Marine Sponge Natural Products with Anticancer Potential: An Updated Review

 , ,
, ,  and
and 
Abstract
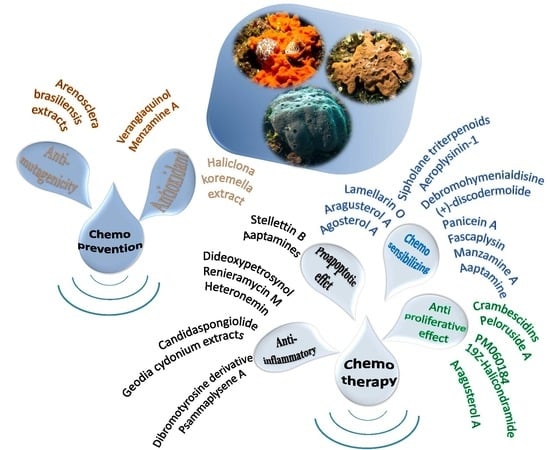
1. Introduction
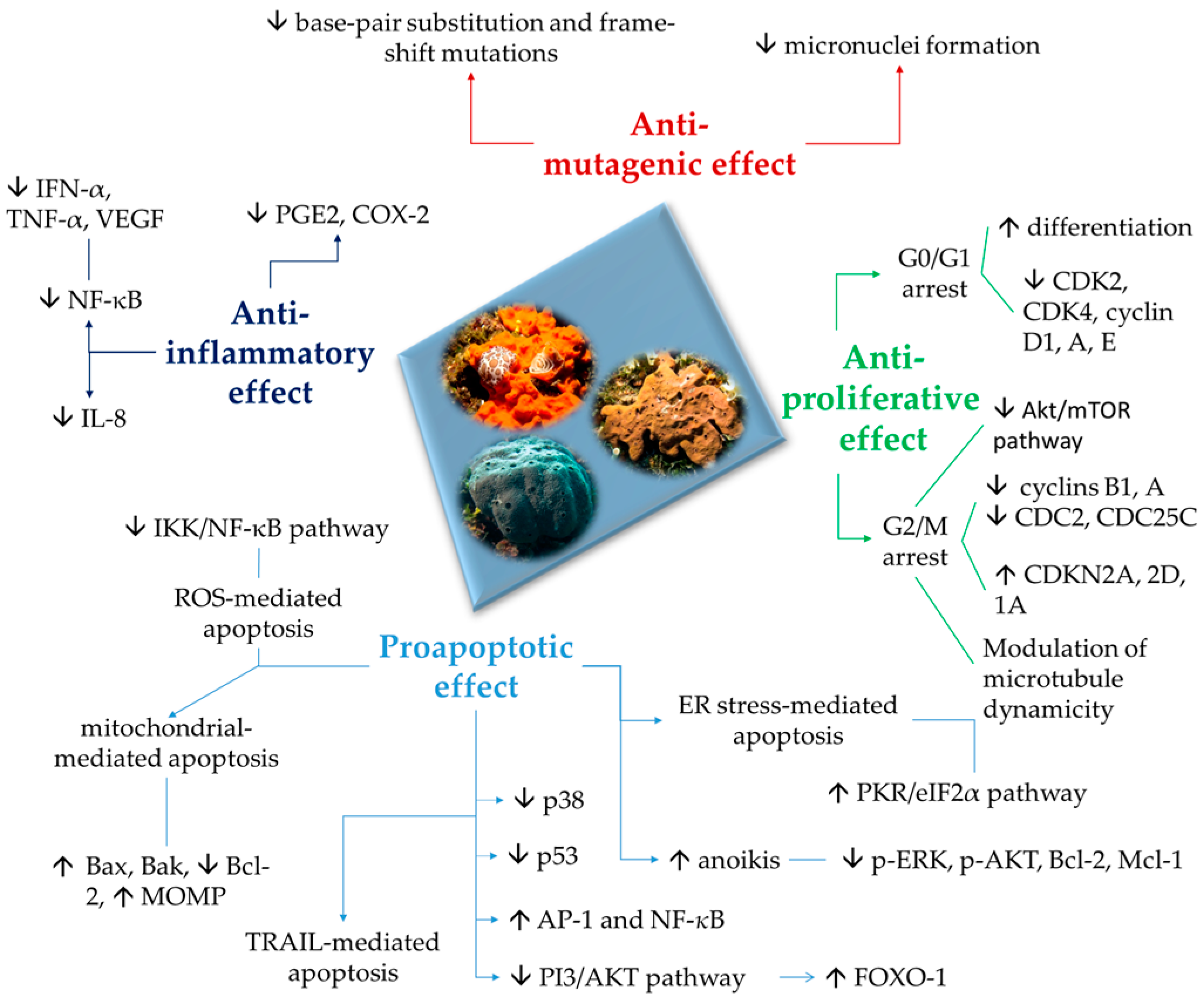
2. Proapoptotic and Anti-Inflammatory Effects
3. Antiproliferative Effects
4. Chemosensitizing Properties
5. Chemoprevention
6. Clinical Studies
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 6 July 2017).
- Amin, A.R.; Kucuk, O.; Khuri, F.R.; Shin, D.M. Perspectives for cancer prevention with natural compounds. J. Clin. Oncol. 2009, 27, 2712–2725. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Lopes, R.M.; Schwartsmann, G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001, 1, 364–369. [Google Scholar] [CrossRef]
- Essack, M.; Bajic, V.B.; Archer, J.A. Recently confirmed apoptosis-inducing lead compounds isolated from marine sponge of potential relevance in cancer treatment. Mar. Drugs 2011, 9, 1580–1606. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Ravindran, J.; Aggarwal, B.B. Nf-κB and cancer: How intimate is this relationship. Mol. Cell. Biochem. 2010, 336, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. Nf-κB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Shubina, L.K.; Kuzmich, A.S.; Bokemeyer, C.; Keller-von Amsberg, G.; Honecker, F. Aaptamines from the marine sponge Aaptos sp. Display anticancer activities in human cancer cell lines and modulate ap-1-, Nf-κB-, and p53-dependent transcriptional activity in mouse jb6 cl41 cells. Biomed. Res. Int. 2014, 2014, 469309. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin a is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs 2015, 33, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Lee, J.Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.M.; Grandjenette, C.; Kim, J.G.; Yoon, A.Y.; et al. Discovery and characterization of isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to trail in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.; Maher, M.; Temkin, A.; Pitts, T.; Wright, A. Spongiatriol inhibits nuclear factor kappa B activation and induces apoptosis in pancreatic cancer cells. Mar. Drugs 2013, 11, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar] [PubMed]
- De Stefano, D.; Tommonaro, G.; Malik, S.A.; Iodice, C.; De Rosa, S.; Maiuri, M.C.; Carnuccio, R. Cacospongionolide and scalaradial, two marine sesterterpenoids as potent apoptosis-inducing factors in human carcinoma cell lines. PLoS ONE 2012, 7, e33031. [Google Scholar] [CrossRef] [PubMed]
- Trisciuoglio, D.; Uranchimeg, B.; Cardellina, J.H.; Meragelman, T.L.; Matsunaga, S.; Fusetani, N.; Del Bufalo, D.; Shoemaker, R.H.; Melillo, G. Induction of apoptosis in human cancer cells by candidaspongiolide, a novel sponge polyketide. J. Natl. Cancer Inst. 2008, 100, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Umeyama, A.; Matsuoka, N.; Mine, R.; Nakata, A.; Arimoto, E.; Matsui, M.; Shoji, N.; Arihara, S.; Takei, M.; Hashimoto, T. Polyacetylene diols with antiproliferative and driving Th1 polarization effects from the marine sponge Callyspongia sp. J. Nat. Med. 2010, 64, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Roel, M.; Rubiolo, J.A.; Guerra-Varela, J.; Silva, S.B.; Thomas, O.P.; Cabezas-Sainz, P.; Sanchez, L.; Lopez, R.; Botana, L.M. Marine guanidine alkaloids crambescidins inhibit tumor growth and activate intrinsic apoptotic signaling inducing tumor regression in a colorectal carcinoma zebrafish xenograft model. Oncotarget 2016, 7, 83071–83087. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Aoki, S.; Sowa, Y.; Sakai, T.; Kobayashi, M. Smenospongine, a sesquiterpene aminoquinone from a marine sponge, induces g1 arrest or apoptosis in different leukemia cells. Mar. Drugs 2008, 6, 480–488. [Google Scholar] [PubMed]
- Shin, D.Y.; Kim, G.Y.; Kim, N.D.; Jung, J.H.; Kim, S.K.; Kang, H.S.; Choi, Y.H. Induction of apoptosis by pectenotoxin-2 is mediated with the induction of DR4/DR5, EGR-1 and NAG-1, activation of caspases and modulation of the Bcl-2 family in p53-deficient Hep3B hepatocellular carcinoma cells. Oncol. Rep. 2008, 19, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Moon, D.O.; Heo, M.S.; Lee, J.D.; Jung, J.H.; Kim, S.K.; Choi, Y.H.; Kim, G.Y. Pectenotoxin-2 abolishes constitutively activated Nf-κB, leading to suppression of Nf-κB related gene products and potentiation of apoptosis. Cancer Lett. 2008, 271, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Kang, S.H.; Lee, K.J.; Heo, M.S.; Choi, K.S.; Choi, Y.H.; Kim, G.Y. Induction of G2/M arrest, endoreduplication, and apoptosis by actin depolymerization agent pextenotoxin-2 in human leukemia cells, involving activation of erk and jnk. Biochem. Pharmacol. 2008, 76, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Ben-Califa, N.; Bishara, A.; Kashman, Y.; Neumann, D. Salarin c, a member of the salarin superfamily of marine compounds, is a potent inducer of apoptosis. Investig. New Drugs 2012, 30, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Tanturli, M.; Ben-Califa, N.; Gozzini, A.; Tusa, I.; Cheloni, G.; Marzi, I.; Cipolleschi, M.G.; Kashman, Y.; Neumann, D.; et al. Salarin C inhibits the maintenance of chronic myeloid leukemia progenitor cells. Cell Cycle 2015, 14, 3146–3154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Ghosh, A.K.; Pommier, Y. Lasonolide a, a potent and reversible inducer of chromosome condensation. Cell Cycle 2012, 11, 4424–4435. [Google Scholar] [CrossRef] [PubMed]
- Cheung, F.W.; Li, C.; Che, C.T.; Liu, B.P.; Wang, L.; Liu, W.K. Geoditin a induces oxidative stress and apoptosis on human colon HT29 cells. Mar. Drugs 2010, 8, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Ho, J.C.; Che, C.T. Apoptotic activity of isomalabaricane triterpenes on human promyelocytic leukemia hl60 cells. Cancer Lett. 2005, 230, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Cheung, F.W.; Che, C.T. Stellettin a induces oxidative stress and apoptosis in hl-60 human leukemia and lncap prostate cancer cell lines. J. Nat. Prod. 2006, 69, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Do, M.T.; Na, M.; Kim, H.G.; Khanal, T.; Choi, J.H.; Jin, S.W.; Oh, S.H.; Hwang, I.H.; Chung, Y.C.; Kim, H.S.; et al. Ilimaquinone induces death receptor expression and sensitizes human colon cancer cells to trail-induced apoptosis through activation of ros-erk/p38 mapk-chop signaling pathways. Food Chem. Toxicol. 2014, 71, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Chung, K.J.; Hwang, I.H.; Gwak, J.; Park, S.; Ju, B.G.; Yun, E.; Kim, D.E.; Chung, Y.H.; Na, M.; et al. Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells. Mar. Drugs 2015, 13, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.H.; Chueh, S.C.; Kung, F.L.; Pan, S.L.; Shen, Y.C.; Guh, J.H. Ilimaquinone, a marine sponge metabolite, displays anticancer activity via gadd153-mediated pathway. Eur. J. Pharmacol. 2007, 556, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Schneiders, U.M.; Schyschka, L.; Rudy, A.; Vollmar, A.M. Bh3-only proteins mcl-1 and bim as well as endonuclease g are targeted in spongistatin 1-induced apoptosis in breast cancer cells. Mol. Cancer Ther. 2009, 8, 2914–2925. [Google Scholar] [CrossRef] [PubMed]
