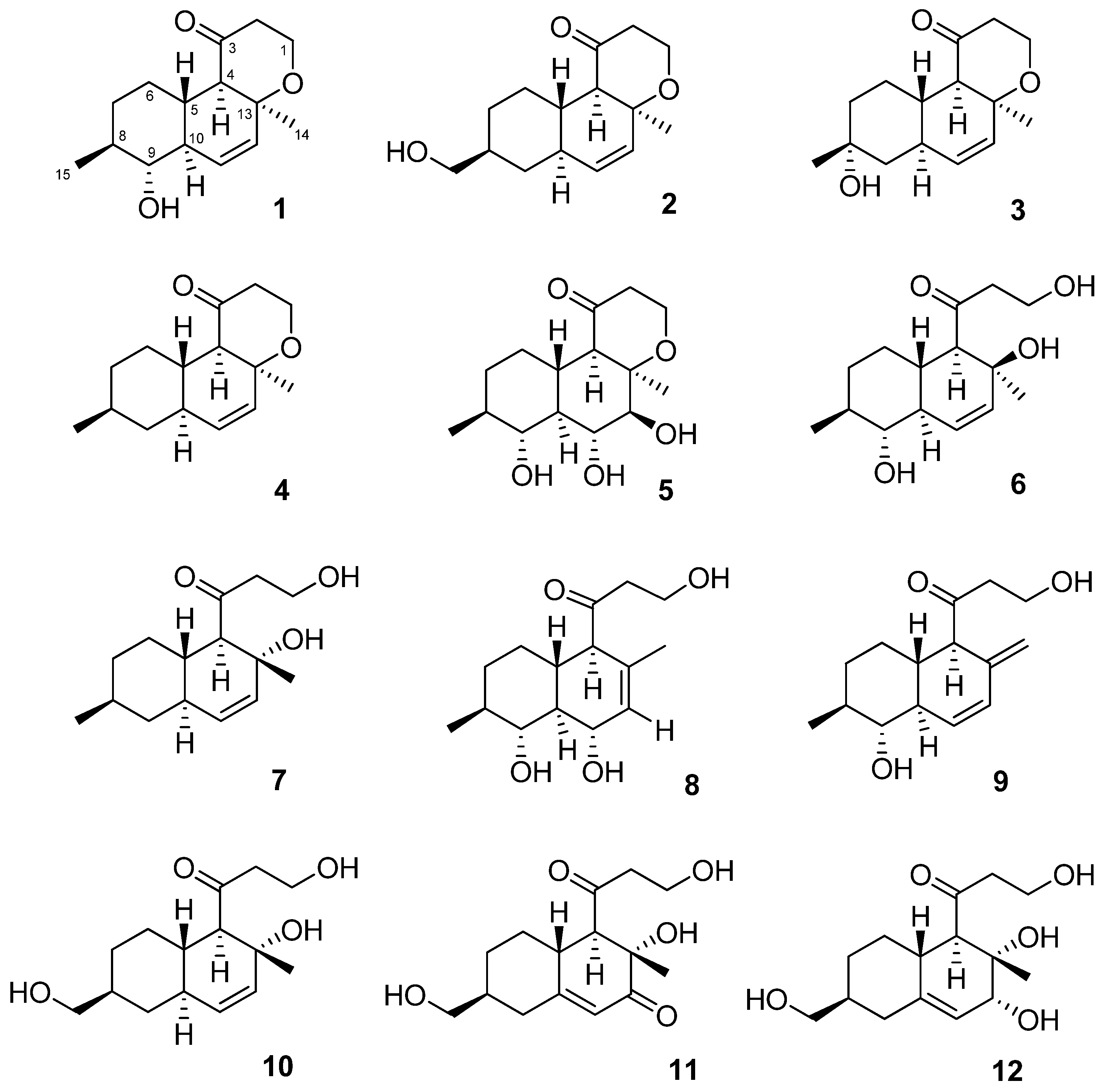
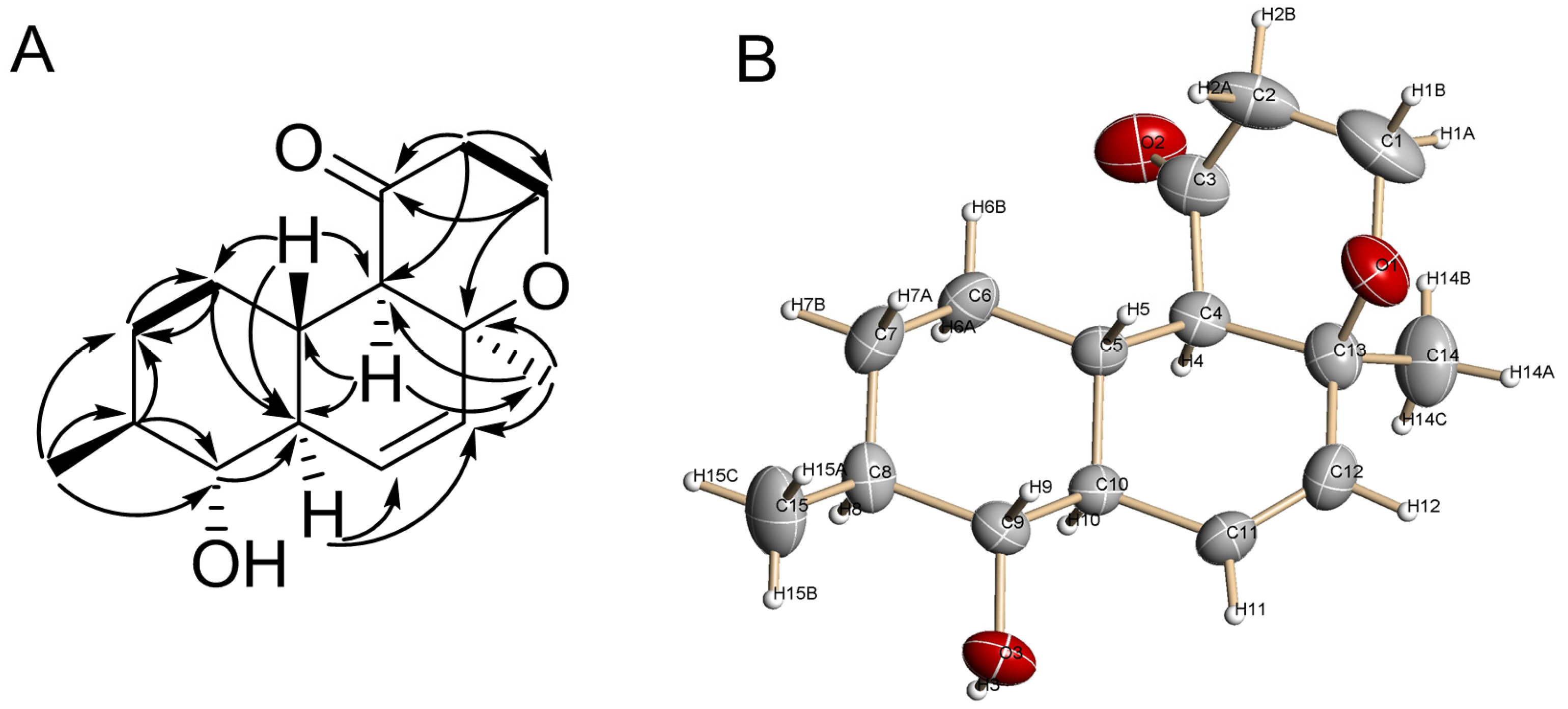
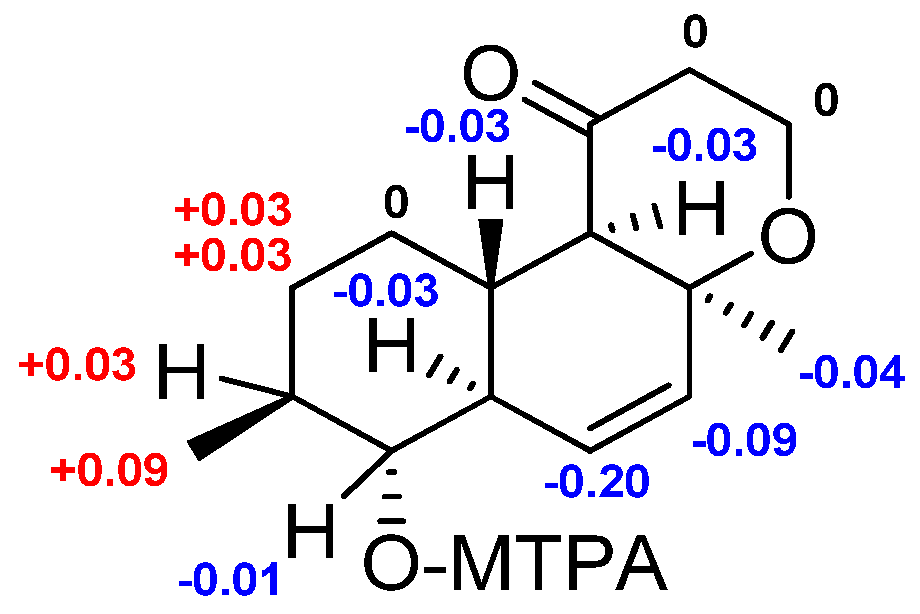
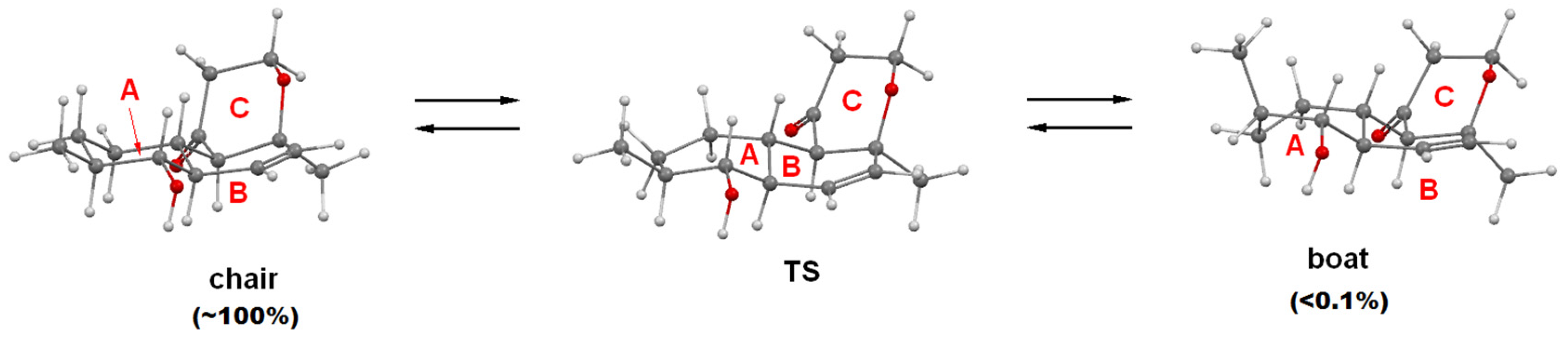
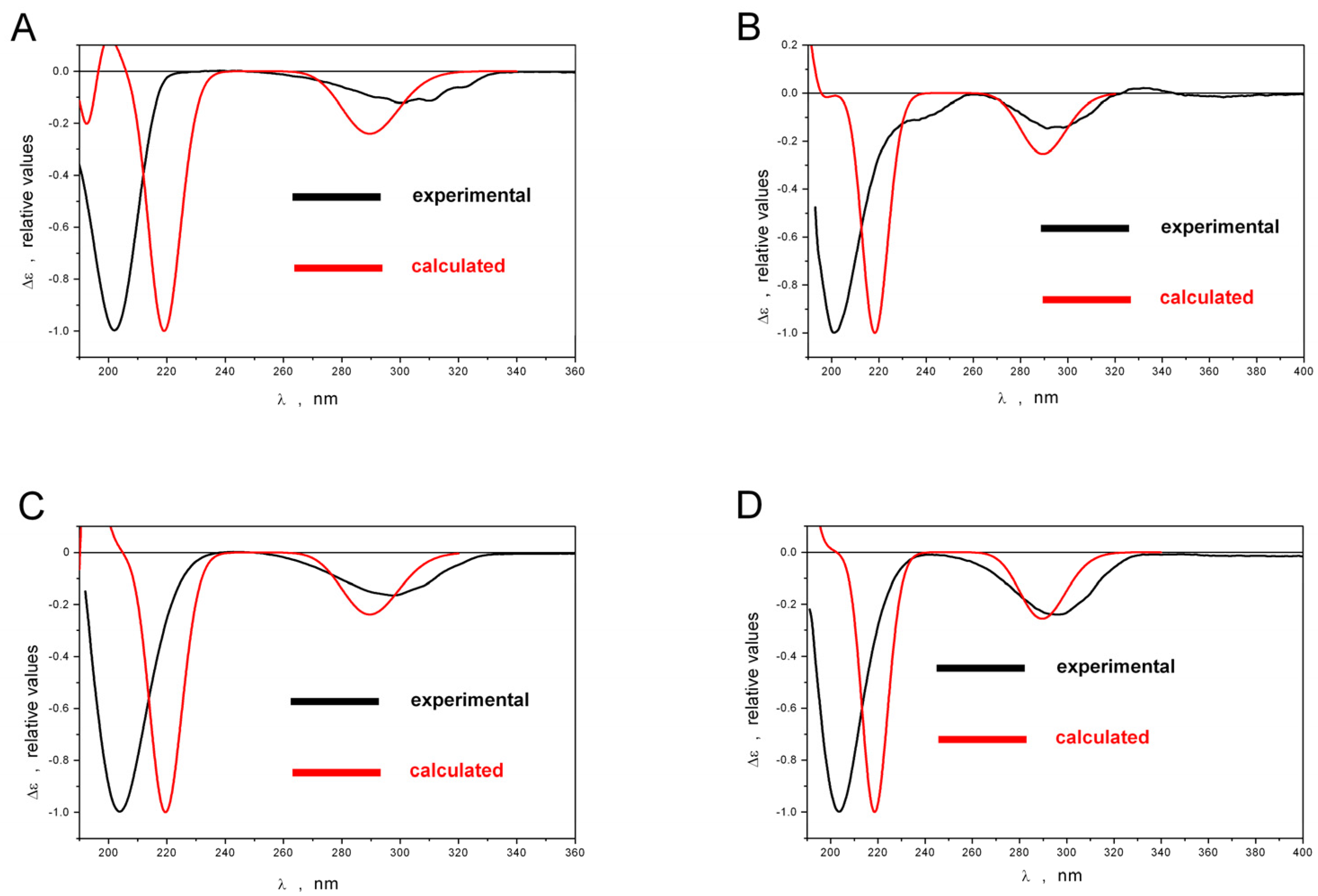
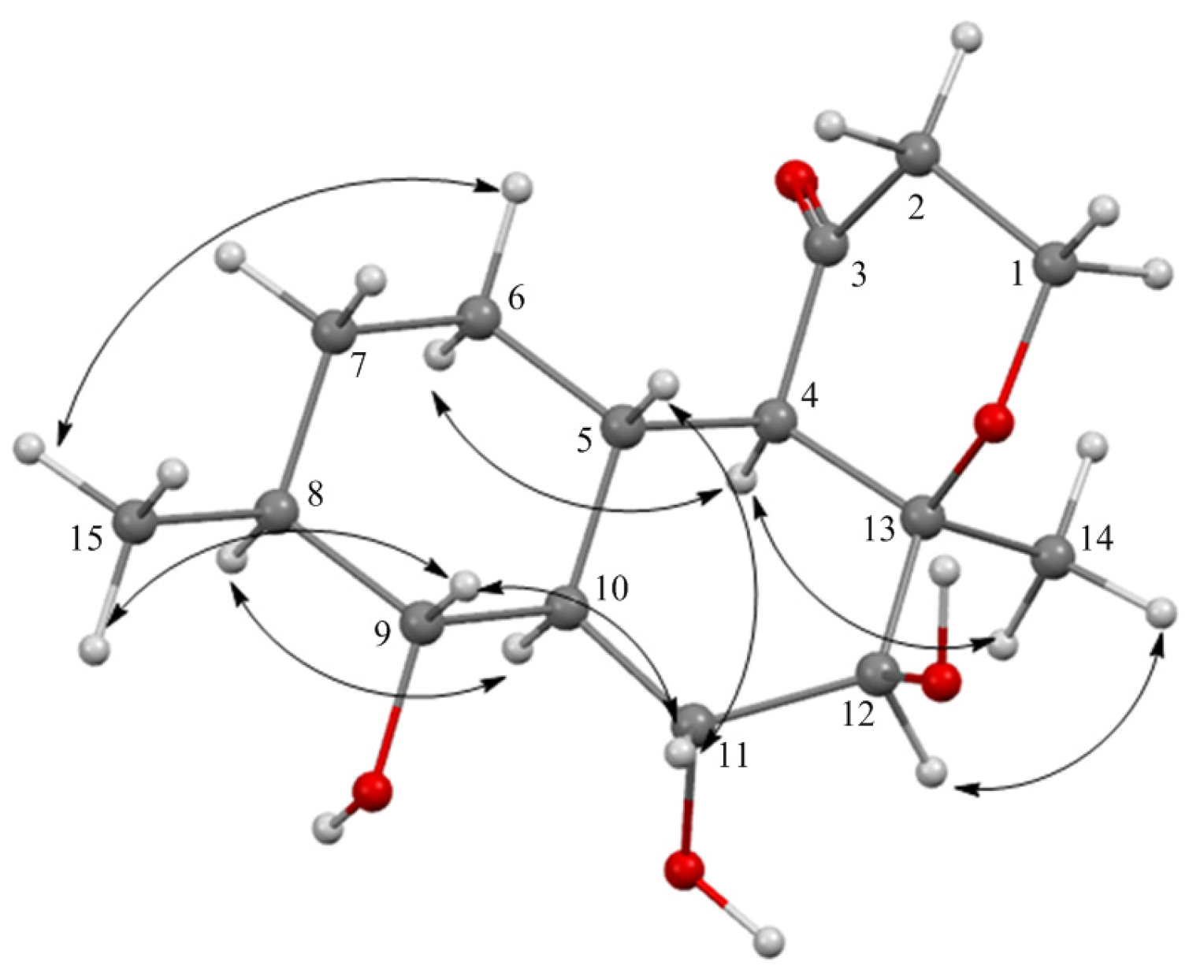
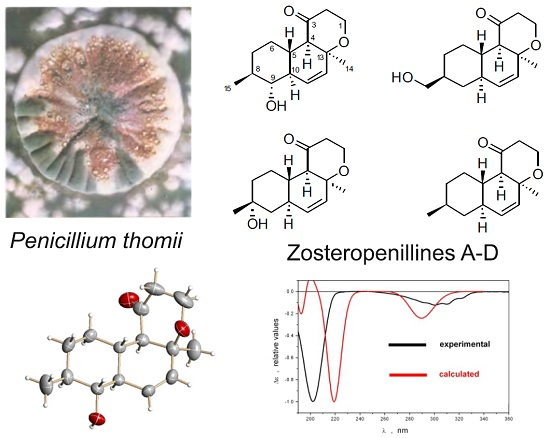
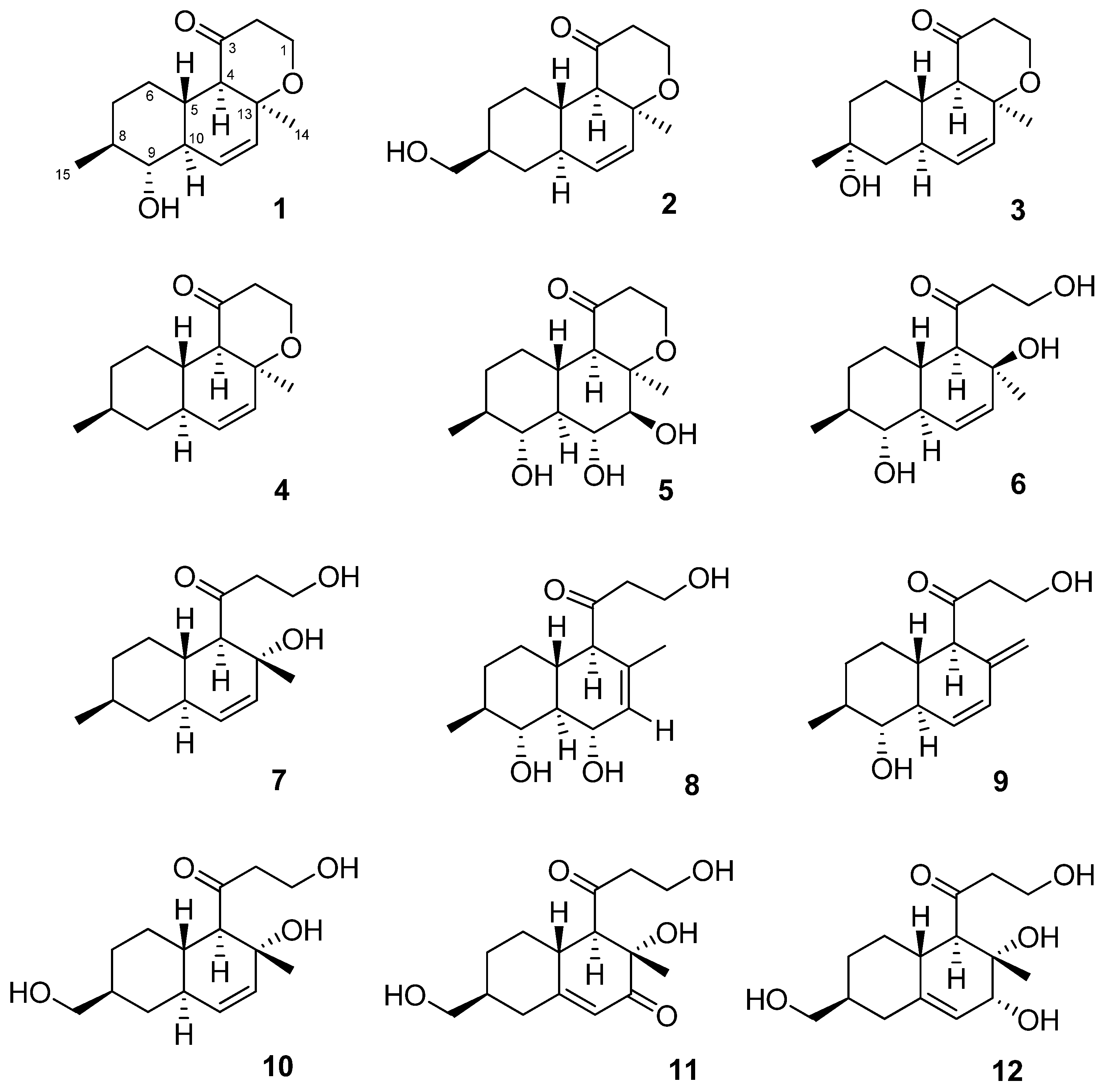
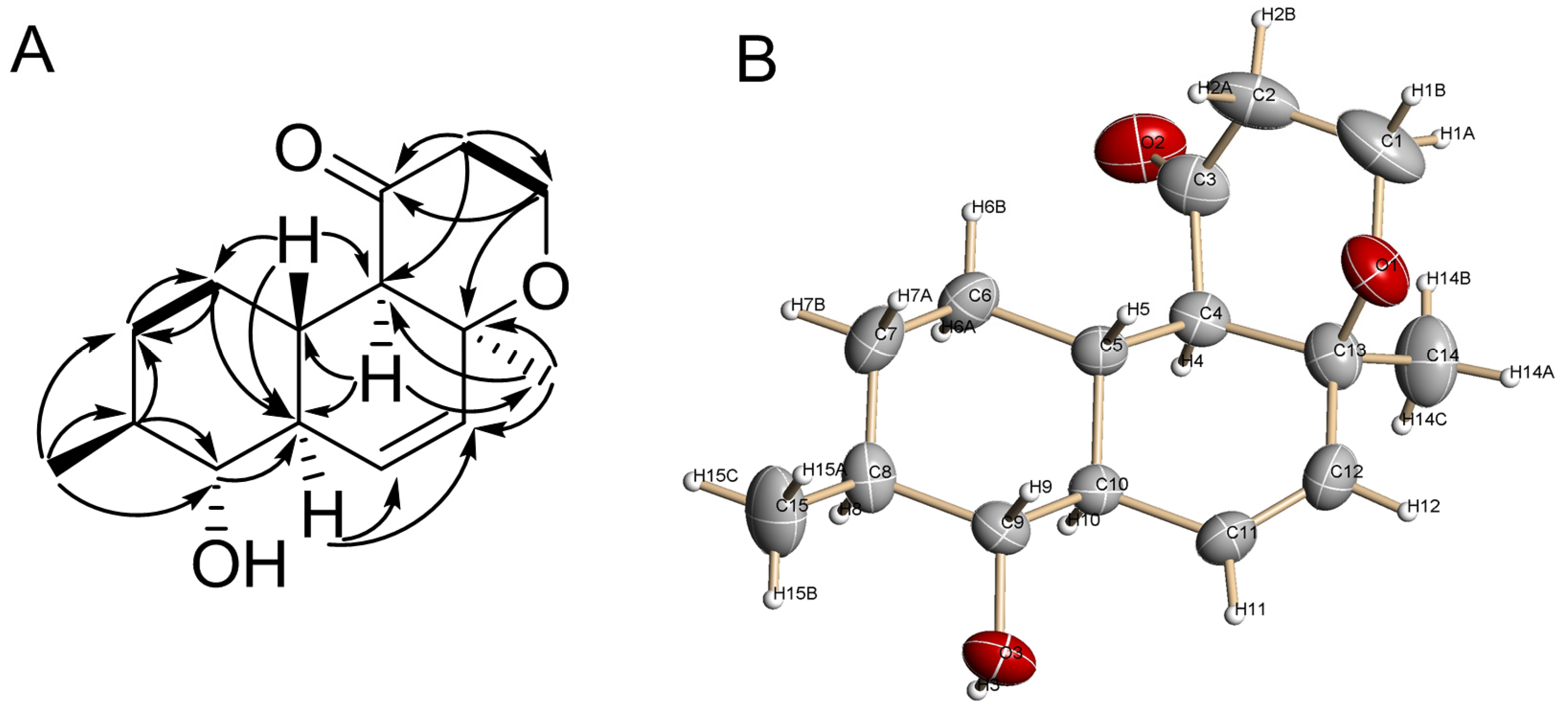
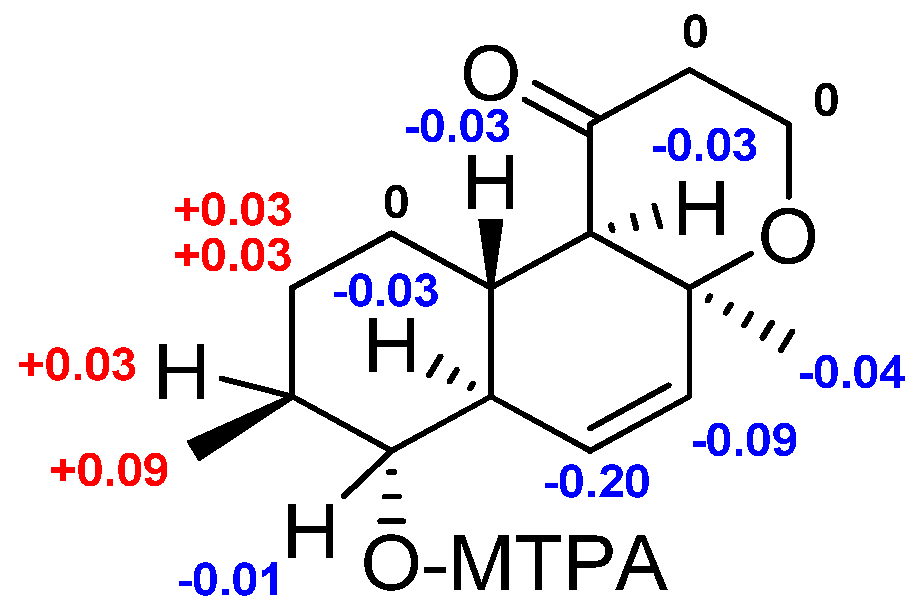
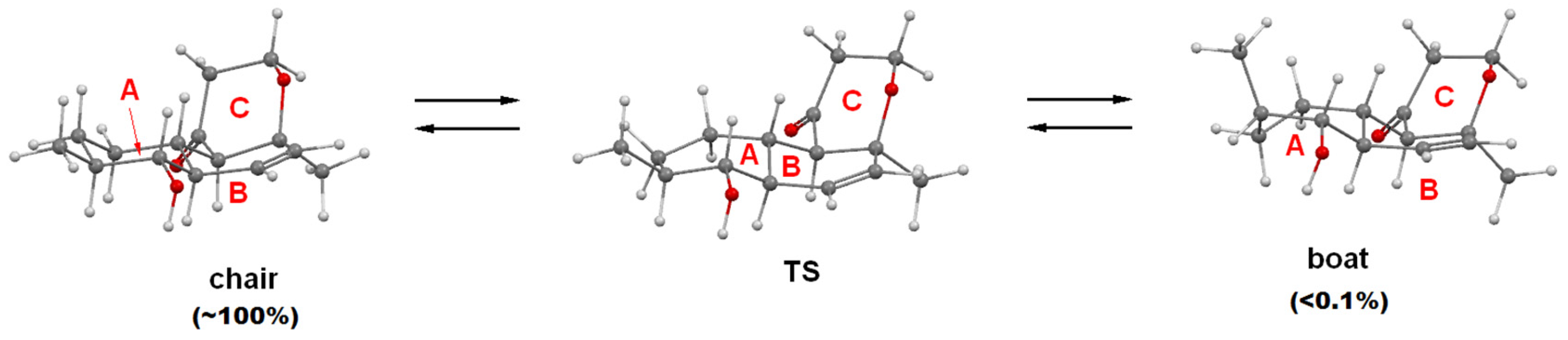
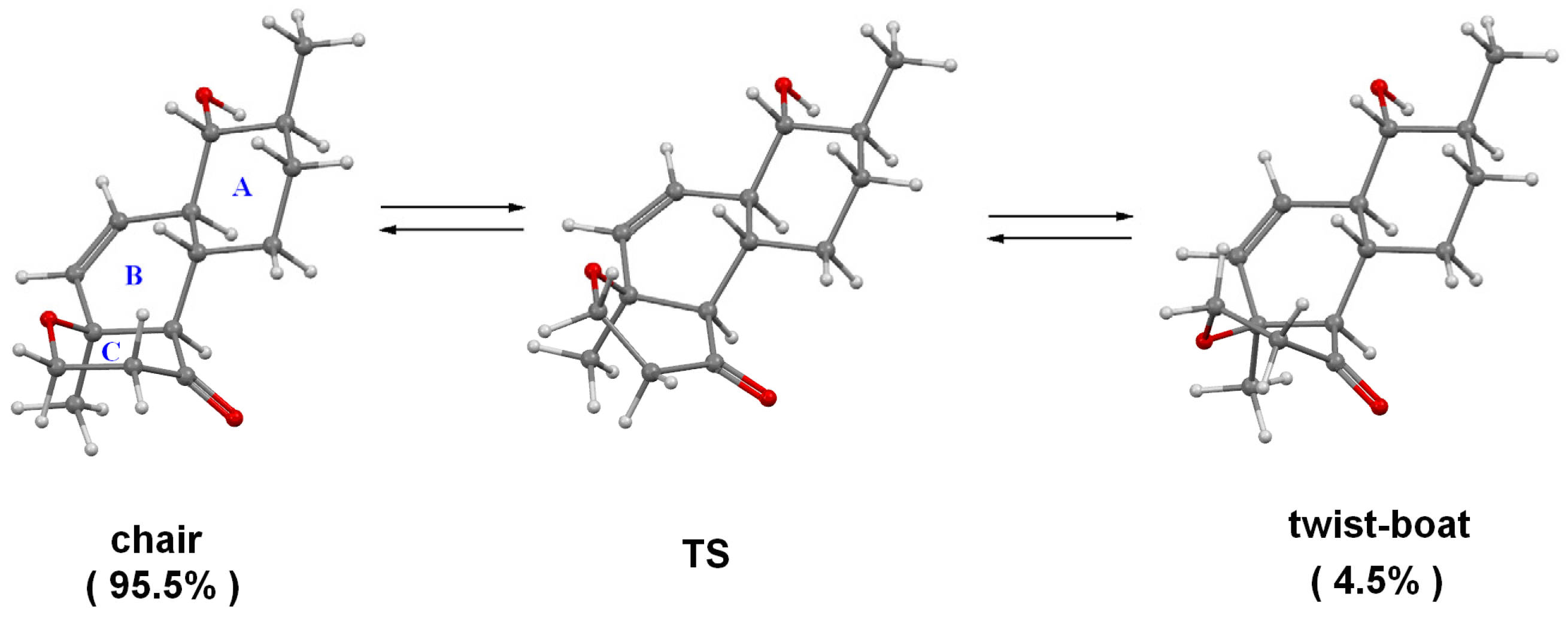
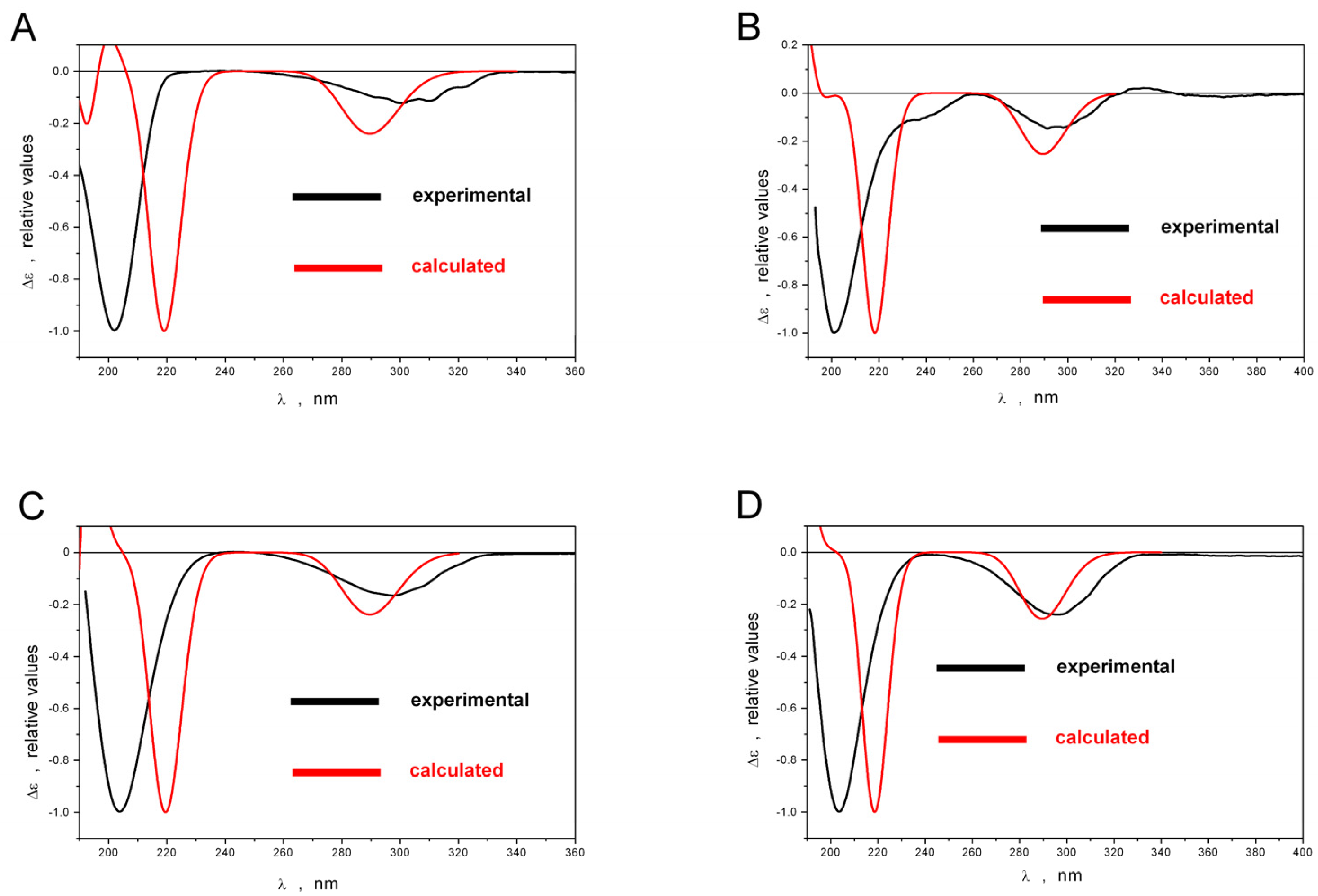
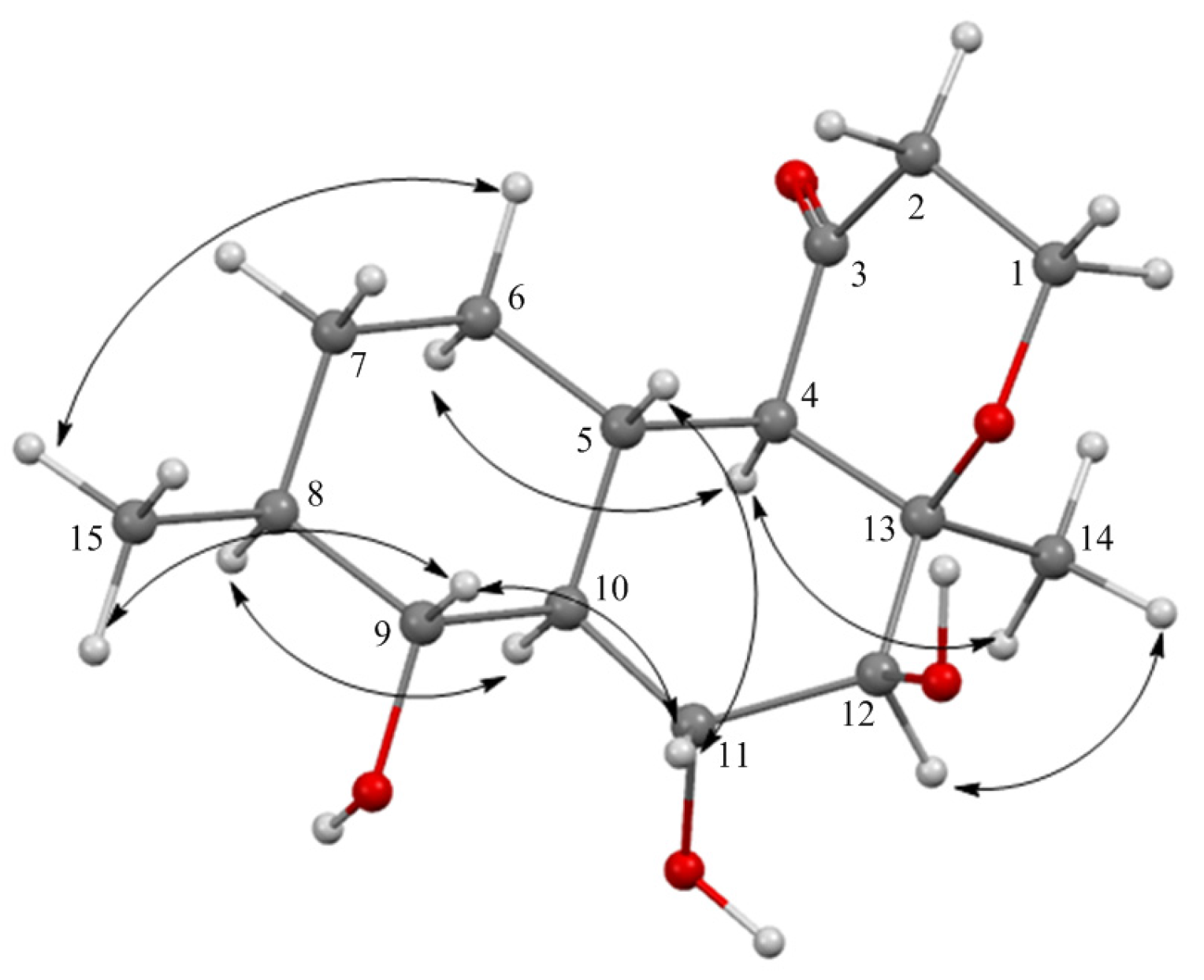
2.1. Structure Elucidation
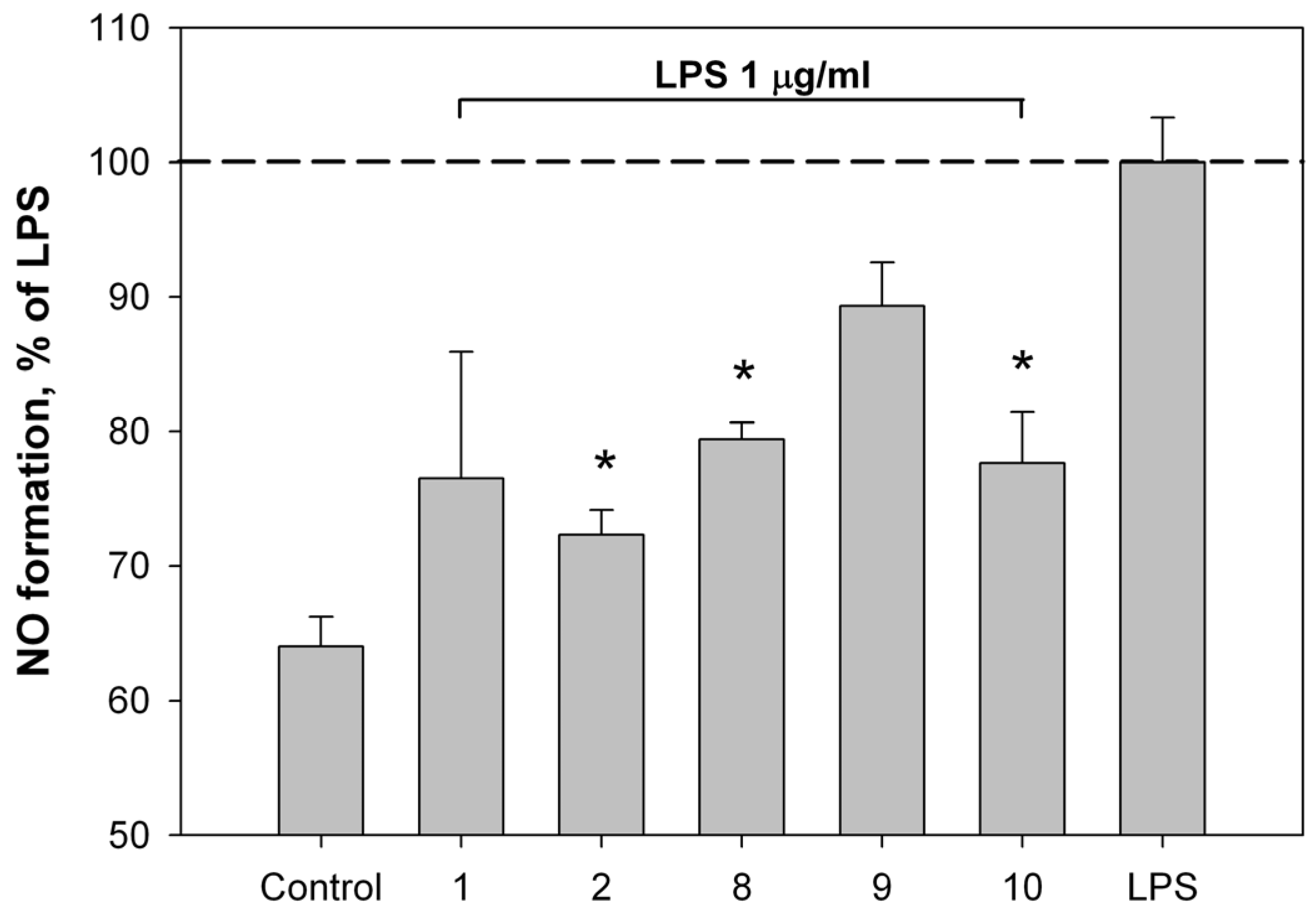
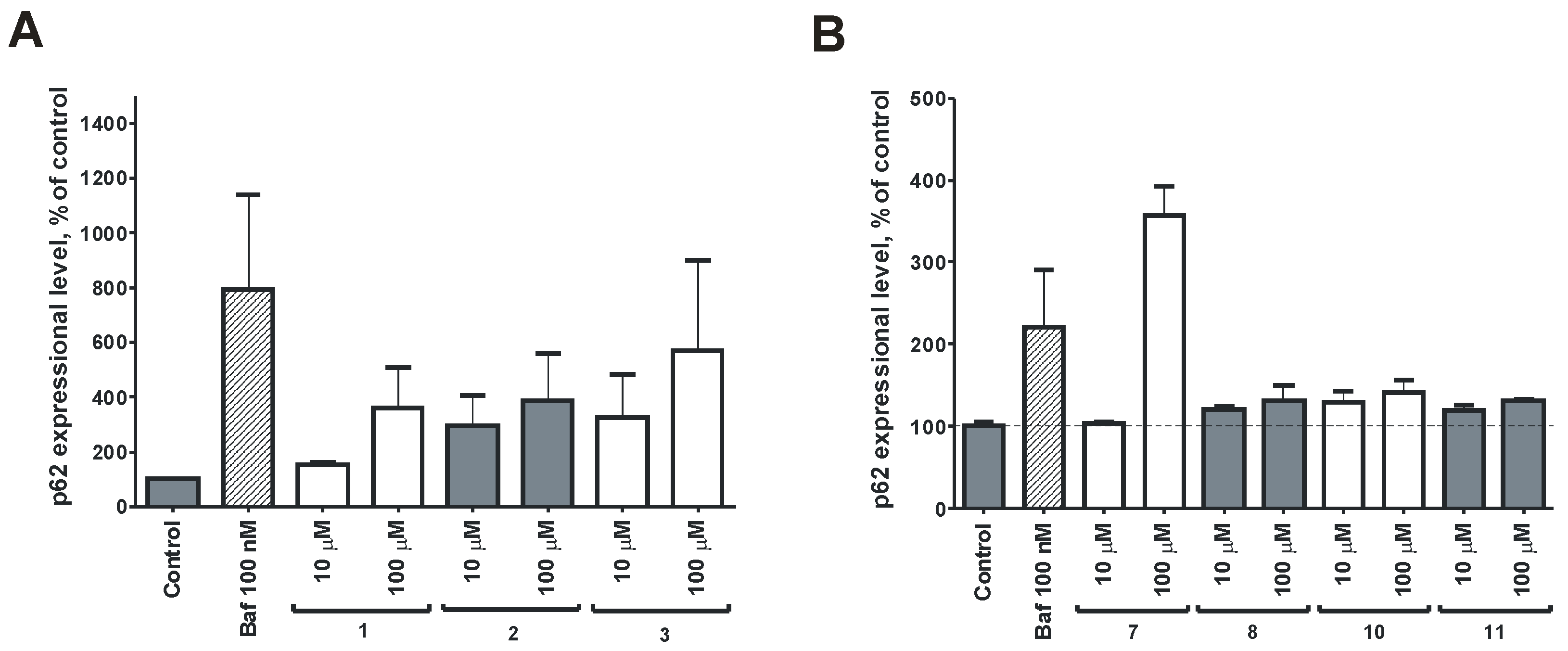
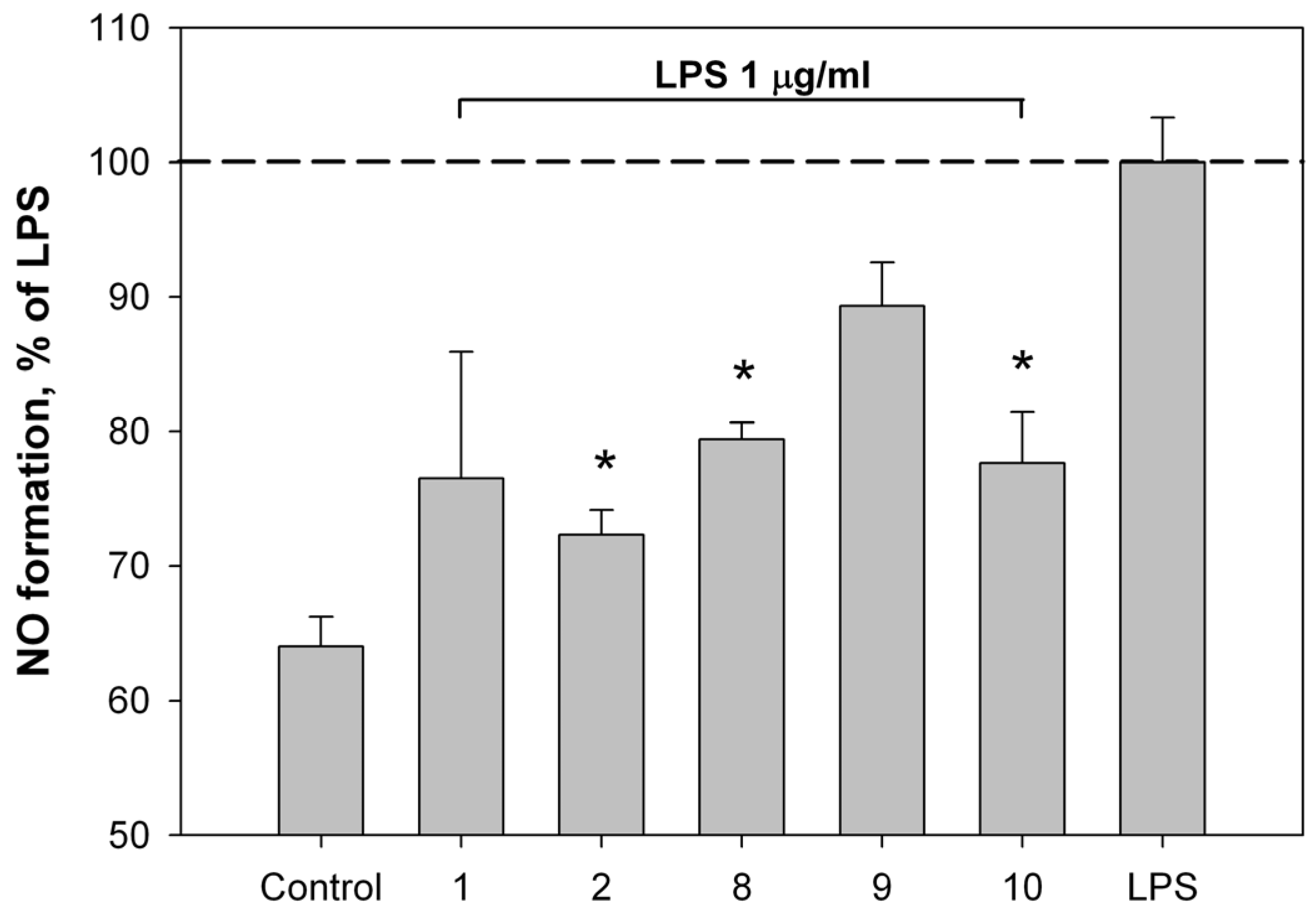
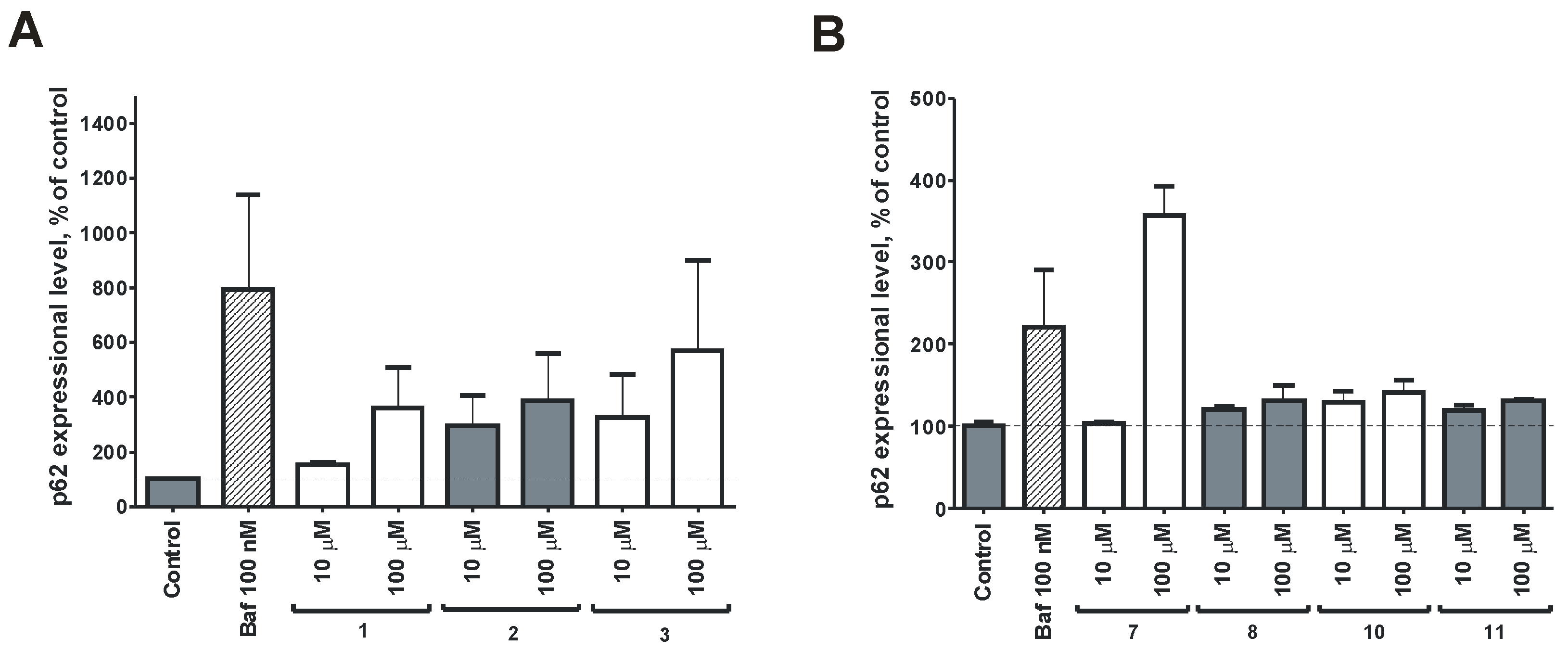
2.2. Bioassay Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Strain
3.3. Cultivation of P. thomii
3.4. Extraction and Isolation
3.5. Spectral Data
3.6. Preparation of (S)-MTPA and (R)-MTPA Esters of 1
3.7. X-ray Crystal Data for 1
3.8. Quantum Chemical Modeling
3.9. Measurement of Nitric Oxide Content
3.10. Statistical Analysis
3.11. Cell Lines and Culture Conditions
3.12. In Vitro MTT-Based Drug Sensitivity Assay
3.13. Western Blotting
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Liming, J.; Chunshan, Q.; Xiyan, H.; Shengdi, F. Potential pharmacological resources: Natural bioactive compounds from marine-derived fungi. Mar. Drugs 2016, 14, 76. [Google Scholar]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the AlgA-Derived Fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Afiyatullov, S.S.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Yurchenko, E.A.; Dyshlovoy, S.A. Sargassopenillines A–G, 6,6-Spiroketals from the AlgA-Derived Fungi Penicillium thomii and Penicillium lividum. Mar. Drugs 2014, 12, 5930–5943. [Google Scholar] [CrossRef] [PubMed]
