Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer


Abstract
1. Introduction
2. Results and Discussion
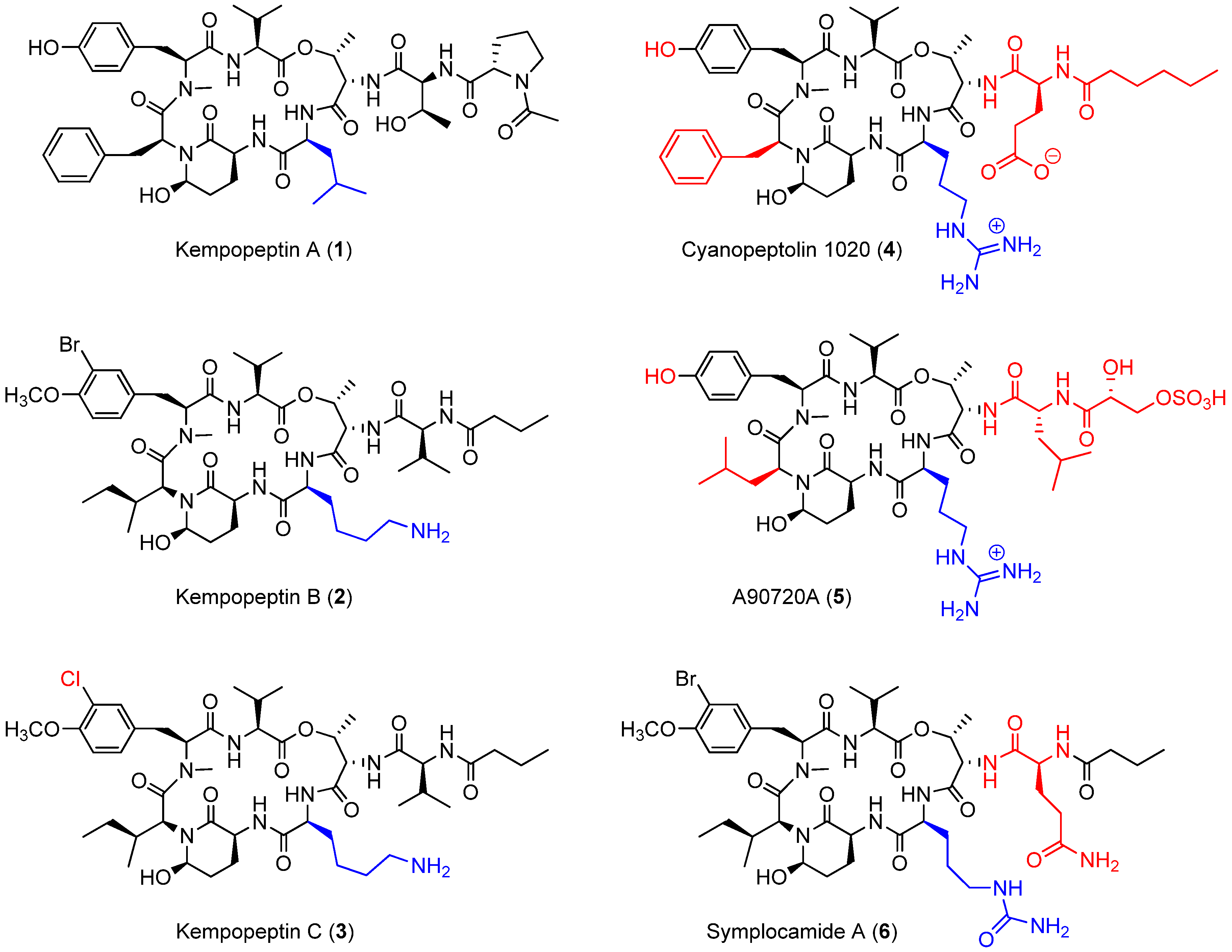
2.1. Isolation and Structure Elucidation