- Schyschka, L.; Rudy, A.; Jeremias, I.; Barth, N.; Pettit, G.R.; Vollmar, A.M. Spongistatin 1: A new chemosensitizing marine compound that degrades xiap. Leukemia 2008, 22, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Schneiders, U.M.; Wiedmann, R.M.; Ischenko, I.; Bruns, C.J.; Rudy, A.; Zahler, S.; Vollmar, A.M. The marine compound spongistatin 1 targets pancreatic tumor progression and metastasis. Int. J. Cancer 2010, 127, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits tnf alpha-induced NF-κB activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Wang, C.T.; Hung, H.C.; Wu, W.J.; Wu, D.C.; Chang, M.C.; Sung, P.J.; Chou, Y.W.; Wen, Z.H.; Tai, M.H. Heteronemin is a novel c-Met/STAT3 inhibitor against advanced prostate cancer cells. Prostate 2016, 76, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Kuo, S.M.; Wu, Y.J.; Su, J.H. Improvement and enhancement of antibladder carcinoma cell effects of heteronemin by the nanosized hyaluronan aggregation. Int. J. Nanomed. 2016, 11, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Sung, P.J.; Chang, Y.L.; Pan, S.L.; Teng, C.M. Heteronemin, a spongean sesterterpene, induces cell apoptosis and autophagy in human renal carcinoma cells. Biomed. Res. Int. 2015, 2015, 738241. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Cho, S.H.; Ono, M.; Kuwano, T.; Nakao, S.; Kuwano, M.; Nakagawa, S.; Gao, J.Q.; Mayumi, T.; Shibuya, M.; et al. Bastadin 6, a spongean brominated tyrosine derivative, inhibits tumor angiogenesis by inducing selective apoptosis to endothelial cells. Anticancer Drugs 2006, 17, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Nagumo, Y.; Watanabe, T.; Imaizumi, T.; Shibuya, M.; Kataoka, T.; Kanoh, N.; Iwabuchi, Y.; Usui, T. Irciniastatin a induces jnk activation that is involved in caspase-8-dependent apoptosis via the mitochondrial pathway. Toxicol. Lett. 2010, 199, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.Y.; Li, M.; Tang, S.A.; Sun, W.; Xu, B.; Cui, J.R.; Lin, W.H. Induction of apoptosis accompanying with g(1) phase arrest and microtubule disassembly in human hepatoma cells by jaspolide b, a new isomalabaricane-type triterpene. Cancer Lett. 2008, 262, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Q.; Zhang, L.; Zhong, Y.; Fan, G.; Zhang, Z.; Wang, R.; Jin, M.; Qiu, Y.; Kong, D. Stellettin b induces apoptosis in human chronic myeloid leukemia cells via targeting pi3k and stat5. Oncotarget 2017, 8, 28906–28921. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin b induces g1 arrest, apoptosis and autophagy in human non-small cell lung cancer A549 cells via blocking PI3K/Akt/mTOR pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.A.; Zhou, Q.; Guo, W.Z.; Qiu, Y.; Wang, R.; Jin, M.; Zhang, W.; Li, K.; Yamori, T.; Dan, S.; et al. In vitro antitumor activity of stellettin b, a triterpene from marine sponge jaspis stellifera, on human glioblastoma cancer sf295 cells. Mar. Drugs 2014, 12, 4200–4213. [Google Scholar] [CrossRef] [PubMed]
- Salma, Y.; Lafont, E.; Therville, N.; Carpentier, S.; Bonnafe, M.J.; Levade, T.; Genisson, Y.; Andrieu-Abadie, N. The natural marine anhydrophytosphingosine, jaspine b, induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009, 78, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Lee, Y.S.; Lee, S.; Kim, S.; Kim, T.Y. Pachastrissamine from Pachastrissa sp. Inhibits melanoma cell growth by dual inhibition of Cdk2 and erk-mediated foxo3 downregulation. Phytother. Res. 2012, 26, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Chau, V.M.; Tran, T.H.; Phan, V.K.; Hoang, T.H.; Nguyen, T.D.; Nguyen, X.N.; Tai, B.H.; Hyun, J.H.; Kang, H.K.; et al. C29 sterols with a cyclopropane ring at C-25 and 26 from the vietnamese marine sponge ianthella sp. In addition, their anticancer properties. Bioorg. Med. Chem. Lett. 2009, 19, 4584–4588. [Google Scholar] [PubMed]
- Guzman, E.A.; Xu, Q.; Pitts, T.P.; Mitsuhashi, K.O.; Baker, C.; Linley, P.A.; Oestreicher, J.; Tendyke, K.; Winder, P.L.; Suh, E.M.; et al. Leiodermatolide, a novel marine natural product, has potent cytotoxic and antimitotic activity against cancer cells, appears to affect microtubule dynamics, and exhibits antitumor activity. Int. J. Cancer 2016, 139, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, D.V.; Modzelewska, K.; Glazar, A.I.; Gray, P.D.; Kaur, M.; Liu, T.; Grossman, D.; Harper, M.K.; Kuwada, S.K.; Moghal, N.; et al. The marine alkaloid naamidine a promotes caspase-dependent apoptosis in tumor cells. Anticancer Drugs 2009, 20, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine alkaloids from the marine sponge monanchora pulchra show cytotoxic properties and prevent egf-induced neoplastic transformation in vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.A.; West, L.M.; Northcote, P.T.; Berridge, M.V.; Miller, J.H. Induction of apoptosis by the marine sponge (mycale) metabolites, mycalamide a and pateamine. Apoptosis 2001, 6, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kikuchi, S.; Ochiai, T.; Ikoma, H.; Kubota, T.; Ichikawa, D.; Fujiwara, H.; Okamoto, K.; Sakakura, C.; Sonoyama, T.; et al. Latrunculin a has a strong anticancer effect in a peritoneal dissemination model of human gastric cancer in mice. Anticancer Res. 2009, 29, 2091–2097. [Google Scholar] [PubMed]
- Kijjoa, A.; Wattanadilok, R.; Campos, N.; Nascimento, M.S.; Pinto, M.; Herz, W. Anticancer activity evaluation of kuanoniamines a and c isolated from the marine sponge oceanapia sagittaria, collected from the gulf of thailand. Mar. Drugs 2007, 5, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Bae, S.J.; Kim, N.D.; Jung, J.H.; Choi, Y.H. Induction of apoptosis by dideoxypetrosynol a, a polyacetylene from the sponge petrosia sp., in human skin melanoma cells. Int. J. Mol. Med. 2004, 14, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A natural histone deacetylase inhibitor, psammaplin a, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.; Hardt, J.L.; Clardy, J.; Lurain, J.R.; Kim, J.J. Induction of apoptosis in endometrial cancer cells by psammaplysene a involves foxo1. Gynecol. Oncol. 2009, 112, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Chen, Y.C.; El-Shazly, M.; Du, Y.C.; Su, C.W.; Tsao, C.W.; Liu, L.L.; Chou, Y.; Chang, W.B.; Su, Y.D.; et al. Towards the small and the beautiful: A small dibromotyrosine derivative from Pseudoceratina sp. sponge exhibits potent apoptotic effect through targeting IKK/NFκB signaling pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.F.; Zhou, J.M.; Zhang, Y.; Deng, R.; Liu, J.N.; Feng, G.K.; Liu, Z.C.; Xiao, D.J.; Deng, S.Z.; Zhu, X.F. Rhabdastrellic acid-a inhibited pi3k/akt pathway and induced apoptosis in human leukemia hl-60 cells. Cell. Biol. Int. 2008, 32, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Shastina, V.; Park, J.I.; Han, J.Y.; Makarieva, T.; Fedorov, S.; Rasskazov, V.; Stonik, V.; Kwak, J.Y. Differential induction of apoptosis of leukemic cells by rhizochalin, two headed sphingolipids from sponge and its derivatives. Biol. Pharm. Bull. 2009, 32, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Kang, B.S.; Yun, H.J.; Cho, H.G.; Makarieva, T.N.; Choi, H.S. Aglycon of rhizochalin from the rhizochalina incrustata induces apoptosis via activation of amp-activated protein kinase in ht-29 colon cancer cells. Biol. Pharm. Bull. 2011, 34, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Makarieva, T.N.; Guzii, A.G.; Shubina, L.K.; Kwak, J.Y.; Stonik, V.A. Marine two-headed sphingolipid-like compound rhizochalin inhibits egf-induced transformation of jb6 p+ cl41 cells. Lipids 2009, 44, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Otte, K.; Alsdorf, W.H.; Hauschild, J.; Lange, T.; Venz, S.; Bauer, C.K.; Bahring, R.; Amann, K.; Mandanchi, R.; et al. Marine compound rhizochalinin shows high in vitro and in vivo efficacy in castration resistant prostate cancer. Oncotarget 2016, 7, 69703–69717. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Choi, Y.H.; Yee, S.B.; Im, E.; Jung, J.H.; Kim, N.D. Ircinin-1 induces cell cycle arrest and apoptosis in sk-mel-2 human melanoma cells. Mol. Carcinog. 2005, 44, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Lateff, A.; Al-Abd, A.M.; Alahdal, A.M.; Alarif, W.M.; Ayyad, S.E.; Al-Lihaibi, S.S.; Hegazy, M.E.; Al Mohammadi, A.; Abdelghany, T.M.; Abdel-Naim, A.B.; et al. Antiproliferative effects of triterpenoidal derivatives, obtained from the marine sponge Siphonochalina sp., on human hepatic and colorectal cancer cells. Z. Naturforsch. C 2016, 71, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sobahi, T.R.A.; Ayyad, S.N.; Abdel-Lateff, A.; Algandaby, M.M.; Alorfi, H.S.; Abdel-Naim, A.B. Cytotoxic metabolites from callyspongia siphonella display antiproliferative activity by inducing apoptosis in hct-116 cells. Pharmacogn. Mag. 2017, 13, S37–S40. [Google Scholar] [PubMed]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides a and b, chlorinated peptide/polyketide hybrids containing a dolapyrrolidinone unit from the caribbean sponge Smenospongia aurea. Evaluation of their role as leads in antitumor drug research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, G.Y.; Kim, W.I.; Hong, S.H.; Park, D.I.; Kim, N.D.; Bae, S.J.; Jung, J.H.; Choi, Y.H. Induction of apoptosis by (z)-stellettic acid c, an acetylenic acid from the sponge Stelletta sp., is associated with inhibition of telomerase activity in human leukemic u937 cells. Chemotherapy 2007, 53, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Halim, H.; Chunhacha, P.; Suwanborirux, K.; Chanvorachote, P. Anticancer and antimetastatic activities of renieramycin m, a marine tetrahydroisoquinoline alkaloid, in human non-small cell lung cancer cells. Anticancer Res. 2011, 31, 193–201. [Google Scholar] [PubMed]