- Afiyatullov, S.S.; Leshchenko, E.V.; Sobolevskaya, M.P.; Denisenko, V.A.; Kirichuk, N.N.; Khudyakova, Y.V.; Hoai, T.P.T.; Dmitrenok, P.S.; Menchinskaya, E.S.; Pislyagin, E.A.; et al. New eudesmane sesquiterpenes from the marine-derived fungus Penicillium thomii. Phytochem. Lett. 2015, 14, 209–214. [Google Scholar] [CrossRef]
- Sobolevskaya, M.P.; Leshchenko, E.V.; Hoai, T.P.T.; Denisenko, V.A.; Dyshlovoy, S.A.; Kirichuk, N.N.; Khudyakova, Y.V.; Kim, N.Y.; Berdyshev, D.V.; Pislyagin, E.A.; et al. Pallidopenillines: Polyketides from the algA-derived fungus Penicillium thomii Maire KMM 4675. J. Nat. Prod. 2016, 79, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Kung, H.J.; Changou, C.; Nguyen, H.G.; Yang, J.C.; Evans, C.P.; Bold, R.J.; Chuang, F. Autophagy and Prostate Cancer Therapeutics. In Prostate Cancer; Tindal, D.J., Ed.; Springer: New York, NY, USA, 2013; Volume 16, pp. 497–518. [Google Scholar]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a modulator and target in prostate cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- APEX2, Bruker AXS Inc.: Madison, WI, USA, 2010.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystalstructure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum—A direct utilation of abinitio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Shiue, T.W.; Chen, Y.H.; Wu, C.M.; Singh, G.; Chen, H.-Y.; Hung, C.-H.; Liaw, W.F.; Wang, Y.M. Nitric Oxide Turn-on Fluorescent Probe Based on Deamination of Aromatic Primary Monoamines. Inorg. Chem. 2012, 51, 5400–5408. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Menchinskaya, E.S.; Venz, S.; Rast, S.; Amann, K.; Hauschild, J.; Otte, K.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; et al. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Int. J. Cancer 2016, 138, 2450–2465. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Venz, S.; Shubina, L.K.; Fedorov, S.N.; Walther, R.; Jacobsen, C.; Stonik, V.A.; Bokemeyer, C.; Balabanov, S.; Honecker, F. Activity of aaptamine and two derivatives, demethyloxyaaptamine and isoaaptamine, in cisplatin-resistant germ cell cancer. J. Proteom. 2014, 96, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kusari, S.; Spireller, M. Natural products containing ‘decalin’ motif in microorganisms. Nat. Prod. Rep. 2014, 31, 1175–1201. [Google Scholar] [CrossRef] [PubMed]
- Stocking, E.M.; Williams, R.M. Chemistry and biology of biosynthetic Diels-Alder reactions. Angew. Chem. Int. Ed. 2003, 42, 3078–3115. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lin, X.; Lu, Z.; Huang, W.; Huang, Y.; Shen, Y. Secondary metabolites of Phomopsis sp. XZ-26, an endophytic fungus from Camptotheca acuminate. Eur. J. Chem. 2009, 18, 2975–2982. [Google Scholar]
- Anisimov, M.M.; Chaikina, E.L.; Afiyatullov, S.S.; Zhuravleva, O.I.; Klykov, A.G.; Kraskovskaja, N.A.; Aminin, D.L. Decumbenones A–C from marine fungus Aspergillus sulphureus as stimulators of the initial stages of development of agricultural plants. Agric. Sci. 2012, 3, 1019–1022. [Google Scholar]
- Jadulco, R.C.; Koch, M.; Kakule, T.B.; Schmidt, E.W.; Orendt, A.; He, H.; Janso, J.E.; Carter, G.T.; Larson, E.C.; Pond, C.; et al. Isolation of pyrrolocins A–C: Cis- and trans-decalin tetramic acid antibiotics from an endophytic fungal-derived pathway. J. Nat. Prod. 2014, 77, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Miura, S.; Yamashita, Y.; Ohta, T. Aspermytin A: A new neurotrophic polyketide isolated from a marine-derived fungus of