
Induction of Diverse Bioactive Secondary Metabolites from the Mangrove Endophytic Fungus Trichoderma sp. (Strain 307) by Co-Cultivation with Acinetobacter johnsonii (Strain B2)



Abstract
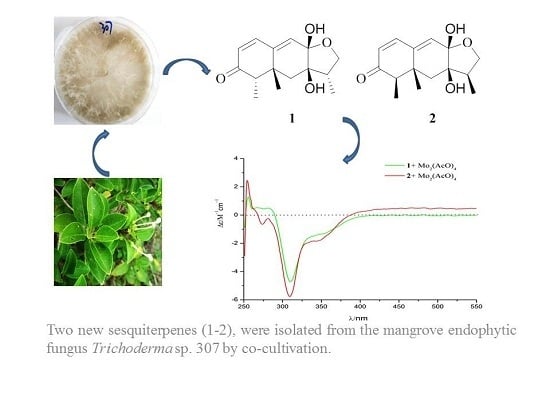
:
1. Introduction
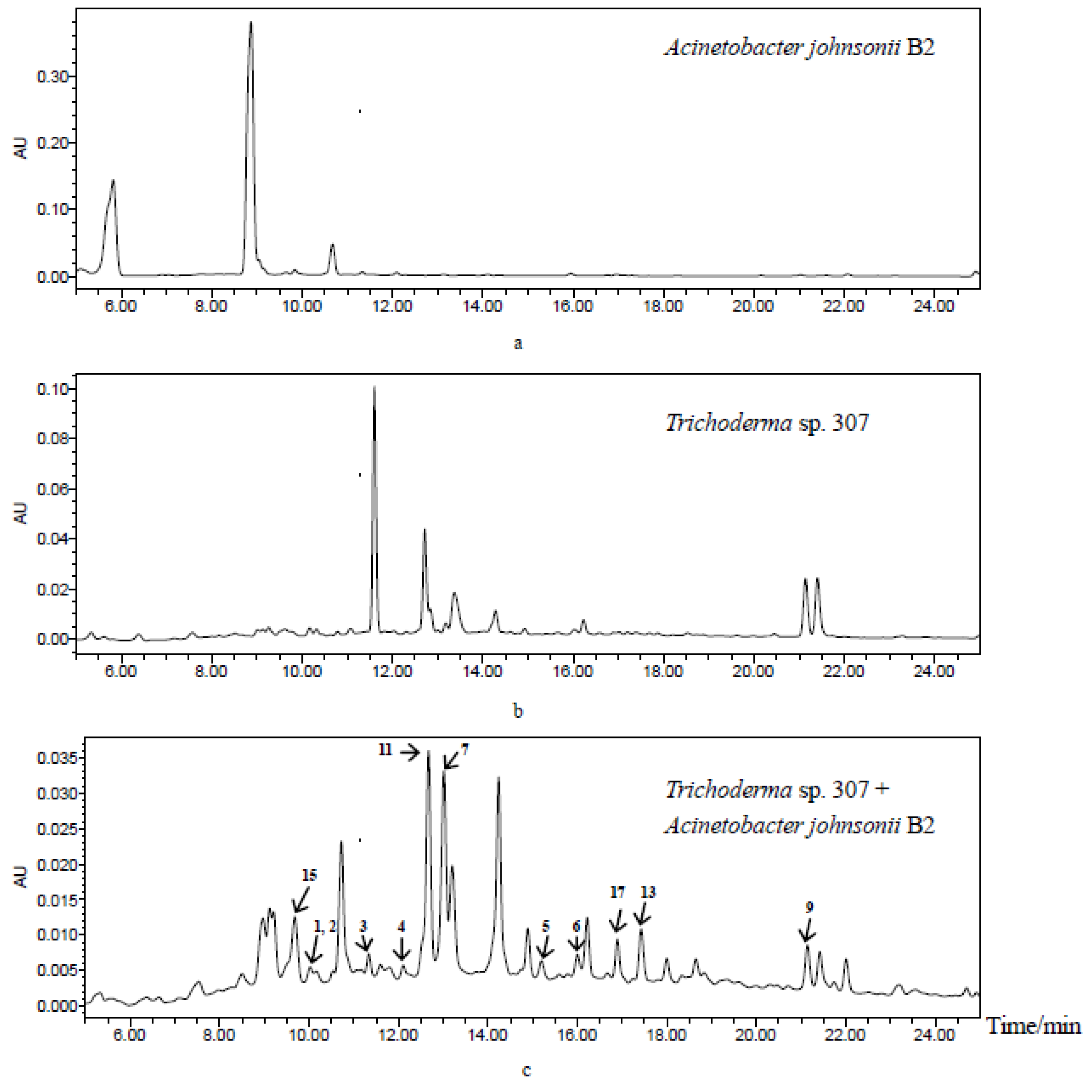
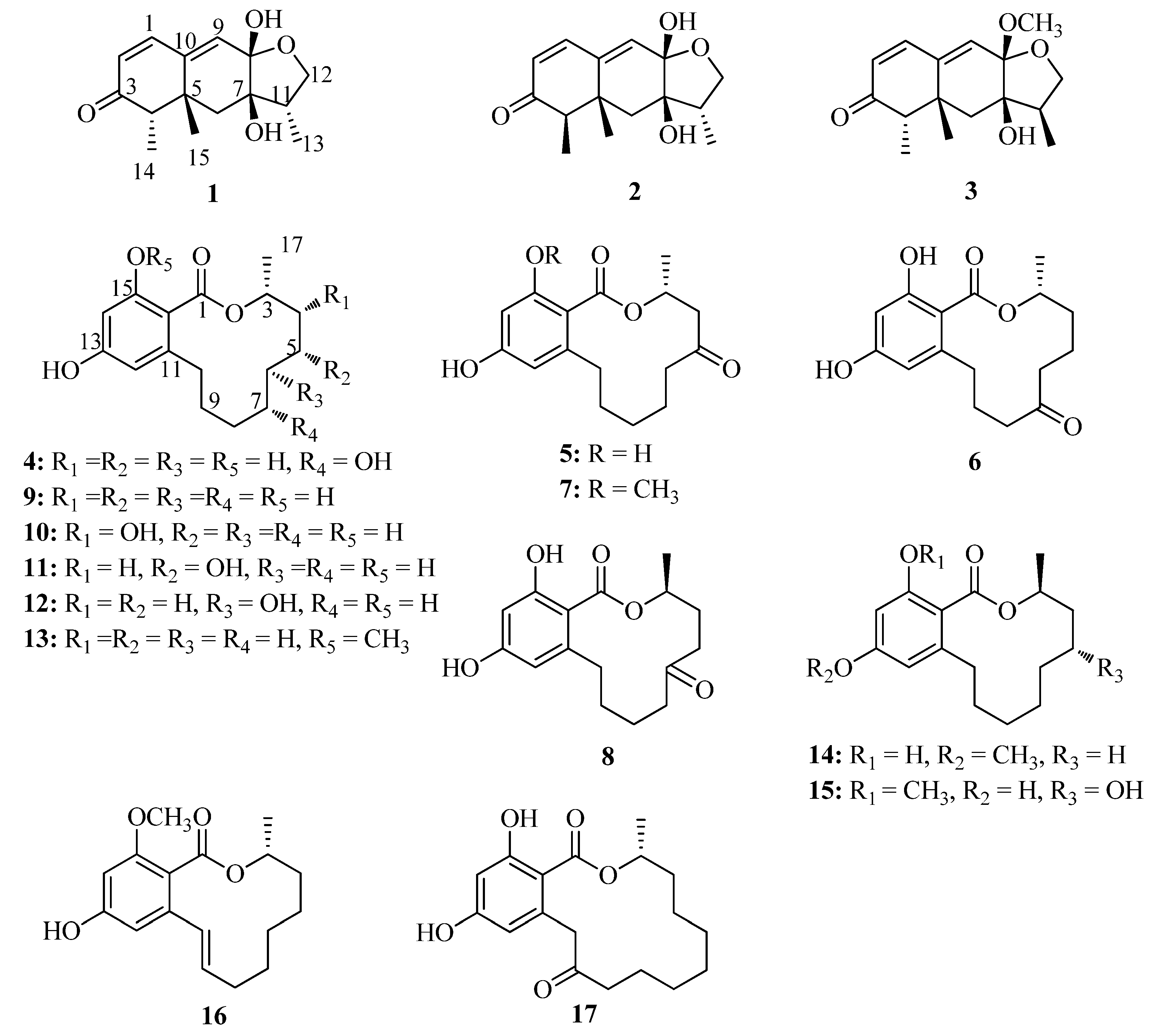
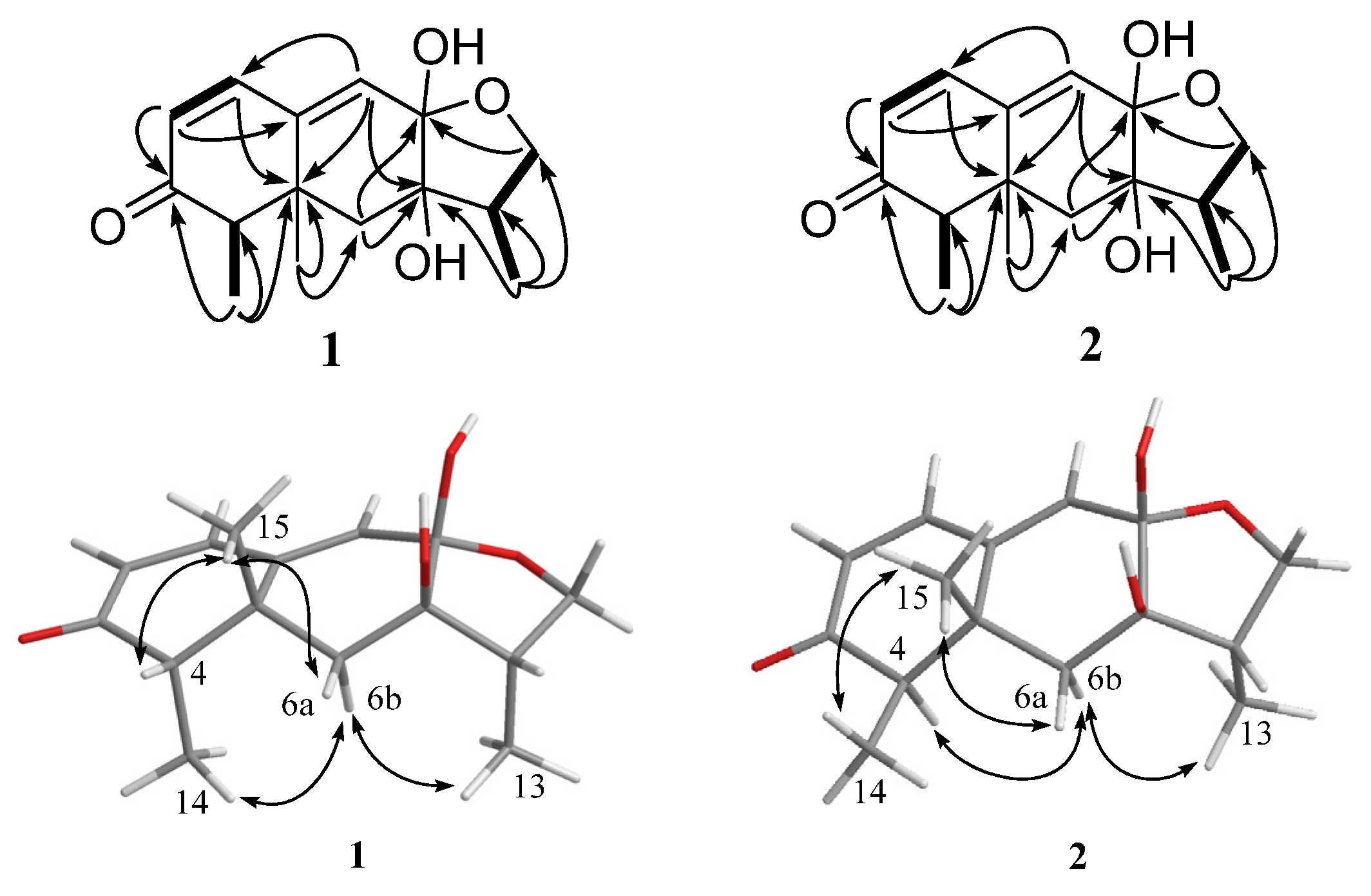
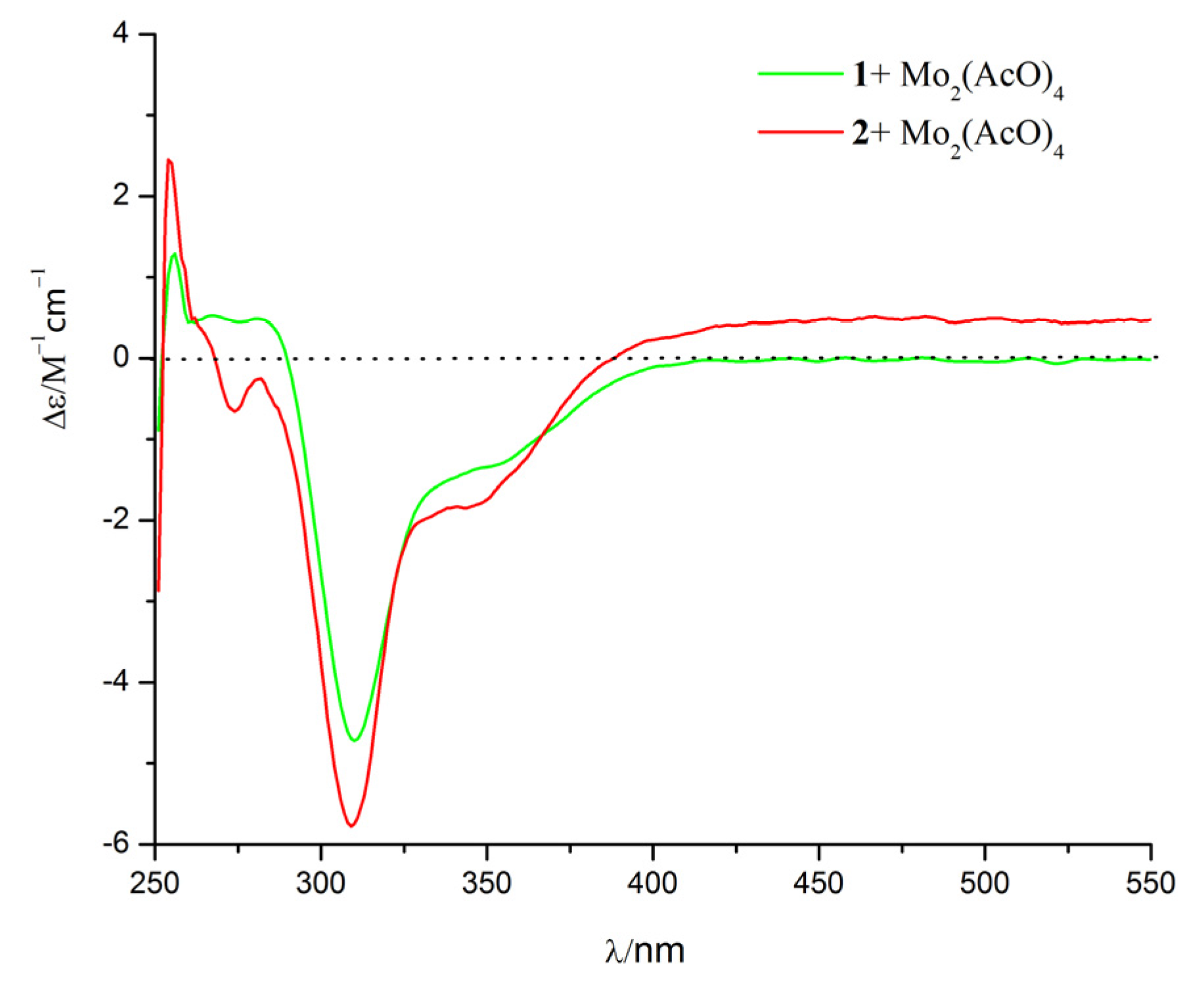
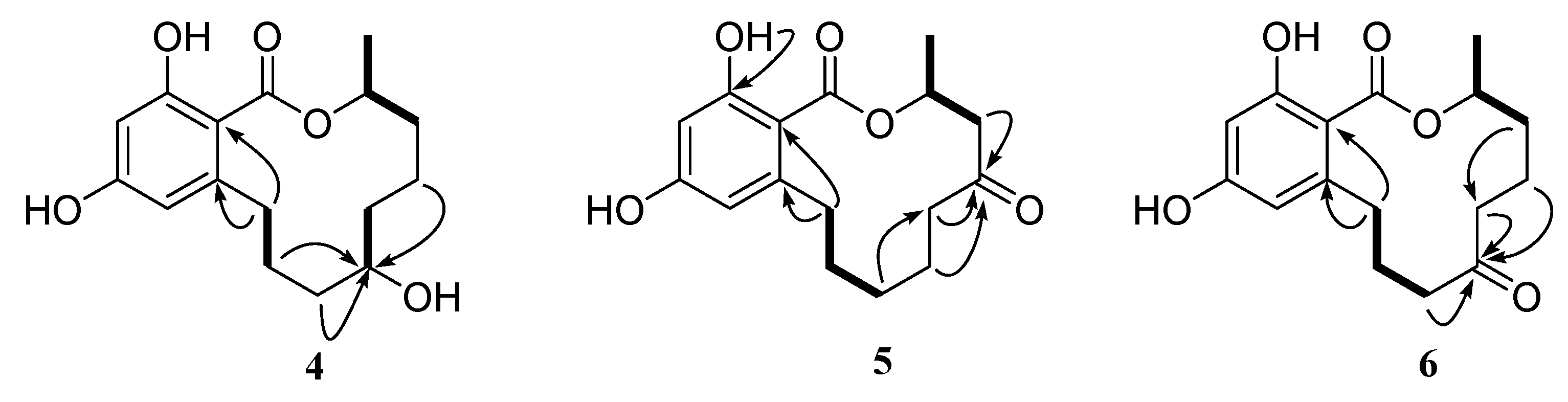
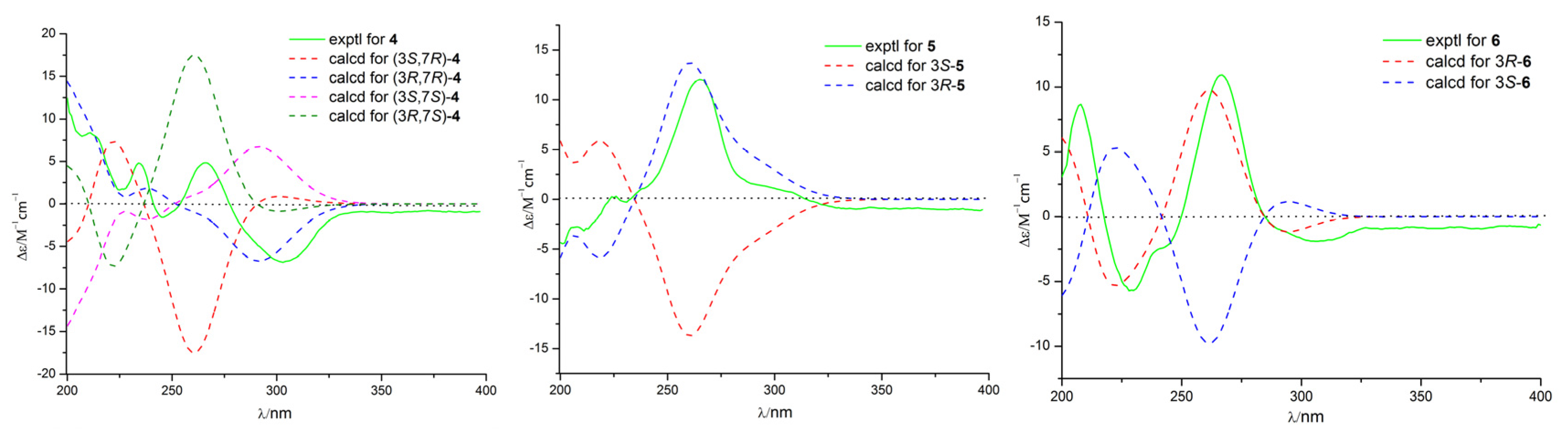
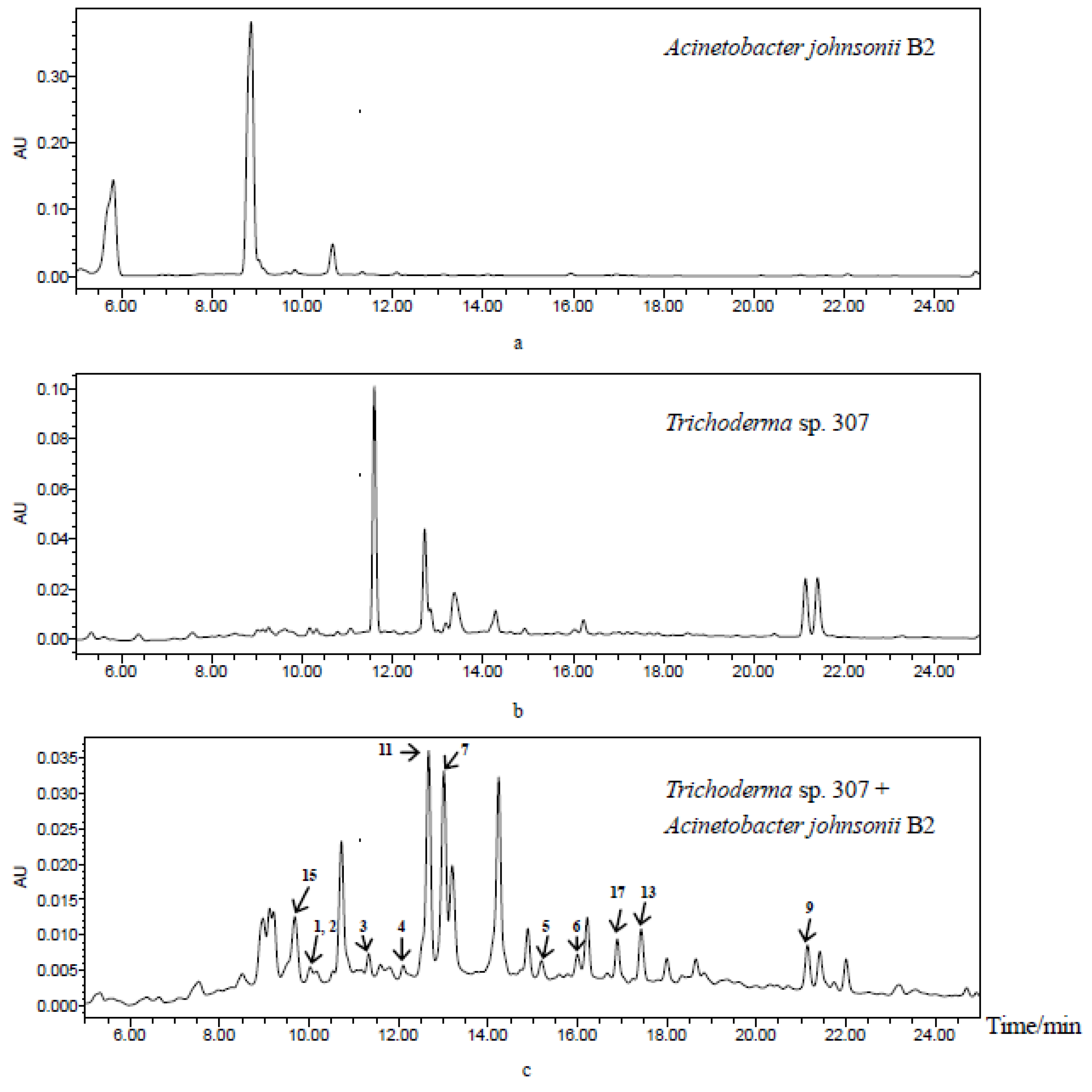
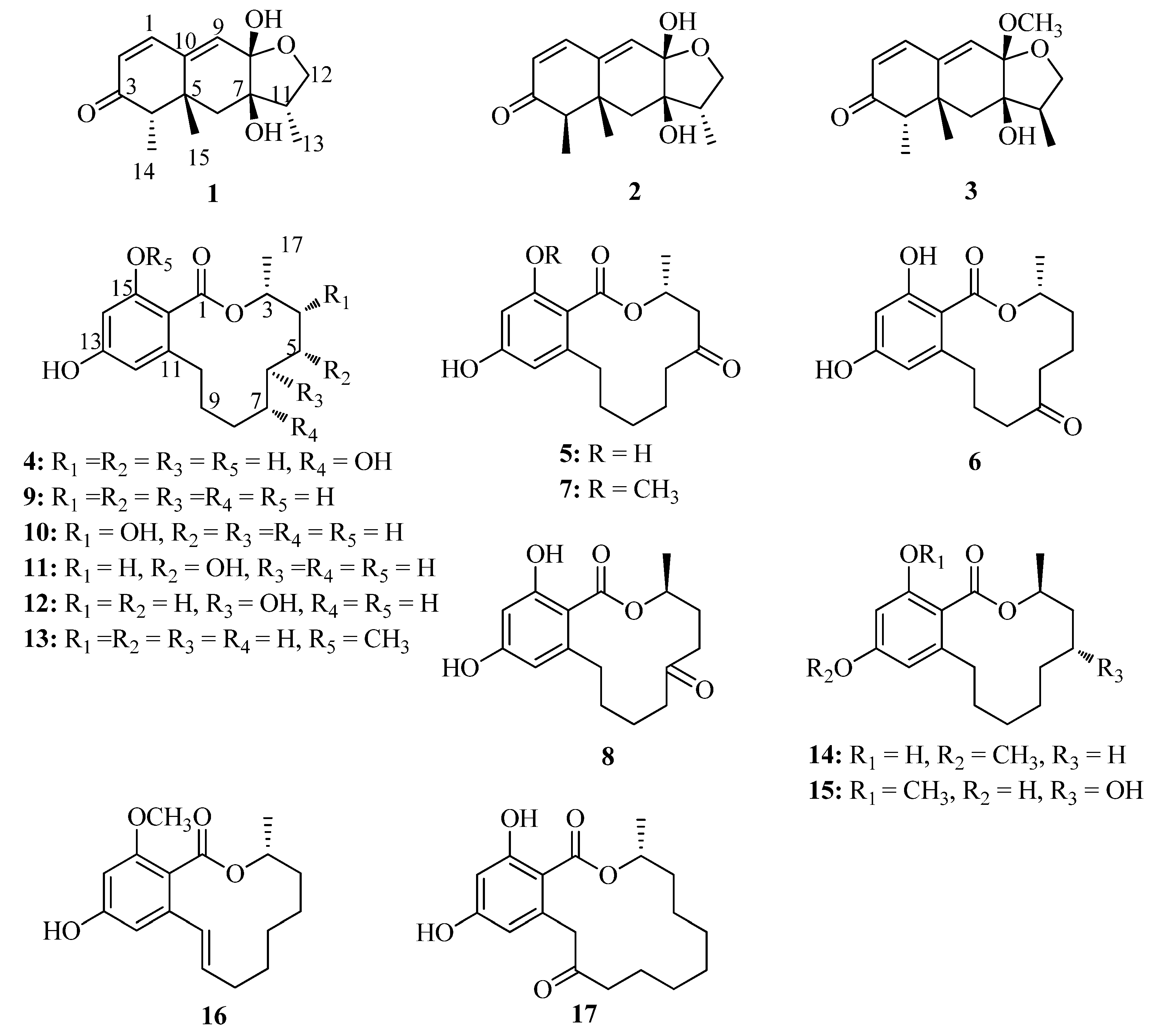
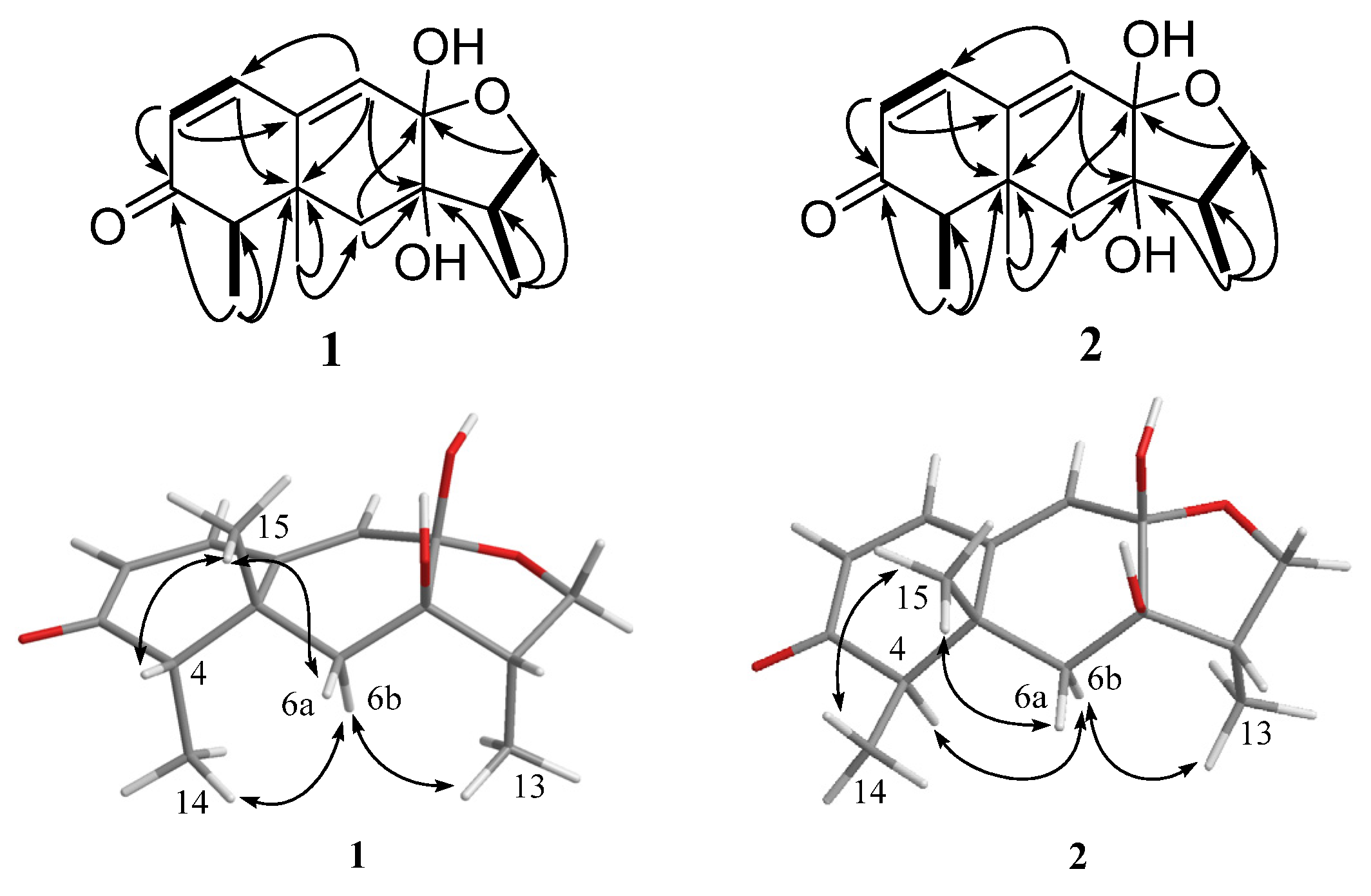
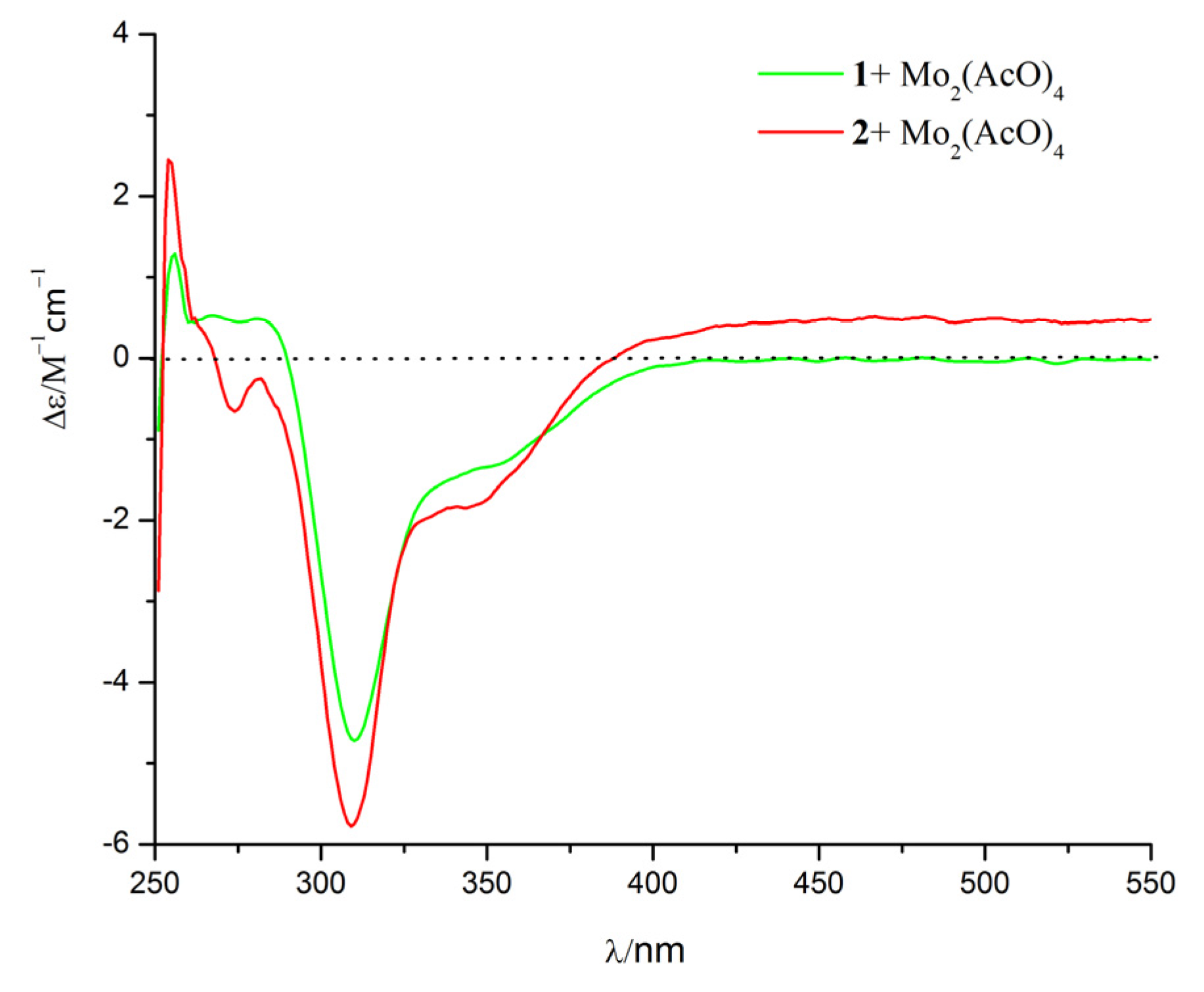
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal and Bacterial Material
3.3. Co-Cultivation, Extraction, and Isolation
3.4. Calculation of ECD Spectra
3.5. α-Glucosidase Inhibitory Activity Assay
3.6. Cytotoxicity Assay
3.7. HPLC Profiles Conditions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ECD | Electronic Circular Dichroism |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| HPLC | High Performance Liquid Chromatography |
References
- Rocha-Martin, J.; Harrington, C.; Dobson, A.D.W.; O’Gara, F. Emerging strategies and integrated systems microbiology technologies for biodiscovery of marine bioactive compounds. Mar. Drugs 2014, 12, 3516–3559. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Chen, S.H.; Liu, W.Y.; Liu, Y.Y.; Huang, X.S.; She, Z.G. Polyketides with immunosuppressive activities from mangrove endophytic fungus Penicillium sp. ZJ-SY2. Mar. Drugs 2016, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.M.; Quan, C.H.; Hou, X.Y.; Fan, S.D. Potential pharmacological resources: Natural bioactive compounds from marine-derived fungi. Mar. Drugs 2016, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, P.J.; Challis, G.L. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Adnani, N.; Vazquez-Rivera, E.; Adibhatla, S.N.; Ellis, G.A.; Braun, D.R.; Bugni, T.S. Investigation of interspecies interactions within marine micromonosporaceae using an improved co-culture approach. Mar. Drugs 2015, 13, 6082–6098. [Google Scholar] [CrossRef] [PubMed]
- Schroeckh, V.; Scherlach, K.; Nützmann, H.W.; Shelest, E.; Schmidt-Heck, W.; Schuemann, J.; Martin, K.; Hertweck, C.; Brakhage, A.A. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 2009, 106, 14558–14563. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Bohni, N.; Schnee, S.; Schumpp, O.; Gindro, K.; Wolfender, J.L. Metabolite induction via microorganism co-culture: A potential way to enhance chemical diversity for drug discovery. Biotechnol. Adv. 2014, 32, 1180–1204. [Google Scholar] [CrossRef] [PubMed]
- Ola, A.R.; Thomy, D.; Lai, D.; Brötz-Oesterhelt, H.; Proksch, P. Inducing secondary metabolite production by the endophytic fungus Fusarium tricinctum through coculture with Bacillus subtilis. J. Nat. Prod. 2013, 76, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Hallyburton, I.; Houssen, W.E.; Bull, A.T.; Goodfellow, M.; Santhanam, R.; Jaspars, M.; Ebel, R. Induction of diverse secondary metabolites in Aspergillus fumigatus by microbial co-culture. RSC Adv. 2013, 3, 14444–14450. [Google Scholar] [CrossRef]