- Tabunoki, H.; Saito, N.; Suwanborirux, K.; Charupant, K.; Satoh, J. Molecular network profiling of u373mg human glioblastoma cells following induction of apoptosis by novel marine-derived anti-cancer 1,2,3,4-tetrahydroisoquinoline alkaloids. Cancer Cell. Int. 2012, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fda Approves New Therapy for Certain Types of Advanced Soft Tissue Sarcoma. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm468832.htm (accessed on 18 September 2017).
- Charupant, K.; Daikuhara, N.; Saito, E.; Amnuoypol, S.; Suwanborirux, K.; Owa, T.; Saito, N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin m-jorunnamycin a analogues. Bioorg. Med. Chem. 2009, 17, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Tanaka, C.; Koizumi, Y.-I.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of renieramycin m into jorumycin and renieramycin j including oxidative degradation products, mimosamycin, renierone, and renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell. Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin m sensitizes anoikis-resistant h460 lung cancer cells to anoikis. Anticancer Res. 2016, 36, 1665–1671. [Google Scholar] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin m attenuates cancer stem cell-like phenotypes in h460 lung cancer cells. Anticancer Res. 2017, 37, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. Ap-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, A.; Tony Kong, A.N. Anticarcinogenesis by dietary phytochemicals: Cytoprotection by nrf2 in normal cells and cytotoxicity by modulation of transcription factors NF-κB and ap-1 in abnormal cancer cells. Food. Chem. Toxicol. 2008, 46, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA damaging agents induce expression of fas ligand and subsequent apoptosis in t lymphocytes via the activation of NF-κB b and ap-1. Mol. Cell. 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Lu, H.; Huang, H. Foxo1: A potential target for human diseases. Curr. Drug Targets 2011, 12, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase b/akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor fkhr1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Tashiro, H.; Blazes, M.S.; Wu, R.; Cho, K.R.; Bose, S.; Wang, S.I.; Li, J.; Parsons, R.; Ellenson, L.H. Mutations in pten are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997, 57, 3935–3940. [Google Scholar] [PubMed]
- Goto, T.; Takano, M.; Albergaria, A.; Briese, J.; Pomeranz, K.M.; Cloke, B.; Fusi, L.; Feroze-Zaidi, F.; Maywald, N.; Sajin, M.; et al. Mechanism and functional consequences of loss of foxo1 expression in endometrioid endometrial cancer cells. Oncogene 2008, 27, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.C.; Hoekstra, A.V.; Blok, L.J.; Hanifi-Moghaddam, P.; Lurain, J.R.; Singh, D.K.; Buttin, B.M.; Schink, J.C.; Kim, J.J. The regulation and function of the forkhead transcription factor, forkhead box O1, is dependent on the progesterone receptor in endometrial carcinoma. Endocrinology 2008, 149, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Hutti, J.E.; Pfefferle, A.D.; Russell, S.C.; Sircar, M.; Perou, C.M.; Baldwin, A.S. Oncogenic pi3k mutations lead to Nf-κB-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012, 72, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The novel Nf-κB inhibitor dhmeq synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ros-dependent mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Madonna, G.; Ullman, C.D.; Gentilcore, G.; Palmieri, G.; Ascierto, P.A. Nf-κB as potential target in the treatment of melanoma. J. Transl. Med. 2012, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Rasheva, V.I.; Domingos, P.M. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009, 14, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and er-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Bayazit, V. Cytotoxic effects of some animal and vegetable extracts and some chemicals on liver and colon carcinoma and myosarcoma. Saudi Med. J. 2004, 25, 156–163. [Google Scholar] [PubMed]
- Costantini, S.; Romano, G.; Rusolo, F.; Capone, F.; Guerriero, E.; Colonna, G.; Ianora, A.; Ciliberto, G.; Costantini, M. Anti-inflammatory effects of a methanol extract from the marine sponge Geodia cydonium on the human breast cancer MCF-7 cell line. Mediat. Inflamm. 2015, 2015, 204975. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Jung, J.H.; Kim, N.D.; Choi, Y.H. Inhibition of cyclooxygenase-2 and telomerase activities in human leukemia cells by dideoxypetrosynol a, a polyacetylene from the marine sponge Petrosia sp. Int. J. Oncol. 2007, 30, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Gately, S.; Kerbel, R. Therapeutic potential of selective cyclooxygenase-2 inhibitors in the management of tumor angiogenesis. Prog. Exp. Tumor. Res. 2003, 37, 179–192. [Google Scholar] [PubMed]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin endoperoxide h synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Prescott, S.M. Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J. Cell. Physiol. 2002, 190, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.A.; Harmody, D.; Pitts, T.P.; Vera-Diaz, B.; Winder, P.L.; Yu, Y.; Wright, A.E. Inhibition of il-8 secretion on bxpc-3 and mia paca-2 cells and induction of cytotoxicity in pancreatic cancer cells with marine natural products. Anticancer Drugs 2017, 28, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Naeth, I.; Venz, S.; Preukschas, M.; Sievert, H.; Jacobsen, C.; Shubina, L.K.; Gesell Salazar, M.; Scharf, C.; Walther, R.; et al. Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with antiproliferative activity. J. Proteome Res. 2012, 11, 2316–2330. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Zhang, P.P.; Wang, P.Q.; Yu, H.B.; Sun, F.; Hu, W.Z.; Wu, W.H.; Zhang, X.; Chen, F.; Chu, Z.Y.; et al. The cytotoxic and mechanistic effects of aaptamine on hepatocellular carcinoma. Anticancer Agents Med. Chem. 2015, 15, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kong, D.; Suna, H.; Sowa, Y.; Sakai, T.; Setiawan, A.; Kobayashi, M. Aaptamine, a spongean alkaloid, activates p21 promoter in a p53-independent manner. Biochem. Biophys. Res. Commun. 2006, 342, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zhao, W.; Zhang, Y.; Kobayashi, M.; Duan, H.; Kong, D. Antiproliferative effect of aaptamine on human chronic myeloid leukemia k562 cells. Int. J. Mol. Sci. 2011, 12, 7352–7359. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Roth, G.P.; Hoffman, J.K.; Divlianska, D.B.; Pechter, D.; Sennett, S.H.; Guzman, E.A.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; et al. Isolation, synthesis, and biological activity of aphrocallistin, an adenine-substituted bromotyramine metabolite from the hexactinellida sponge Aphrocallistes beatrix. J. Nat. Prod. 2009, 72, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, K.; Yamagishi, T.; Ichihara, T.; Nakaike, S.; Iguchi, K.; Yamada, Y.; Fukumoto, H.; Yoneda, T.; Samata, K.; Ikeya, H.; et al. Mechanism of action of aragusterol a (yta0040), a potent anti-tumor marine steroid targeting the g(1) phase of the cell cycle. Int. J. Cancer 2000, 88, 810–819. [Google Scholar] [CrossRef]