the genus Aspergillus. Bioorg. Med. Chem. Lett. 2004, 14, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, O.I.; Afiyatullov, S.S.; Vishchuk, O.S.; Denisenko, V.A.; Slinkina, N.N.; Smetanina, O.F. Decumbenone C, a new cytotoxic decaline derivative from the marine fungus Aspergillus sulphureus KMM 4640. Arch. Pharm. Res. 2012, 35, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Feng, T.; Li, Z.; Liu, J. Five new polyketides from the basidiomycete Craterellus odoratus. Nat. Prod. Bioprospect. 2012, 2, 170–173. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 * | 2 * | 3 * | 4 ** | 5 ** | 6 * |
|---|---|---|---|---|---|---|
| 1 | 60.9, CH2 | 60.9, CH2 | 60.9, CH2 | 60.9, CH2 | 60.9, CH2 | 57.6, CH2 |
| 2 | 39.9, CH2 | 39.9, CH2 | 39.9, CH2 | 39.9, CH2 | 37.9, CH2 | 48.6, CH2 |
| 3 | 210.2, C | 210.4, C | 210.3, C | 210.9, C | 206.0, C | 217.6, C |
| 4 | 62.8, CH | 62.9, CH | 62.7, CH | 63.0, CH | 63.7, CH | 61.8, CH |
| 5 | 37.5, CH | 39.5, CH | 38.9, CH | 39.3. CH | 38.5, CH | 37.4, CH |
| 6 | 28.7, CH2 | 28.8, CH2 | 38.7, CH2 | 29.3, CH2 | 29.0, CH2 | 29.4, CH2 |
| 7 | 32.7, CH2 | 29.1, CH2 | 24.9, CH2 | 34.9, CH2 | 31.8, CH2 | 32.9, CH2 |
| 8 | 40.2, CH | 40.5, CH | 69.7, C | 32.8, CH | 39.2, CH | 40.6, CH |
| 9 | 77.9, CH | 35.2, CH2 | 44.5, CH2 | 40.9, CH2 | 79.2, CH | 77.8, CH |
| 10 | 48.6, CH | 41.4, CH | 36.9, CH | 41.9, CH | 53.5, CH | 49.1, CH |
| 11 | 130.1, CH | 134.4, CH | 134.4, CH | 130.5, CH | 67.3, CH | 128.6, CH |
| 12 | 131.6, CH | 130.9, CH | 131.1, CH | 134.8, CH | 72.7, CH | 133.0, CH |
| 13 | 74.7, C | 75.0, C | 75.0, C | 75.2, C | 77.5, C | 69.1, C |
| 14 | 23.2, CH3 | 23.3, CH3 | 23.3, CH3 | 23.3, CH3 | 22.0, CH3 | 28.8, CH3 |
| 15 | 18.4, CH3 | 68.1, CH2 | 31.5, CH3 | 22.3, CH3 | 19.1, CH3 | 18.4, CH3 |