- Whitt, J.; Shipley, S.M.; Newman, D.J.; Zuck, K.M. Tetramic acid analogues produced by coculture of Saccharopolyspora erythraea with Fusarium pallidoroseum. J. Nat. Prod. 2014, 77, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.P.; Zhuang, Y.B.; Kong, F.D.; Zhang, C.X.; Zhu, W.M. Phenolic polyketides from the co-cultivation of marine-derived Penicillium sp. WC-29–5 and Streptomyces fradiae 007. Mar. Drugs 2014, 12, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Schumpp, O.; Bohni, N.; Monod, M.; Gindro, K.; Wolfender, J.L. De novo production of metabolites by fungal co-culture of Trichophyton rubrum and Bionectria ochroleuca. J. Nat. Prod. 2013, 76, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ding, W.J.; Li, C.Y.; Cox, D.G. Two new cyclopeptides from the co-culture broth of two marine mangrove fungi and their antifungal activity. Pharmacogn. Mag. 2014, 10, 410–414. [Google Scholar] [PubMed]
- Zhu, F.; Chen, G.Y.; Wu, J.S.; Pan, J.H. Structure revision and cytotoxic activity of marinamide and its methyl ester, novel alkaloids produced by co-cultures of two marine-derived mangrove endophytic fungi. Nat. Prod. Res. 2013, 27, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Kossuga, M.H.; Ferreira, A.G.; Sette, L.D.; Berlinck, R.G. Two polyketides from a co-culture of two marine-derived fungal strains. Nat. Prod. Commun. 2013, 8, 721–724. [Google Scholar]
- Li, H.X.; Huang, H.B.; Shao, C.L.; Huang, H.R.; Jiang, J.Y.; Zhu, X.; Lin, Y.Y.; Liu, L.; Lu, Y.J.; Li, M.F.; et al. Cytotoxic norsesquiterpene peroxides from the endophytic fungus Talaromyces flavus isolated from the mangrove plant Sonneratia apetala. J. Nat. Prod. 2011, 74, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.X.; Qiao, L.T.; Wang, J.J.; Zeng, H.M.; She, Z.G.; Miao, C.D.; Hong, K.; Gu, Y.C.; Liu, L.; Lin, Y.C. Two new meroterpenes from the mangrove endophytic fungus Aspergillus sp. 085241B. Helv. Chim. Acta 2011, 94, 1875–1880. [Google Scholar] [CrossRef]