- Guzman, E.A.; Johnson, J.D.; Carrier, M.K.; Meyer, C.I.; Pitts, T.P.; Gunasekera, S.P.; Wright, A.E. Selective cytotoxic activity of the marine-derived batzelline compounds against pancreatic cancer cell lines. Anticancer Drugs 2009, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Edelson, J.R.; Brautigan, D.L. The discodermia calyx toxin calyculin a enhances cyclin d1 phosphorylation and degradation, and arrests cell cycle progression in human breast cancer cells. Toxins 2011, 3, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid differentiation in k562 chronic myelogenous cells induced by crambescidin 800, a pentacyclic guanidine alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Isbrucker, R.A.; Cummins, J.; Pomponi, S.A.; Longley, R.E.; Wright, A.E. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem. Pharmacol. 2003, 66, 75–82. [Google Scholar] [CrossRef]
- Park, C.; Kim, G.Y.; Kim, G.D.; Lee, W.H.; Cheong, J.H.; Kim, N.D.; Bae, S.J.; Jung, J.H.; Choi, Y.H. Suppression of u937 human monocytic leukemia cell growth by dideoxypetrosynol a, a polyacetylene from the sponge Petrosia sp., via induction of cdk inhibitor p16 and down-regulation of prb phosphorylation. Oncol. Rep. 2006, 16, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, E.; Kowalski, R.J.; Hamel, E.; Lin, C.M.; Longley, R.E.; Gunasekera, S.P.; Rosenkranz, H.S.; Day, B.W. Discodermolide, a cytotoxic marine agent that stabilizes microtubules more potently than taxol. Biochemistry 1996, 35, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Honore, S.; Kamath, K.; Braguer, D.; Wilson, L.; Briand, C.; Jordan, M.A. Suppression of microtubule dynamics by discodermolide by a novel mechanism is associated with mitotic arrest and inhibition of tumor cell proliferation. Mol. Cancer Ther. 2003, 2, 1303–1311. [Google Scholar] [PubMed]
- Rangel, M.; Prado, M.P.; Konno, K.; Naoki, H.; Freitas, J.C.; Machado-Santelli, G.M. Cytoskeleton alterations induced by geodia corticostylifera depsipeptides in breast cancer cells. Peptides 2006, 27, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Kim, G.D.; Jeon, J.E.; Shin, J.; Lee, S.K. Anti-proliferative effect of (19Z)-halichondramide, a novel marine macrolide isolated from the sponge Chondrosia corticata, is associated with G2/M cell cycle arrest and suppression of mtor signaling in human lung cancer cells. Toxicol In Vitro 2013, 27, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.J.; Coleman, J.E.; Andersen, R.J.; Roberge, M. Cytotoxic peptides hemiasterlin, hemiasterlin a and hemiasterlin b induce mitotic arrest and abnormal spindle formation. Cancer Chemother. Pharmacol. 1997, 39, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Dalby, S.M.; Roberts, J.C.; Naylor, G.J.; Guzman, E.A.; Isbrucker, R.; Pitts, T.P.; Linley, P.; Divlianska, D.; Reed, J.K.; et al. Leiodermatolide, a potent antimitotic macrolide from the marine sponge leiodermatium sp. Angew. Chem. Int. Ed. Engl. 2011, 50, 3219–3223. [Google Scholar] [CrossRef] [PubMed]
- Mailhol, D.; Willwacher, J.; Kausch-Busies, N.; Rubitski, E.E.; Sobol, Z.; Schuler, M.; Lam, M.H.; Musto, S.; Loganzo, F.; Maderna, A.; et al. Synthesis, molecular editing, and biological assessment of the potent cytotoxin leiodermatolide. J. Am. Chem. Soc. 2014, 136, 15719–15729. [Google Scholar] [CrossRef] [PubMed]
- Sangrajrang, S.; Zidane, M.; Berda, P.; More, M.T.; Calvo, F.; Fellous, A. Different microtubule network alterations induced by pachymatismin, a new marine glycoprotein, on two prostatic cell lines. Cancer Chemother. Pharmacol. 2000, 45, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Zidane, M.; Pondaven, P.; Roussakis, C.; More, M.T. Effects in vitro of pachymatismin, a glycoprotein from the marine sponge Pachymatisma johnstonii, on a non-small-cell bronchopulmonary carcinoma line (nsclc-n6). Anticancer Res. 1996, 16, 2805–2812. [Google Scholar] [PubMed]
- Zidane, M.; More, M.T.; Pondaven, P.; Jaquot, C.; Riou, D.; Roussakis, C. In vivo effect of pachymatismin, a new marine glycoprotein, on a human non-small-cell lung carcinoma. In Vivo 1997, 11, 185–188. [Google Scholar] [PubMed]
- Hood, K.A.; West, L.M.; Rouwe, B.; Northcote, P.T.; Berridge, M.V.; Wakefield, S.J.; Miller, J.H. Peloruside a, a novel antimitotic agent with paclitaxel-like microtubule- stabilizing activity. Cancer Res. 2002, 62, 3356–3360. [Google Scholar] [PubMed]
- Chan, A.; Andreae, P.M.; Northcote, P.T.; Miller, J.H. Peloruside a inhibits microtubule dynamics in a breast cancer cell line MCF7. Investig. New Drugs 2011, 29, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Higuchi, K.; Isozumi, N.; Matsui, K.; Miyamoto, Y.; Itoh, N.; Tanaka, K.; Kobayashi, M. Differentiation in chronic myelogenous leukemia cell k562 by spongean sesterterpene. Biochem. Biophys. Res. Commun. 2001, 282, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ahn, E.Y.; Ryu, S.H.; Kim, D.K.; Park, J.S.; Kang, S.W.; You, S.; Lee, B.J.; Jung, J.H. Mechanism of cell cycle arrest by (8e, 13z, 20z)-strobilinin/(7e, 13z, 20z)-felixinin from a marine sponge Psammocinia sp. Oncol. Rep. 2005, 14, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Pera, B.; Barasoain, I.; Pantazopoulou, A.; Canales, A.; Matesanz, R.; Rodriguez-Salarichs, J.; Garcia-Fernandez, L.F.; Moneo, V.; Jimenez-Barbero, J.; Galmarini, C.M.; et al. New interfacial microtubule inhibitors of marine origin, pm050489/pm060184, with potent antitumor activity and a distinct mechanism. ACS Chem. Biol. 2013, 8, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Diez, M.; Guillen-Navarro, M.J.; Pera, B.; Bouchet, B.P.; Martinez-Leal, J.F.; Barasoain, I.; Cuevas, C.; Andreu, J.M.; Garcia-Fernandez, L.F.; Diaz, J.F.; et al. Pm060184, a new tubulin binding agent with potent antitumor activity including p-glycoprotein over-expressing tumors. Biochem. Pharmacol. 2014, 88, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Angawi, R.F.; Saqer, E.; Abdel-Lateff, A.; Badria, F.A.; Ayyad, S.E. Cytotoxic neviotane triterpene-type from the red sea sponge Siphonochalina siphonella. Pharmacogn. Mag. 2014, 10, S334–341. [Google Scholar] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Smenospongine, a spongean sesquiterpene aminoquinone, induces erythroid differentiation in k562 cells. Anticancer Drugs 2004, 15, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.; Tamrakar, S.; Ludlow, J.W. Protein phosphatase type 1, the product of the retinoblastoma susceptibility gene, and cell cycle control. Front. Biosci. 1998, 3, D1209–1219. [Google Scholar] [PubMed]
- Robinson, J.; Sieff, C.; Delia, D.; Edwards, P.A.; Greaves, M. Expression of cell-surface HLA-DR, HLA-ABC and glycophorin during erythroid differentiation. Nature 1981, 289, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.L.; Raines, M.; Vera, J.; Lacomis, L.; Tempst, P.A.; Golde, D. Identification of crkl as the constitutively phosphorylated 39-kd tyrosine phosphoprotein in chronic myelogenous leukemia cells. Blood 1994, 84, 2912–2918. [Google Scholar] [PubMed]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calo, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Waight, A.B.; Senter, P.D. A previously undescribed tubulin binder. Proc. Natl. Acad. Sci. USA 2014, 111, 13684–13685. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.H.; Singh, A.J.; Northcote, P.T. Microtubule-stabilizing drugs from marine sponges: Focus on peloruside a and zampanolide. Mar. Drugs 2010, 8, 1059–1079. [Google Scholar] [CrossRef] [PubMed]
- Gnanambal, K.M.; Lakshmipathy, S.V. Dictyoceratidan poisons: Defined mark on microtubule-tubulin dynamics. Life Sci. 2016, 148, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.J.; Giannakakou, P.; Gunasekera, S.P.; Longley, R.E.; Day, B.W.; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–622. [Google Scholar] [PubMed]
- Gaitanos, T.N.; Buey, R.M.; Diaz, J.F.; Northcote, P.T.; Teesdale-Spittle, P.; Andreu, J.M.; Miller, J.H. Peloruside a does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004, 64, 5063–5067. [Google Scholar] [CrossRef] [PubMed]
- Northcote, P.T.; Miller, J.H.; Hood, K.A.; West, L.M. Bioactive Compound. U.S. Patent 6,790,862 B, 14 September 2004. [Google Scholar]
- Rowe, M.R. Resistance to Microtubule-Stabilising Agents Following Point Mutation of Human βi-Tubulin. Master’s Thesis, Victoria University of Wellington, Wellington, New Zealand, June 2015. [Google Scholar]