| Position | 1 ** | 2 * | 3 * | 4 ** | 5 ** | 6 * |
|---|---|---|---|---|---|---|
| 1 | a: 4.11, ddd (12.0, 8.0, 1.2) b: 3.89, td (12.0, 3.0) | a: 4.11, ddd (11.8, 8.0, 1.0) b: 3.89 td (12.0, 3.0) | a: 4.11, ddd (11.8, 8.0, 1.0) b: 3.89, td (12.0, 2.9) | a: 4.11, ddd (11.7, 8.0, 1.4) b: 3.89, td (12.0, 3.0) | a: 4.22, dd (12.0, 9.0) b: 3.91, td (12.0, 4.0) | a: 3.91, ddd (11.7, 6.2, 4.2) b: 3.87, ddd (11.7, 6.2, 4.2) |
| 2 | a: 2.64, ddd (14.0, 12.0, 8.0) b: 2.21, ddt (14.0, 3.0, 1.2) | a: 2.66, ddd (14.1, 12.1, 8.0) b: 2.21, ddt (14.1, 3.0, 1.3) | a: 2.66, ddd (14.0, 12.1, 8.0) b: 2.21, ddt (13.9, 2.9, 1.3) | a: 2.68, ddd (14.0, 12.0, 3.0) b: 2.21, ddt (14.0, 3.0, 1.5) | a: 2.67, ddd (15.0, 12.0, 9.0) b: 2.21, dd (15.0, 4.0) | a: 2.86, ddd (18.4, 6.2, 4.2) b: 2.76, ddd (18.4, 6.2, 4.4) |
| 4 | 2.09, dd (11.8, 1.2) | 2.07, dd (11.3, 1.2) | 2.10, dd (11.3, 1.2) | 2.04, dd (11.5, 1.6) | 2.11, d (11.7) | 2.59, d (11.4) |
| 5 | 1.89, tdd (11.8, 10.6, 3.2) | 1.78, m | 1.75, m | 1.75, m | 1.95, m | 1.93, tdd (11.4, 10.4, 3.0) |
| 6 | a: 1.28, m b: 1.14, qd (13.0, 3.3) | a: 1.41, dq (13.2, 3.2) b: 1.14, qd (13.2, 3.5) | a: 1.69, m b: 1.41, m | a: 1.34, dq (13.3, 3.6) b: 1.11, qd (13.0, 3.8) | a: 1.33, m b: 1.11, m | a: 1.50, m b: 1.15, qd (12.7, 3.2) |
| 7 | a: 1.78, dq (12.7, 3.3) b: 1.08, qd (12.7, 3.6) | a: 1.85, dq (12.9, 3.4) b: 1.01, qd (12.9, 3.7) | a: 1.45, m b: 1.19, m | a: 1.75, m b: 0.96, m | a: 1.73, m b: 1.03, m | a: 1.77, m b: 1.12, qd (13.0, 3.3) |
| 8 | 1.41, m | 1.63, m | 1.48, m | 1.48, m | 1.42, m | |
| 9 | 2.92, td (9.9, 5.8) | a: 1.91, dq (12.6, 3.5) b: 0.93, q (12.3) | a: 1.75, m b: 1.28, m | a: 1.77, m b: 0.87, q (12.2) | 3.29, t (9.0) | 2.92, t (9.9) |
| 10 | 1.65, tt (10.4, 2.3) | 1.74, m | 2.15, ddd (13.3, 10.2, 3.5) | 1.71, m | 1.61, td (10.8, 9.0) | 1.73, tt (10.3, 2.3) |