- Ma, Y.H.; Li, J.; Huang, M.X.; Liu, L.; Wang, J.; Lin, Y.C. Six new polyketide decalin compounds from mangrove endophytic fungus Penicillium aurantiogriseum 328#. Mar. Drugs 2015, 13, 6306–6318. [Google Scholar] [PubMed]
- Huang, M.X.; Li, J.; Liu, L.; Yin, S.; Wang, J.; Lin, Y.C. Phomopsichin A–D; four new chromone derivatives from mangrove endophytic fungus Phomopsis sp. 33#. Mar. Drugs 2016, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, L.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef] [PubMed]
- Górecki, M.; Jablonska, E.; Kruszewska, A.; Suszczynska, A.; Urbanczyk-Lipkowska, Z.; Gerards, M.; Morzycki, J.W.; Szczepek, W.J.; Frelek, J. Practical method for the absolute configuration assignment of tert/tert 1,2-diols using their complexes with Mo2(OAc)4. J. Org. Chem. 2007, 72, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, D.; Si, Y.K.; Lv, H.J.; Wu, X.F.; Li, Y.; Liu, Y.Y.; Yu, S.S. Application of dimolybdenum reagent Mo2(OAc)4 for determination of the absolute configurations of vic-diols. Chin. J. Org. Chem. 2010, 30, 1270–1278. [Google Scholar]
- Rudiyansyah; Garson, M.J. Secondary metabolites from the wood bark of Durio zibethinus and Durio kutejensis. J. Nat. Prod. 2006, 69, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Nakamori, K.; Omer, E.A.; Hatakeyama, C.; Yoshihara, T.; Ichihara, A. Three lasiodiplodins from Lasiodiplodia theobromae IFO 31059. Phytochemistry 1998, 49, 579–584. [Google Scholar] [CrossRef]