- Meyer, C.J.; Krauth, M.; Wick, M.J.; Shay, J.W.; Gellert, G.; De Brabander, J.K.; Northcote, P.T.; Miller, J.H. Peloruside a inhibits growth of human lung and breast tumor xenografts in an athymic nu/nu mouse model. Mol. Cancer Ther. 2015, 14, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Northcote, P.T.; Miller, J.H. Peloruside a: A lead non-taxoid-site microtubule-stabilizing agent with potential activity against cancer, neurodegeneration, and autoimmune disease. Nat. Prod. Rep. 2016, 33, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Eras, J.; Northcote, P.T.; Cabral, F.; Miller, J.H. Resistance to peloruside a and laulimalide: Functional significance of acquired betai-tubulin mutations at sites important for drug-tubulin binding. Curr. Cancer Drug Targets 2014, 14, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Rawson, P.; Northcote, P.T.; Miller, J.H. Acquired resistance to peloruside a and laulimalide is associated with downregulation of vimentin in human ovarian carcinoma cells. Pharm. Res. 2012, 29, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of atp-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Abolhoda, A.; Wilson, A.E.; Ross, H.; Danenberg, P.V.; Burt, M.; Scotto, K.W. Rapid activation of mdr1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin. Cancer Res. 1999, 5, 3352–3356. [Google Scholar] [PubMed]
- Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Remiao, F. Cellular models and in vitro assays for the screening of modulators of p-gp, mrp1 and bcrp. Molecules 2017, 22, 600. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming abc transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.X.; Sun, Y.H.; He, J.G.; Cao, H.; Jiang, G.Q. Increased activity of chk enhances the radioresistance of MCF-7 breast cancer stem cells. Oncol. Lett. 2015, 10, 3443–3449. [Google Scholar] [CrossRef] [PubMed]
- Martello, L.A.; McDaid, H.M.; Regl, D.L.; Yang, C.P.; Meng, D.; Pettus, T.R.; Kaufman, M.D.; Arimoto, H.; Danishefsky, S.J.; Smith, A.B.; et al. Taxol and discodermolide represent a synergistic drug combination in human carcinoma cell lines. Clin. Cancer Res. 2000, 6, 1978–1987. [Google Scholar] [PubMed]
- Honore, S.; Kamath, K.; Braguer, D.; Horwitz, S.B.; Wilson, L.; Briand, C.; Jordan, M.A. Synergistic suppression of microtubule dynamics by discodermolide and paclitaxel in non-small cell lung carcinoma cells. Cancer Res. 2004, 64, 4957–4964. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.S.; Lopez-Barcons, L.; Freeze, B.S.; Smith, A.B., 3rd; Goldberg, G.L.; Horwitz, S.B.; McDaid, H.M. Potentiation of taxol efficacy and by discodermolide in ovarian carcinoma xenograft-bearing mice. Clin. Cancer Res. 2006, 12, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G. Cytotoxic effects of fascaplysin against small cell lung cancer cell lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, L.; Tribalat, M.A.; Sauvard, L.; Cazareth, J.; Lalli, E.; Broutin, I.; Thomas, O.P.; Mus-Veteau, I. Natural paniceins from mediterranean sponge inhibit the multidrug resistance activity of patched and increase chemotherapy efficiency on melanoma cells. Oncotarget 2015, 6, 22282–22297. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, A.; Bargh, K.; Kelly, C.; Northcote, P.T.; Miller, J.H. Peloruside a synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol. Pharm. 2007, 4, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Xiao, X.; Zhang, Y.K.; Talele, T.T.; Salim, A.A.; Chen, Z.S.; Capon, R.J. Lamellarin o, a pyrrole alkaloid from an australian marine sponge, Ianthella sp., reverses bcrp mediated drug resistance in cancer cells. Mar Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Chen, Z.S.; Higasiyama, K.; Setiawan, A.; Akiyama, S.; Kobayashi, M. Reversing effect of agosterol a, a spongean sterol acetate, on multidrug resistance in human carcinoma cells. Jpn. J. Cancer Res. 2001, 92, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Aoki, S.; Komatsu, M.; Ueda, K.; Sumizawa, T.; Furukawa, T.; Okumura, H.; Ren, X.Q.; Belinsky, M.G.; Lee, K.; et al. Reversal of drug resistance mediated by multidrug resistance protein (mrp) 1 by dual effects of agosterol a on mrp1 function. Int. J. Cancer 2001, 93, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.P.; Khanfar, M.A.; Kuang, Y.; Dai, C.L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse p-glycoprotein (abcb1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Jain, S.; Kim, I.W.; Peng, X.X.; Abraham, I.; Youssef, D.T.; Fu, L.W.; El Sayed, K.; Ambudkar, S.V.; Chen, Z.S. Sipholenol a, a marine-derived sipholane triterpene, potently reverses p-glycoprotein (abcb1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007, 98, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Bidet, M.; Tomico, A.; Martin, P.; Guizouarn, H.; Mollat, P.; Mus-Veteau, I. The hedgehog receptor patched functions in multidrug transport and chemotherapy resistance. Mol. Cancer Res. 2012, 10, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Rifai, Y.; Arai, M.A.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. Terpenoids and a flavonoid glycoside from acacia pennata leaves as hedgehog/gli-mediated transcriptional inhibitors. J. Nat. Prod. 2010, 73, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Segraves, N.L.; Lopez, S.; Johnson, T.A.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla—Fascaplysinopsis sponges and didemnum tunicates. Tetrahedron Lett. 2003, 44, 3471–3475. [Google Scholar] [CrossRef]
- Radwan, M.; Hanora, A.; Khalifa, S.; Abou-El-Ela, S.H. Manzamines: A potential for novel cures. Cell Cycle 2012, 11, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.A.; Johnson, J.D.; Linley, P.A.; Gunasekera, S.E.; Wright, A.E. A novel activity from an old compound: Manzamine a reduces the metastatic potential of aspc-1 pancreatic cancer cells and sensitizes them to trail-induced apoptosis. Investig. New Drugs 2011, 29, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Waissbluth, S.; Peleva, E.; Daniel, S.J. Platinum-induced ototoxicity: A review of prevailing ototoxicity criteria. Eur. Arch. Otorhinolaryngol. 2017, 274, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed]
- Shippee, B.M.; Bates, J.S.; Richards, K.L. The role of screening and monitoring for bleomycin pulmonary toxicity. J. Oncol. Pharm. Pract. 2016, 22, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.D.; Hartmann, R.; Muller, W.E.; de Voogd, N.; Lai, D.; Proksch, P. Aaptamine derivatives from the indonesian sponge Aaptos suberitoides. J. Nat. Prod. 2013, 76, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ebel, R.; Brenzinger, M.; Kunze, A.; Gross, H.J.; Proksch, P. Wound activation of protoxins in marine sponge Aplysina aerophoba. J. Chem. Ecol. 1997, 23, 1451–1462. [Google Scholar] [CrossRef]
- Funk, F.; Kruger, K.; Henninger, C.; Watjen, W.; Proksch, P.; Thomale, J.; Fritz, G. Spongean alkaloids protect rat kidney cells against cisplatin-induced cytotoxicity. Anticancer Drugs 2014, 25, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.S.; Tummala, H.; Chakarov, S.; Zhelev, N.; Lane, D.P. Targeting atm pathway for therapeutic intervention in cancer. Biodiscovery 2012. [Google Scholar] [CrossRef]
- Curman, D.; Cinel, B.; Williams, D.E.; Rundle, N.; Block, W.D.; Goodarzi, A.A.; Hutchins, J.R.; Clarke, P.R.; Zhou, B.B.; Lees-Miller, S.P.; et al. Inhibition of the g2 DNA damage checkpoint and of protein kinases chk1 and chk2 by the marine sponge alkaloid debromohymenialdisine. J. Biol. Chem. 2001, 276, 17914–17919. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Ward, T.H.; Dive, C. Oxaliplatin responses in colorectal cancer cells are modulated by chk2 kinase inhibitors. Br. J. Pharmacol. 2010, 159, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Vergani, V.; Drudis, T.; Borsotti, P.; Pitelli, M.R.; Viale, G.; Giavazzi, R.; Taraboletti, G. The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin. Cancer Res. 1996, 2, 1843–1849. [Google Scholar] [PubMed]