| 11 | 6.24, dd (10.0, 2.4) | 5.67, dd (9.8, 1.5) | 5.63, brs | 5.59, dd (9.9, 2.5) | 4.38, t (10.5) | 6.11, dd (10.0, 2.0) |
| 12 | 5.69, dd (10.0, 2.6) | 5.61, dd (9.8, 2.5) | 5.63, brs | 5.65, dd (9.8, 1.0) | 3.78, d (10.5) | 5.64, dd (10.0, 2.6) |
| 14 | 1.26, s | 1.26, s | 1.26, s | 1.25, s | 1.36, s | 1.30, s |
| 15 | 1.03, d (6.5) | a: 3.50, dd (10.3, 6.3) b: 3.47, dd (10.3, 6.3) | 1.25, s | 0.91, d (6.6) | 1.06, d (6.5) | 1.03, d (6.5) |
| 9-OH | 1.55, d (6.3) |
| Position | 7 * | 8 ** | 9 * | 10 ** | 11 * | 12 ** |
|---|---|---|---|---|---|---|
| 1 | 58.1, CH2 | 57.9, CH2 | 58.0, CH2 | 58.0, CH2 | 57.9, CH2 | 58.0, CH2 |
| 2 | 49.3, CH2 | 43.6, CH2 | 42.1, CH2 | 49.2, CH2 | 48.9, CH2 | 49.5, CH2 |
| 3 | 214.3, C | 214.0, C | 213.5, C | 214.1, C | 211.2, C | 213.2, C |
| 4 | 63.5, CH | 61.7, CH | 59.6, CH | 63.4, CH | 61.6, CH | 57.7, CH |
| 5 | 41.3, CH | 37.9, CH | 40.3, CH | 41.5, CH | 39.4, CH | 40.3, CH |
| 6 | 29.4, CH2 | 30.5, CH2 | 29.9, CH2 | 28.7, CH2 | 32.4, CH2 | 32.1, CH2 |
| 7 | 34.8, CH2 | 31.9, CH2 | 32.6, CH2 | 28.9, CH2 | 27.9, CH2 | 28.1, CH2 |
| 8 | 32.9, CH | 39.2, CH | 40.4, CH | 40.6, CH | 41.1, CH | 41.1, CH |
| 9 | 40.9, CH2 | 82.3, CH | 78.3, CH | 35.2, CH2 | 38.3, CH2 | 37.2, CH2 |
| 10 | 42.1, CH | 49.6, CH | 47.2, CH | 41.5, CH | 166.3, C | 144.5, C |
| 11 | 131.9, CH | 73.2, CH | 130.6, CH | 131.5, CH | 120.0, CH | 118.8, CH |
| 12 | 133.7, CH | 129.0, CH | 129.3, CH | 134.0, CH | 200.3, C | 68.0, CH |
| 13 | 73.0, C | 131.5, C | 140.8, C | 72.9, C | 74.2, C | 72.4, C |
| 14 | 25.9, CH3 | 20.9, CH3 | 112.8, CH2 | 25.9, CH3 | 21.8, CH3 | 20.5, CH3 |
| 15 | 22.3, CH3 | 17.8, CH3 | 18.4, CH3 | 68.1, CH2 | 67.4, CH2 | 67.8, CH2 |
| Position | 7 * | 8 ** | 9 * | 10 ** | 11 * | 12 *** |