- Höller, U.; König, G.M.; Wright, A.D. Three new metabolites from marine-derived fungi of the genera Coniothyrium and Microsphaeropsis. J. Nat. Prod. 1999, 62, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Li, C.Y.; Lin, Y.C.; Peng, G.T.; She, Z.G.; Zhou, S.N. Lactones from a brown alga endophytic fungus (No. ZZF36) from the South China Sea and their antimicrobial activities. Bioorg. Med. Chem. Lett. 2006, 16, 4205–4208. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.M.; Zheng, C.J.; Han, C.R.; Chen, G.Y.; Dai, C.Y.; Song, X.P.; Zhang, J.C.; Chen, W.H. Lactones from Ficus auriculata and their effects on the proliferation function of primary mouse osteoblasts in vitro. Bioorg. Med. Chem. Lett. 2014, 24, 3952–3955. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Asai, M.; Matsuura, H.; Yoshihara, T. Potato micro-tuber inducing hydroxylasiodiplodins from Lasiodiplodia theobromae. Phytochemistry 2000, 54, 489–494. [Google Scholar] [CrossRef]
- Leet, K.H.; Hayashi, N.; Okano, M.; Hall, I.H.; Wu, R.Y.; Mcphailti, A.T. Lasiodiplodin, a potent antileukemic macrolide from Euphorbia splendens. Phytochemistry 1982, 21, 1119–1121. [Google Scholar] [CrossRef]
- Abreu, P.J.; Liu, Y. Ozoroalide, a new macrolide from Ozoroa insignis. Fitoterapia 2007, 78, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Aver, W.A.; Peña-Rodriguez, L. Minor metabolites of Monocillium nordinii. Phytochemistry 1987, 26, 1353–1355. [Google Scholar] [CrossRef]
- Chen, S.H.; Liu, Y.Y.; Liu, Z.M.; Cai, R.L.; Lu, Y.J.; Huang, X.S.; She, Z.G. Isocoumarins and benzofurans from the mangrove endophytic fungus Talaromyces amestolkiae possess α-glucosidase inhibitory and antibacterial activities. RSC Adv. 2016, 6, 26412–26420. [Google Scholar] [CrossRef]