- Shaw, S.J. The structure activity relationship of discodermolide analogues. Mini Rev. Med. Chem. 2008, 8, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Tangutur, A.D.; Kumar, D.; Krishna, K.V.; Kantevari, S. Microtubule targeting agents as cancer chemotherapeutics: An overview of molecular hybrids as stabilising and destabilising agents. Curr. Top. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Lenzi, M.; Hrelia, P. Chemoprevention of cancer by isothiocyanates and anthocyanins: Mechanisms of action and structure-activity relationship. Curr. Med. Chem. 2008, 15, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Aiub, C.; Giannerini, A.; Ferreira, F.; Mazzei, J.; Stankevicins, L.; Lobo-Hajdu, G.; Guimaraes, P.; Hajdu, E.; Felzenszwalb, I. Genotoxic evaluation of extracts from aplysina fulva, a brazilian marine sponge. Mutat. Res. 2006, 611, 34–41. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Bagnasco, M.; Bennicelli, C.; Camoirano, A.; Bojnemirski, A.; Kurelec, B. Biotransformation of genotoxic agents in marine sponges. Mechanisms and modulation. Mutagenesis 1995, 10, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Stankevicins, L.; Aiub, C.; Maria, L.C.; Lobo-Hajdu, G.; Felzenszwalb, I. Genotoxic and antigenotoxic evaluation of extracts from arenosclera brasiliensis, a brazilian marine sponge. Toxicol. In Vitro 2008, 22, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Aqil, F.; Zahin, M.; El Sayed, K.A.; Ahmad, I.; Orabi, K.Y.; Arif, J.M. Antimicrobial, antioxidant, and antimutagenic activities of selected marine natural products and tobacco cembranoids. Drug Chem. Toxicol. 2011, 34, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R. Antimutagens as cancer chemopreventive agents in the diet. Mutat. Res. 1994, 307, 395–410. [Google Scholar] [CrossRef]
- Nogueira, M.E.; Passoni, M.H.; Biso, F.I.; Longo Mdo, C.; Cardoso, C.R.; Santos, L.C.; Varanda, E.A. Investigation of genotoxic and antigenotoxic activities of melampodium divaricatum in salmonella typhimurium. Toxicol. In Vitro 2006, 20, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Steele, V.E.; Kelloff, G.J. Development of cancer chemopreventive drugs based on mechanistic approaches. Mutat. Res. 2005, 591, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Dobias, L.; Cerna, M.; Rossner, P.; Sram, R. Genotoxicity and carcinogenicity of metronidazole. Mutat. Res. 1994, 317, 177–194. [Google Scholar] [CrossRef]
- Hickman, J.A.; Graeser, R.; de Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; van der Kuip, H.; Consortium, I.P. Three-dimensional models of cancer for pharmacology and cancer cell biology: Capturing tumor complexity in vitro/ex vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Drugs.com. Fda Approves Halaven. Available online: https://www.drugs.com/newdrugs/fda-approves-halaven-late-stage-breast-cancer-2406.html (accessed on 20 July 2017).
- Ro, J.; Cheng, F.T.; Sriuranpong, V.; Villalon, A.; Smruti, B.K.; Tsang, J.; Yap, Y.S.; Asian Working Group for Eribulin Clinical. Patient management with eribulin in metastatic breast cancer: A clinical practice guide. J. Breast Cancer 2016, 19, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Partridge, A.H.; Rumble, R.B.; Carey, L.A.; Come, S.E.; Davidson, N.E.; Di Leo, A.; Gralow, J.; Hortobagyi, G.N.; Moy, B.; Yee, D.; et al. Chemotherapy and targeted therapy for women with human epidermal growth factor receptor 2-negative (or unknown) advanced breast cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2014, 32, 3307–3329. [Google Scholar] [CrossRef] [PubMed]
- Towle, M.J.; Salvato, K.A.; Wels, B.F.; Aalfs, K.K.; Zheng, W.; Seletsky, B.M.; Zhu, X.; Lewis, B.M.; Kishi, Y.; Yu, M.J.; et al. Eribulin induces irreversible mitotic blockade: Implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions. Cancer Res. 2011, 71, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin b. Cancer Res. 2001, 61, 1013–1021. [Google Scholar] [PubMed]
- Kanthou, C.; Tozer, G.M. Microtubule depolymerizing vascular disrupting agents: Novel therapeutic agents for oncology and other pathologies. Int. J. Exp. Pathol. 2009, 90, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, S.I.; Kawano, S.; Taylor, N.; Oestreicher, J.; Matsui, J.; Chow, J.; Oda, Y.; Funahashi, Y. Eribulin mesylate exerts specific gene expression changes in pericytes and shortens pericyte-driven capillary network in vitro. Vasc. Cell 2014, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Dybdal-Hargreaves, N.F.; Risinger, A.L.; Mooberry, S.L. Eribulin mesylate: Mechanism of action of a unique microtubule-targeting agent. Clin. Cancer Res. 2015, 21, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Steinestel, K.; Eder, S.; Schrader, A.J.; Steinestel, J. Clinical significance of epithelial-mesenchymal transition. Clin. Transl. Med. 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (emt) to mesenchymal-epithelial transition (met) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Billups, C.A.; Desjardins, C.; et al. Initial testing (stage 1) of eribulin, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2013, 60, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Towle, M.J.; Nomoto, K.; Asano, M.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Broad spectrum preclinical antitumor activity of eribulin (halaven(r)): Optimal effectiveness under intermittent dosing conditions. Anticancer Res. 2012, 32, 1611–1619. [Google Scholar] [PubMed]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Dieras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (embrace): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Kaufman, P.A.; Awada, A.; Twelves, C.; Yelle, L.; Perez, E.A.; Velikova, G.; Olivo, M.S.; He, Y.; Dutcus, C.E.; Cortes, J. Phase iii open-label randomized study of eribulin mesylate versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 2015, 33, 594–601. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fda Approves First Drug to Show Survival Benefit in Liposarcoma. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm483714.htm (accessed on 17 August 2017).
- Schoffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=breast+cancer&term=eribulin+mesylate&cntry1=&state1=&Search=Search (accessed on 20 July 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?term=eribulin+mesylate&type=&rslt=&age_v=&gndr=&cond=Cancer&intr=&titles=combination&outc=&spons=&lead=&id=&cntry1=&state1=&cntry2=&state2=&cntry3=&state3=&locn=&rcv_s=&rcv_e=&lup_s=&lup_e= (accessed on 23 July 2017).
- Talpir, R.; Benayahu, Y.; Kashman, Y.; Pannell, L.; Schleyer, M. Hemiasterlin and geodiamolide ta; two new cytotoxic peptides from the marine sponge Hemiasterella minor (kirkpatrick). Tetrahedron Lett. 1994, 35, 4453–4456. [Google Scholar] [CrossRef]
- Kuznetsov, G.; TenDyke, K.; Towle, M.J.; Cheng, H.; Liu, J.; Marsh, J.P.; Schiller, S.E.; Spyvee, M.R.; Yang, H.; Seletsky, B.M.; et al. Tubulin-based antimitotic mechanism of e7974, a novel analogue of the marine sponge natural product hemiasterlin. Mol. Cancer Ther. 2009, 8, 2852–2860. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, G.; Towle, M.J.; Liu, J.; Cheng, H.; Tendyke, K.; Kowalczyk, J.J.; Campagna, S.; Littlefield, B.A. Tubulin-based antimitotic mechanism of novel hemiasterlin analog E7974. In Proceedings of the 96th Annual Meeting 2005; AACR: Anaheim, CA, USA, 2005; Volume 65, p. 810. [Google Scholar]
- Rocha-Lima, C.M.; Bayraktar, S.; Macintyre, J.; Raez, L.; Flores, A.M.; Ferrell, A.; Rubin, E.H.; Poplin, E.A.; Tan, A.R.; Lucarelli, A.; et al. A phase 1 trial of e7974 administered on day 1 of a 21-day cycle in patients with advanced solid tumors. Cancer 2012, 118, 4262–4270. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Boni, V.; Tolcher, A.; Smith, L.; Cubillo, A.; Rasco, D.; Calvo, E.; Amaya, A.; Ordoñez, E.; Patnaik, A.; et al. 361 phase i, open-label, dose-escalating clinical and pharmacokinetic study of the novel antimicrotubulin agent pm060184 administered over 10 minutes on days 1–3 and 15–17 every 28 days to patients with advanced malignant solid tumors. Eur. J. Cancer 2015, 51, S74. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry. Available online: https://www.fda.gov/downloads/Drugs/Guidances/ucm085389.pdf (accessed on 25 July 2017).