|---|---|---|---|---|---|---|
| 1 | a: 3.90, ddd (11.7, 7.1, 3.7) b: 3.86, ddd (11.7, 6.6, 3.7) | 3.85, m (2H) | a: 3.91, ddd (11.5, 7.1, 3.9) b: 3.85, ddd (11.5, 6.8, 4.2) | a: 3.91, ddd (11.2, 6.9, 3.6) b: 3.87, ddd (11.2, 6.7, 3.7) | a: 3.96, ddd (11.5, 7.7, 3.7) b: 3.82, ddd (11.5, 6.7, 3.9) | a: 3.95, ddd (11.3, 7.5, 3.5) b: 3.82, ddd (11.3, 6.4, 3.6) |
| 2 | a: 3.04, ddd (18.0, 6.8, 3.8) b: 2.60, ddd (18.0, 7.1, 3.8) | a: 2.69, ddd (18.2, 6.6, 4.3) b: 2.64, ddd (18.2, 6.3, 4.3) | a: 2.83, ddd (18.3, 6.8, 3.8) b: 2.65, ddd (18.3, 6.8, 4.0) | a: 3.06, ddd (18.0, 7.1, 3.7) b: 2.61, ddd (18.0, 7.0, 3.7) | a: 3.14, ddd (18.0, 6.5, 3.6) b: 2.66, ddd (18.0, 7.6, 3.9) | a: 3.05, ddd (18.1, 6.5, 3.6) b: 2.69, ddd (18.0, 7.6, 3.7) |
| 4 | 2.89, d (11.5) | 2.92, m | 3.15, dtd (12.1, 2.6, 0.9) | 2.92, d (11.6) | 3.01, d (9.8) | 3.09, d (10.0) |
| 5 | 1.48, m | 1.69, m | 1.68, m | 1.52, dq (11.7, 2.7) | 2.89, m | 2.55, m |
| 6 | a: 1.70, m b: 0.93, m | a: 1.57, dq (13.2, 3.5) b: 1.17, qd (13.1, 3.5) | a: 1.53, m b: 1.14, m | a: 1.79, m b: 0.95, qd (12.3, 3.6) | a: 1.91, dq (12.9, 3.0) b: 1.15, qd (13.1, 3.3) | a: 1.74, m b: 1.12, m |
| 7 | a: 1.71, m b: 0.96, m | a: 1.70, m b: 1.03, qd (13.2, 3.5) | a: 1.77, m b: 1.10, m | a: 1.79, m b: 1.02, qd (12.4, 3.6) | a: 1.84, dq (13.1, 3.2) b: 1.29, m | a: 1.79, m b: 1.12, m |
| 8 | 1.46, m | 1.47, m | 1.43, m | 1.61, m | 1.74, m | 1.64, m |
| 9 | a: 1.73, m b: 0.77, q (12.4) | 3.25, t (9.3) | 2.89, t (9.9) | a: 1.87, m b: 0.82, q (13.0) | a: 2.60, dq (14.0, 2.5) b: 2.01, m | a: 2.39, dq (13.5, 2.5) b: 1.77, m |
| 10 | 1.84, m | 1.28, dt (11.9, 9.3) | 1.85, t (10.0) | 1.87, m | ||
| 11 | 5.42, brs | 4.32, dq (8.9, 2.1) | 6.20, brs | 5.44, brs | 5.90, brs | 5.60, dt (5.9, 2.1) |