- Chen, S.H.; Liu, Z.M.; Li, H.X.; Xia, G.P.; Lu, Y.J.; He, L.; Huang, S.D.; She, Z.G. β-Resorcylic acid derivatives with α-glucosidase inhibitory activity from Lasiodiplodia sp. ZJ-HQ1, an endophytic fungus in the medicinal plant Acanthus ilicifolius. Phytochem. Lett. 2015, 13, 141–146. [Google Scholar] [CrossRef]
- Li, J.; Xue, Y.Y.; Yuan, J.; Lu, Y.J.; Zhu, X.; Lin, Y.C.; Liu, L. Lasiodiplodins from mangrove endophytic fungus Lasiodiplodia sp. 318#. Nat. Prod. Res. 2016, 30, 755–760. [Google Scholar] [PubMed]
- Huang, J.G.; Xu, J.Y.; Wang, Z.; Khan, D.; Niaz, S.I.; Zhu, Y.H.; Lin, Y.C.; Li, J.; Liu, L. New lasiodiplodins from mangrove endophytic fungus Lasiodiplodia sp. 318#. Nat. Prod. Res. 2016, 31, 326–332. [Google Scholar] [PubMed]
- Wang, X.; Tan, T.; Mao, Z.G.; Lei, N.; Wang, Z.M.; Hu, B.; Chen, Z.Y.; She, Z.G.; Zhu, Y.H.; Wang, H.J. The marine metabolite SZ-685C induces apoptosis in primary human nonfunctioning pituitary adenoma cells by inhibition of the Akt pathway in vitro. Mar. Drugs 2015, 13, 1569–1580. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 146.6 | 7.04 (d, J = 9.8) | 146.1 | 7.01 (d, J = 9.8) |
| 2 | 126.3 | 5.84 (d, J = 9.8) | 128.4 | 5.93 (d, J = 9.8) |
| 3 | 206.8 | 203.4 | ||
| 4 | 55.0 | 2.24 (q, J = 7.3) | 54.4 | 2.24 (q, J = 6.9) |
| 5 | 39.9 | 40.4 | ||
| 6a | 34.6 | 1.79 (d, J = 14.1) | 38.9 | 1.87 (d, J = 14.0) |
| 6b | 1.44 (d, J = 14.1) | 1.51 (d, J = 14.0) | ||
| 7 | 77.8 | 77.6 | ||
| 8 | 100.5 | 100.3 | ||
| 9 | 135.3 | 6.10 (s) | 132.2 | 5.94 (s) |
| 10 | 139.4 | 142.4 | ||
| 11 | 43.9 | 2.58 (ddq, J = 8.3, 7.2, 6.4) | 43.9 | 2.54 (ddq, J = 7.8, 6.9, 6.4) |
| 12a | 71.8 | 4.07 (dd, J = 8.8, 8.3) | 71.6 | 4.04 (dd, J = 8.3, 7.8) |