- Cavalcanti, B.C.; Sombra, C.M.; de Oliveira, J.H.; Berlinck, R.G.; de Moraes, M.O.; Pessoa, C. Cytotoxicity and genotoxicity of ingenamine g isolated from the brazilian marine sponge Pachychalina alcaloidifera. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2008, 147, 409–415. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compounds | Sponge | Cell Line | Concentration Range (μM a) | Chromatin Condensation/DNA Fragmentation | Phosphatidylserin Externalization | Caspase Activation | PARP Cleavage | Reference |
|---|---|---|---|---|---|---|---|---|
| Aaptamine | Aaptos sp. | THP-1 | 50–200 | √ | [14] | |||
| Demethyl(oxy)aaptamine | Aaptos sp. | THP-1 | 10–25 | √ | [14] | |||
| Isoaaptamine | Aaptos sp. | THP-1 | 10–25 | √ | [14] | |||
| Microsclerodermin A | Amphibleptula sp. | AsPC-1 | 2.4 | √ | 3 and 7 | [15] | ||
| BxPC-3 | 2.4 | √ | 3 and 7 | |||||
| PANC-1 | 2.4 | √ | 3 and 7 | |||||
| Isofistularin-3 | Aplysina aerophoba | Raji | 50 | √ | 3 and 7 | √ | [16] | |
| U937 | 50 | √ | 3 and 7 | √ | ||||
| Spongiatriol | Australian spongia sp. | AsPC-1 | 6.8 | √ | 3 and 7 | [17] | ||
| PANC-1 | 6.8 | √ | 3 and 7 | |||||
| MIA PaCa-2 | 6.8 | 3 and 7 | ||||||
| BxPC-3 | 6.8 | 3 and 7 | ||||||
| Laulimalide | Cacospongia mycofijiensis | MDA-MB-435 | 0.1 | 3 | √ | [18] | ||
| Scalaradial | Cacospongia scalaris | HeLa | 10 μg/mL | 3 | [19] | |||
| T47D | 10 μg/mL | √ | ||||||
| Candidaspongiolide | Candidaspongia sp. | U251 HCT116 | 0.05–0.10 | √ | 3 and 12 | √ | [20] | |
| Callyspongidiol | Callyspongia sp. | HL-60 | 31.0–77.5 | √ | [21] | |||
| Crambescidin 800 | Crambe crambe | HepG2 | 0.5–2.5 | √ | 3 | [22] | ||
| Crambescidin 816 | Crambe crambe | HepG2 | 0.5–2.5 | √ | 3 | [22] | ||
| Crambescidin 830 | Crambe crambe | HepG2 | 0.5–2.5 | √ | 3 | [22] | ||
| Smenospongine | Dactylospongia elegans | U937 | 5–15 | √ | [23] | |||
| HL-60 | ||||||||
| Pectenotoxin-2 | Dinophysis fortii and Dinophysis acuminata | Hep3B | 0.01 μg/mL | √ | 3, 8 and 9 | [24] | ||
| U937 | 0.008–0.010 μg/mL | √ | √ | 3 | √ | [25,26] | ||
| Salarin C | Fascaplysinopsis sp. | K562 normoxic and hypoxic conditions | 0.01–0.2 | √ | √ | 3 and 9 | √ | [27,28] |
| Cacospongionolide | Fasciospongia cavernosa | Hela | 10 μg/mL | 3 | [19] | |||
| T47D | 10 μg/mL | √ | ||||||
| Lasonolide A | Forcepia sp. | CA46, Ramos, Daudi, HL-60, MDA-MD-231, MCF-7, HCT-116, HT-29 | 0.1 | √ | [29] | |||
| Geoditin A | Geodia japonica | HL-60 | 1.6 to 25 μg/mL | √ | √ | 3 | [30,31] | |
| HT-29 | 5–30 | √ | ||||||
| Stellettin A | Geodia japonica | HL-60 | 4 μg/mL | 3 | [32] | |||
| Ilimaquinone | Hippospongia metachromia | HCT116 | 2.5–10 | √ | √ | 3 and 8 | √ | [33,34] |
| PC-3 | 2–10 | √ | [35] | |||||
| Spongistatin 1 | Spirastrella spinispirulifera, Hyrtios erecta | MCF-7 | 0.0002–0.0005 | √ | slight activation of 2, 3, 6, 7, 8 and 9 | [36] | ||
| Jurkat | 0.0002 | √ | 2, 3, 7, 8 and 9 | √ | [37] | |||
| L3.6pl | 0.00001–0.01 | √ | [38] | |||||
| Heteronemin | Hyrtios sp. | K562 | 1.4–5.6 | √ | √ | 3, 8 and 9 | √ | [39] |
| DU145 | 0.01–1 μg/mL | √ | 3, 8 and 9 | [40] | ||||
| PC-3 | 0.01–1 μg/mL | √ | 3, 8 and 9 | |||||
| LNCaP | 0.01 μg/mL | √ | ||||||
| T24 | 0.1–0.8 μg/mL | √ | √ | 3 and 9 | √ | [41] | ||
| A498 | 0.5–3 | √ | 3, 8 and 9 | √ | [42] | |||
| Bastadin 6 | lanthella sp. | HUVEC | 0.01–1 | √ | 3 and 7 | [43] | ||
| Irciniastatin A | Ircinia ramose, Psammocinia sp. | Jurkat | 0.01 | √ | 3, 8 and 9 | [44] | ||
| Jaspolide B | Jaspis sp. | Bel-7402 HepG2 | 0.5 10–20 | √ √ | [45] | |||
| Stellettin B | Jaspis stellifera | K562 | 0.012–0.054 | √ | 3 and 9 | √ | [46] | |
| A549 | 0.02–1 | √ | 3 and 7 | √ | [47] | |||
| SF295 | 0.04–1 | √ | √ | [48] | ||||
| Jaspine B or Pachastrissamine | Jaspis sp. Pachastrissa sp. | B16 HaCaT | 5 5 μg/mL | √ | √ | 3 and 9 3 | √ | [49,50] |
| Petrosterol-3,6-dione | Lanthella sp. | HL-60 | 19.9 | √ | [51] | |||
| 5α,6α-epoxy-petrosterol | Lanthella sp. | HL-60 | 21.3 | √ | [51] | |||
| petrosterol | Lanthella sp. | HL-60 | 21.5 | √ | [51] | |||
| Leiodermatolide | Leiodermatium sp. | AsPC-1 | 0.01 | √ | [52] | |||
| BxPC-3 | 0.01 | √ | 3 | |||||
| MIA PaCa-2 | 0.01 | √ | 3 | |||||
| Naamidine A | Leucetta chagosensis | √ | 3, 8 and 9 | √ | [53] | |||
| Monanchocidin A | Monanchora pulchra | HeLa | 1.39 2.01 | √ | √ | 3 and 7 | [54] | |
| Monanchocidin B | Monanchora pulchra | HeLa | 0.58 1.36 | √ | 3 and 7 | [54] | ||
| Monanchocidin C | Monanchora pulchra | HeLa | 1.84 1.31 | √ | 3 and 7 | [54] | ||
| Ptilomycalin A | Monanchora pulchra | HeLa | 1.1 0.5 | √ | 3 and 7 | [54] | ||
| Monanchomycalin B | Monanchora pulchra | HeLa | 1.5 1.72 | √ | 3 and 7 | [54] | ||
| Normonanchocidin D | Monanchora pulchra | HeLa | 2.1 5.2 | √ | 3 and 7 | [54] | ||
| Urupocidin A | Monanchora pulchra | HeLa | 28.7 27 | √ | [54] | |||
| Pulchranin A | Monanchora pulchra | HeLa | 51 58 | √ | 3 and 7 | [54] | ||
| Pateamine | Mycale sp. | 32D | 0.1 | √ | [55] | |||
| Mycalamide A | Mycale sp. | 32D | 0.1 | √ | [55] | |||
| Latrunculin A | Negombata magnifica | MKN45 NUGC-4 | −10 0.01–10 | 3 and 7 | [56] | |||
| Kuanoniamines A | Oceanapia sagittaria | MCF-7 | 0.5–2.5 | √ | [57] | |||
| Kuanoniamines C | Oceanapia sagittaria | MCF-7 | 1.0–2.5 | √ | [57] | |||
| Dideoxypetrosynol A | Petrosia sp. | SK-MEL-2 | 0.1–0.3 μg/mL | √ | 3 and 9 | √ | [58] | |
| Psammaplin A | Psammaplysilla sp. | Human endometrial Ishikawa | √ | [59] | ||||
| Psammaplysene A | Psammaplysilla | Ishikawa ECC1 | 1 1 | √ | [60] | |||
| (1′R,5′S,6′S)-2-(3′,5′-dibromo-1′,6′-dihydroxy-4′-oxocyclohex-2′-enyl) acetonitrile | Pseudoceratina sp. | K562 | 7.7–30.8 | √ | 3 and 9 | √ | [61] | |
| 13E,17E-globostellatic acid X methyl ester | Rhabdastrella globostellata | HUVEC | 1–10 | 3 and 7 | [51] | |||
| Rhabdastrellic acid-A | Rhabdastrella globostellata | HL-60 | √ | 3 | √ | [62] | ||
| Rhizochalin or Rhizocalinin | Rhizochalina incrustata | HL-60 | 10–25 | √ | 3, 8 and 9 | [63] | ||
| HT-29 | 1–6 | √ | √ | 3 | √ | [64] | ||
| THP-1 | 1–10 | √ | √ | [65] | ||||
| PC-3 | 0.5–4 | √ | 8 | √ | [66] | |||
| DU-145 | 0.5–4 | √ | 8 | √ | ||||
| 22Rv1 | 0.5–4 | √ | 8 | √ | ||||
| VCaP | 0.5–4 | √ | 8 | √ | ||||
| Ircinin-1 | Sarcotragus | SK-MEL-2 | 25–50 | √ | 3 and 9 | √ | [67] | |
| Sipholenol A | Siphonochalina sp. | HepG2 | 17.18 | √ | 3 | [68] | ||
| HCT-116 | 14.8 | √ | 3 | [69] | ||||
| Sipholenol L | Siphonochalina sp. | HepG2 | 24 | √ | 3 | [68] | ||
| HCT-116 | 19.8 | √ | 3 | [69] | ||||
| Smenamides A and B | Smenospongia aurea | Calu-1 | 0.05–0.1 | √ | [70] | |||
| (Z)-stellettic acid C | Stelletta sp. | U937 | 17.2–103.3 | √ | 3, 8 and 9 | [71] | ||