| 12 | 5.42, brs | 5.53, q (1.8) | 6.20, brs | 5.44, brs | 4.36, dd (5.9, 1.7) | |
| 14 | 1.20, s | 1.60, brs | a: 4.99, dd (2.8, 1.3) b: 4.59, dt (2.3, 0.9) | 1.20, s | 1.19, s | 1.20, s |
| 15 | 0.89, d (6.5) | 1.02, d (6.5) | 1.05, d (6.5) | a: 3.47, dd (10.6, 6.6) b: 3.45, dd (10.6, 6.6) | a: 3.58, dd (10.5, 5.8) b: 3.54, dd (10.5, 5.8) | a: 3.50, dd (10.5, 5.9) b: 3.48, dd (10.4, 6.2) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afiyatullov, S.S.; Leshchenko, E.V.; Berdyshev, D.V.; Sobolevskaya, M.P.; Antonov, A.S.; Denisenko, V.A.; Popov, R.S.; Pivkin, M.V.; Udovenko, A.A.; Pislyagin, E.A.; et al. Zosteropenillines: Polyketides from the Marine-Derived Fungus Penicillium thomii. Mar. Drugs 2017, 15, 46. https://doi.org/10.3390/md15020046
Afiyatullov SS, Leshchenko EV, Berdyshev DV, Sobolevskaya MP, Antonov AS, Denisenko VA, Popov RS, Pivkin MV, Udovenko AA, Pislyagin EA, et al. Zosteropenillines: Polyketides from the Marine-Derived Fungus Penicillium thomii. Marine Drugs. 2017; 15(2):46. https://doi.org/10.3390/md15020046
Chicago/Turabian StyleAfiyatullov, Shamil Sh., Elena V. Leshchenko, Dmitrii V. Berdyshev, Maria P. Sobolevskaya, Alexandr S. Antonov, Vladimir A. Denisenko, Roman S. Popov, Mikhail V. Pivkin, Anatoly A. Udovenko, Evgeny A. Pislyagin, and et al. 2017. "Zosteropenillines: Polyketides from the Marine-Derived Fungus Penicillium thomii" Marine Drugs 15, no. 2: 46. https://doi.org/10.3390/md15020046
APA StyleAfiyatullov, S. S., Leshchenko, E. V., Berdyshev, D. V., Sobolevskaya, M. P., Antonov, A. S., Denisenko, V. A., Popov, R. S., Pivkin, M. V., Udovenko, A. A., Pislyagin, E. A., Von Amsberg, G., & Dyshlovoy, S. A. (2017). Zosteropenillines: Polyketides from the Marine-Derived Fungus Penicillium thomii. Marine Drugs, 15(2), 46. https://doi.org/10.3390/md15020046