| 12b | 3.34 (dd, J = 8.8, 6.4) | 3.31 (dd, J = 8.3, 6.4) | ||
| 13 | 9.2 | 1.01 (d, J = 7.2) | 9.2 | 1.01 (d, J = 6.9) |
| 14 | 14.8 | 0.95 (d, J = 7.3) | 7.5 | 1.08 (d, J = 6.9) |
| 15 | 28.0 | 1.35 (s) | 20.9 | 1.16 (s) |
| Position | 4 a | 5 b | 6 b | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 172.0 | 171.0 | 171.8 | |||
| 3 | 73.7 | 5.37 (m) | 69.3 | 5.69 (ddq, J = 10.8, 6.3, 2.3) | 74.9 | 5.28 (ddq, J = 12.8, 6.3, 3.3) |
| 4 | 31.4 | 1.94 (m) | 46.1 | 3.31 (dd, J = 16.4, 10.8) | 31.7 | 1.94 (dd, J = 12.8, 7.7) |
| 1.65 (m) | 2.39 (dd, J = 16.4, 2.3) | 1.84 (dd, J = 7.7, 3.3) | ||||
| 5 | 30.2 | 1.74 (m) | 210.7 | 19.2 | 2.08 (m) | |
| 1.28 (m) | 1.76 (m) | |||||
| 6 | 32.5 | 2.12 (m) | 46.1 | 2.61 (m) | 38.6 | 2.64 (m) |
| 1.83 (m) | 2.30 (m) | 2.55 (m) | ||||
| 7 | 68.0 | 4.27 (m) | 21.8 | 2.01 (m) | 211.4 | |
| 1.86 (m) | ||||||
| 8 | 24.1 | 1.92 (m) | 29.9 | 1.48 (m) | 42.1 | 2.44 (m) |
| 1.67 (m) | 1.25 (m) | 2.35 (m) | ||||
| 9 | 31.1 | 2.06 (m) | 30.3 | 1.66 (m) | 28.9 | 2.20 (m) |
| 1.76 (m) | 1.57 (m) | 1.75 (m) | ||||
| 10 | 33.7 | 3.22 (m) | 36.8 | 3.45 (m) | 33.6 | 3.14 (m) |
| 2.59 (m) | 2.11 (m) | 2.61 (m) | ||||
| 11 | 148.3 | 149.0 | 148.0 | |||
| 12 | 111.9 | 6.74 (d, J = 2.4) | 111.1 | 6.17 (d, J = 2.6) | 110.8 | 6.24 (d, J = 2.6) |
| 13 | 163.9 | 160.5 | 160.5 | |||
| 14 | 102.3 | 6.84 (d, J = 2.4) | 101.8 | 6.26 (d, J = 2.6) | 102.0 | 6.30 (d, J = 2.6) |
| 15 | 164.9 | 166.1 | 165.8 | |||
| 16 | 107.6 | 104.8 | 106.0 | |||
| 17 | 18.2 | 1.38 (d, J = 6.3) | 19.9 | 1.44 (d, J = 6.3) | 19.6 | 1.35 (d, J = 6.3) |
| 15-OH | 12.42 (s) | 11.97 (s) | 12.05(s) | |||
| Compounds | IC50 (µM) | Compounds | IC50 (µM) |
|---|---|---|---|
| 1 | >200 | 10 | 60.3 ± 0.7 |