| Renieramycin M | Xestospongia sp. | H460 | 5–40 | √ | [72,73] | |||
| U373MG | 0.0031 | 3 | √ |
| Compounds | Sponges | Cell Lines | Concentrations (μM a) | Phase of Cell-Cycle Arrest | Molecular Targets | Reference |
|---|---|---|---|---|---|---|
| Aaptamine | Aaptos aaptos | NT2 | 1–50 | G2/M | / | [106] |
| HepG2 | 50–100 | G2/M | ↓ cyclins D and E, CDK2 ↑ p21 | [107] | ||
| HCC-LM3 | S | |||||
| Aaptos suberitoides | MG63 | 30 μg/mL | G2/M | ↑ p21 | [108] | |
| K562 | 20–100 | G2/M | ↑ p21 | [109] | ||
| Aphrocallistin | Aphrocallistes beatrix beatrix | Panc-1 | ≤46.5 | G0/G1 | / | [110] |
| Aragusterol A | Xestospongia sp. | A549 | 1–10 | Late G1 | ↓ CDK2, CDK4 ↓ cyclins D1, A, E ↓ pRb | [111] |
| Batzelline A and B Isobatzelline A, C, D Secobatzelline A and B | Batzella sp. | AsPC-1 | 5 or 25 μg/mL | S | Intercalate into DNA and/or inhibit Topoisomerase II activity | [112] |
| Isobatzelline E | G2/M | |||||
| Calyculin A | Discodermia calyx | MDA-MB-468 MCF-7 MDA-MB-231 | 0.01 | G0/G1 | ↓ cyclin D1 | [113] |
| Crambescidin 800 | Monanchora ungiculata | K562 | 0.15–1.5 | S | ↑ p21 | [114] |
| Crambescidin 800, 816 and 830 | Crambe crambe | HepG2 | 2.5 | G0/G1 | ↓ cyclins A, D ↓ CDK2, 6, 1 ↑ CDKN2A, 2D, 1A | [22] |
| Dictyostatin-1 | Corallistidae sp. | A549 | 0.01–1 | G2/M | ↑ micronuclei, asters and abnormal mitotic spindles formation | [115] |
| Dideoxypetrosynol A | Petrosia sp. | U937 | 0.2–1 μg/mL | G0/G1 | ↑ cyclin D1 ↓ cyclin E ↑ pRB-E2F1 complex and p16 | [116] |
| (+)-Discodermolide | Discodermia dissoluta | MCF-7, CA46 | 0.01–1 | G2/M | Stabilize microtubules | [117] |
| A549 | 0.07–0.166 | G2/M | abnormal mitotic spindles ↓ microtubules dynamicity | [118] | ||
| Geodiamolide A, B, H and I | Geodia corticostylifera | T47D, MCF7 | 50 ng/mL | Not investigated | ↑ disorganization of actin filaments | [119] |
| (19Z)-Halichondramide | Chondrosia corticata | A549 | 0.025–0.1 | G2/M | ↑ p53, GADD45 ↓ CDC2, CDC25C, cyclin B1, cyclin A | [120] |
| Hemiasterlin, Hemiasterlin A and B | Hemiasterella minor | MCF-7 | 0.0005–0.01 | G2/M | ↑ abnormal mitotic spindles formation | [121] |
| Jaspolide B | Jaspis sp. | Bel-7402 HepG2 | 20 | G0/G1 | ↑ microtubule disassembly | [45] |
| Laulimalide | Cacospongia mycofijiensis | MDA-MB-435 | 0.02 | G2/M | Microtubule stabilization | [18] |
| A-10 SK-OV-3 | 0.02–2 | ↑ micronuclei and abnormal mitotic spindles formation | ||||
| Leiodermatolide | Leiodermatium sp. | PANC-1 | 0.01–0.1 | G2/M | ↓ mitotic spindles formation and microtubule elongation | [52] |
| A549 | 0.01–1 | G2/M | ↓ mitotic spindles formation | [122] | ||
| U2OS | 0.018–0.23 | G2/M | Tubulin disruption Centrosome amplification Micronuclei formation | [123] | ||
| Pachymatismin | Pachymatisma johnstonii | DU145 | 4–16 | Microtubules depolymerization | [124] | |
| NSCLC-N6 | 2–20 μg/mL | G0/G1 | [125] | |||
| NSCLC-N6 subcutaneous xenografts | 0.5–5 mg/kg | ↓ tumor growth | [126] | |||
| Peloruside A | Mycale hentscheli | H441 | 0.01–1 | G2/M | Microtubule stabilization, ↑ micronuclei and abnormal mitotic spindles formation | [127] |
| MCF-7 | 0.025–0.1 | G2/M | ↓ microtubule dynamicity (growth rate, growth length, time spent growing) | [128] | ||
| PHC-1 | Phyllospongia chondrodes | K562 | 0.1–5 μg/mL | G0/G1 | ↑ haemoglobin, glycophorin A and enucleation | [129] |
| PM050489, PM060184 | Lithoplocamia lithistoides | A549 | 0.25–1 × 10−3 | G2/M [130] | ↓ microtubules formation binding αβ tubulin dimers | [131] |
| PM060184 | 0.001 | ↑ abnormal mitotic spindles formation, ↓ CDK1, ↑ p21 | [132] | |||
| HCT116 | 0.01 | ↑ formation of multinucleated cells | ||||
| MDA-MB-231 subcutaneous xenografts | 16 mg/kg | |||||
| Sipholenol-A | Siphonochalina siphonella | PC-3 | 7.9 | G0/G1 | / | [133] |
| Smenospongine | Dactylospongia elegans | K562 | 3–15 | G0/G1 | ↑ p21, ↓ p57, ↓ pRb; ↑ haemoglobin, glycophorin A | [23,134] |
| (8E,13Z,20Z)-strobilinin/(7E,13Z,20Z)-felixinin 1:1 | Psammocinia sp. | HeLa | 10–50 | S | ↓ topoisomerase I and polymerase alpha-primase activities | [130] |
| Drug Associations | Sponge | Cell Line | Concentrations (μM a) | CI | Biological Effect | Reference |
|---|---|---|---|---|---|---|
| Debromohymenialdisine (DBH) + Radiotherapy | Stylissa flabeliformis | MCF-7 | 3 (DBH) + 2–5 Gy | ↓ pChk1/2, survival rate and cancer stem cell subpopulation | [156] | |
| (+)-Discodermolide (D) + Taxol (T) | Discodermia dissoluta | A549 | 0.1–5 (T) + 0.5–25 (D) (1:5 molar ratio) | 0.396 b | ↑ antiproliferative effect and aneuploidy | [157] |
| MCF-7 | 0.273 b | |||||
| SKOV-3 | 0.476 b | |||||
| (+)-Discodermolide + Paclitaxel (PT) | A549 | 0.07 (D) + 0.02 (PT) | 0.59 ± 0.04 | Microtubules stabilization G2/M arrest apoptosis | [158] | |
| (+)-Discodermolide + Taxol | SKOV-3 | 0.001 (D) + 0.02 (T) or 0.02 (D) + 0.001 (T) | ≤0.7 | ↑ antiproliferative effect and aneuploidy | [159] | |
| SKOV-3 xenograft-bearing athymic (nu/nu) female mice | 5 mg/kg (D) + 20 mg/kg (T) | ↓ tumor volume and vascularization | ||||
| Fascaplysin (F) + Camptothecin (C) 10-hydroxycamptothecin (HC) | Fascaplysinopsis Bergquist sp. | NCI-H417 | 0.5 (F) + 0.5 (C) | 0.53 | [160] | |
| 1 (F) + 2 (HC) | 0.82 | |||||
| Panicein A (PA) + Doxorubicin (Doxo) | Haliclona (Soestella) mucosa | MEWO | 10 (PA) + 2 (Doxo) | ↓ IC50 and Doxorubicin cellular efflux ↑ apoptosis | [161] | |
| A375 | 25 (PA) + 1.5 (Doxo) | |||||
| Peloruside A (P) + Paclitaxel (PT) | Mycale hentscheli | 1A9 | 0.005–0.02 (P)+ 0.005–0.015 (PT) | 0.48–0.96 | ↑ G2/M arrest | [162] |
| HL-60 | 0.015–0.03 (P) + 0.02–0.04 (PT) | 0.16–0.87 | ||||
| Peloruside A (P) + Epothilone A (E) | 1A9 | 0.005–0.025 (P) + 0.005–0.01 (E) | 0.41–0.96 | ↑ G2/M arrest | ||
| HL-60 | 0.02–0.125 (P) + 0.01–0.02 (E) | 0.08–1.04 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine Sponge Natural Products with Anticancer Potential: An Updated Review. Mar. Drugs 2017, 15, 310. https://doi.org/10.3390/md15100310
Calcabrini C, Catanzaro E, Bishayee A, Turrini E, Fimognari C. Marine Sponge Natural Products with Anticancer Potential: An Updated Review. Marine Drugs. 2017; 15(10):310. https://doi.org/10.3390/md15100310
Chicago/Turabian StyleCalcabrini, Cinzia, Elena Catanzaro, Anupam Bishayee, Eleonora Turrini, and Carmela Fimognari. 2017. "Marine Sponge Natural Products with Anticancer Potential: An Updated Review" Marine Drugs 15, no. 10: 310. https://doi.org/10.3390/md15100310
APA StyleCalcabrini, C., Catanzaro, E., Bishayee, A., Turrini, E., & Fimognari, C. (2017). Marine Sponge Natural Products with Anticancer Potential: An Updated Review. Marine Drugs, 15(10), 310. https://doi.org/10.3390/md15100310