| 2 | 188.7 ± 1.2 | 11 | >200 |
| 3 | >200 | 12 | >200 |
| 4 | 25.8 ± 0.2 | 13 | >200 |
| 5 | 54.6 ± 0.5 | 14 | 198.1 ± 1.5 |
| 6 | 178.5 ± 1.1 | 15 | ˃200 |
| 7 | 176.8 ± 1.4 | 16 | 101.3 ± 0.9 |
| 8 | 64.2 ± 0.5 | 17 | 105.7 ± 1.0 |
| 9 | 48.9 ± 0.4 | Acarbose b | 703.8 ± 2.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Niaz, S.I.; Khan, D.; Wang, Z.; Zhu, Y.; Zhou, H.; Lin, Y.; Li, J.; Liu, L. Induction of Diverse Bioactive Secondary Metabolites from the Mangrove Endophytic Fungus Trichoderma sp. (Strain 307) by Co-Cultivation with Acinetobacter johnsonii (Strain B2). Mar. Drugs 2017, 15, 35. https://doi.org/10.3390/md15020035
Zhang L, Niaz SI, Khan D, Wang Z, Zhu Y, Zhou H, Lin Y, Li J, Liu L. Induction of Diverse Bioactive Secondary Metabolites from the Mangrove Endophytic Fungus Trichoderma sp. (Strain 307) by Co-Cultivation with Acinetobacter johnsonii (Strain B2). Marine Drugs. 2017; 15(2):35. https://doi.org/10.3390/md15020035
Chicago/Turabian StyleZhang, Liuhong, Shah Iram Niaz, Dilfaraz Khan, Zhen Wang, Yonghong Zhu, Haiyun Zhou, Yongcheng Lin, Jing Li, and Lan Liu. 2017. "Induction of Diverse Bioactive Secondary Metabolites from the Mangrove Endophytic Fungus Trichoderma sp. (Strain 307) by Co-Cultivation with Acinetobacter johnsonii (Strain B2)" Marine Drugs 15, no. 2: 35. https://doi.org/10.3390/md15020035
APA StyleZhang, L., Niaz, S. I., Khan, D., Wang, Z., Zhu, Y., Zhou, H., Lin, Y., Li, J., & Liu, L. (2017). Induction of Diverse Bioactive Secondary Metabolites from the Mangrove Endophytic Fungus Trichoderma sp. (Strain 307) by Co-Cultivation with Acinetobacter johnsonii (Strain B2). Marine Drugs, 15(2), 35. https://doi.org/10.3390/md15020035