2.2. Enzyme Inhibition Assays
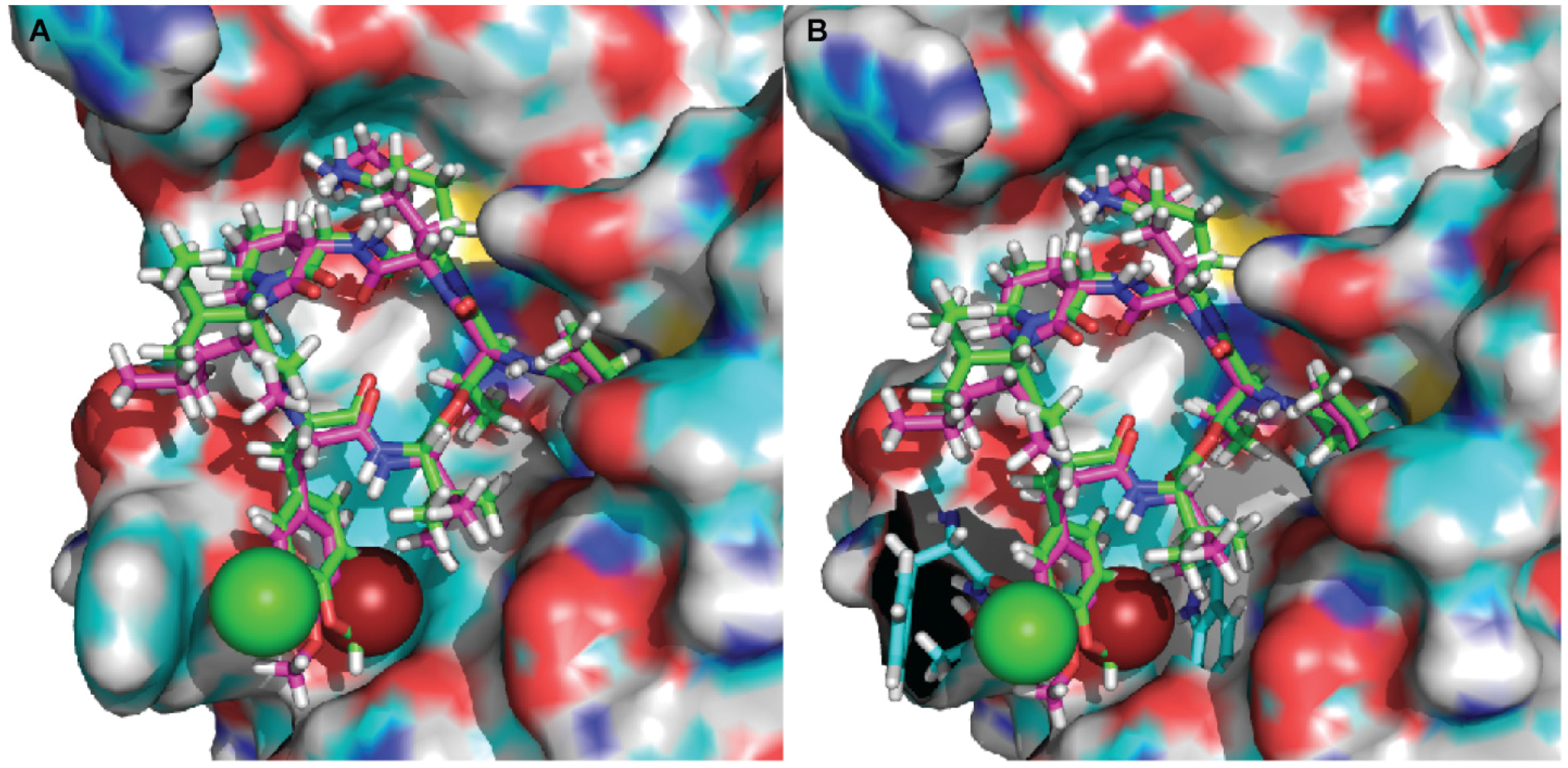
2.3. Molecular Docking
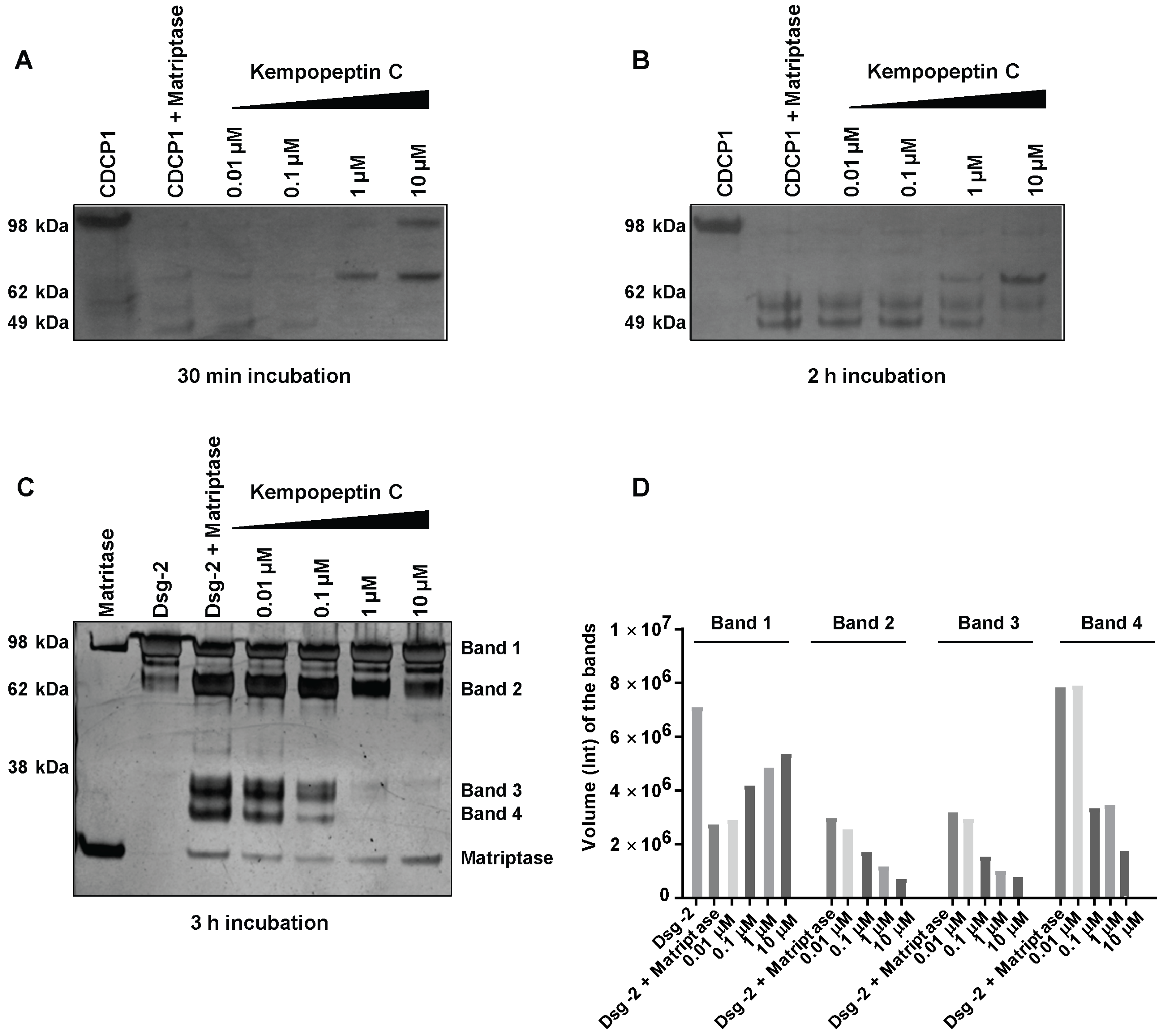
2.4. In Vitro Cleavage of CDCP1 by Matriptase
2.5. In Vitro Cleavage of Desmoglein 2 by Matriptase
2.6. Level of Dsg-2 and CDCP1 in Breast Cancer Cell Lines
2.7. Effects of Kempopeptins B and C on the Viability of MDA-MB-231
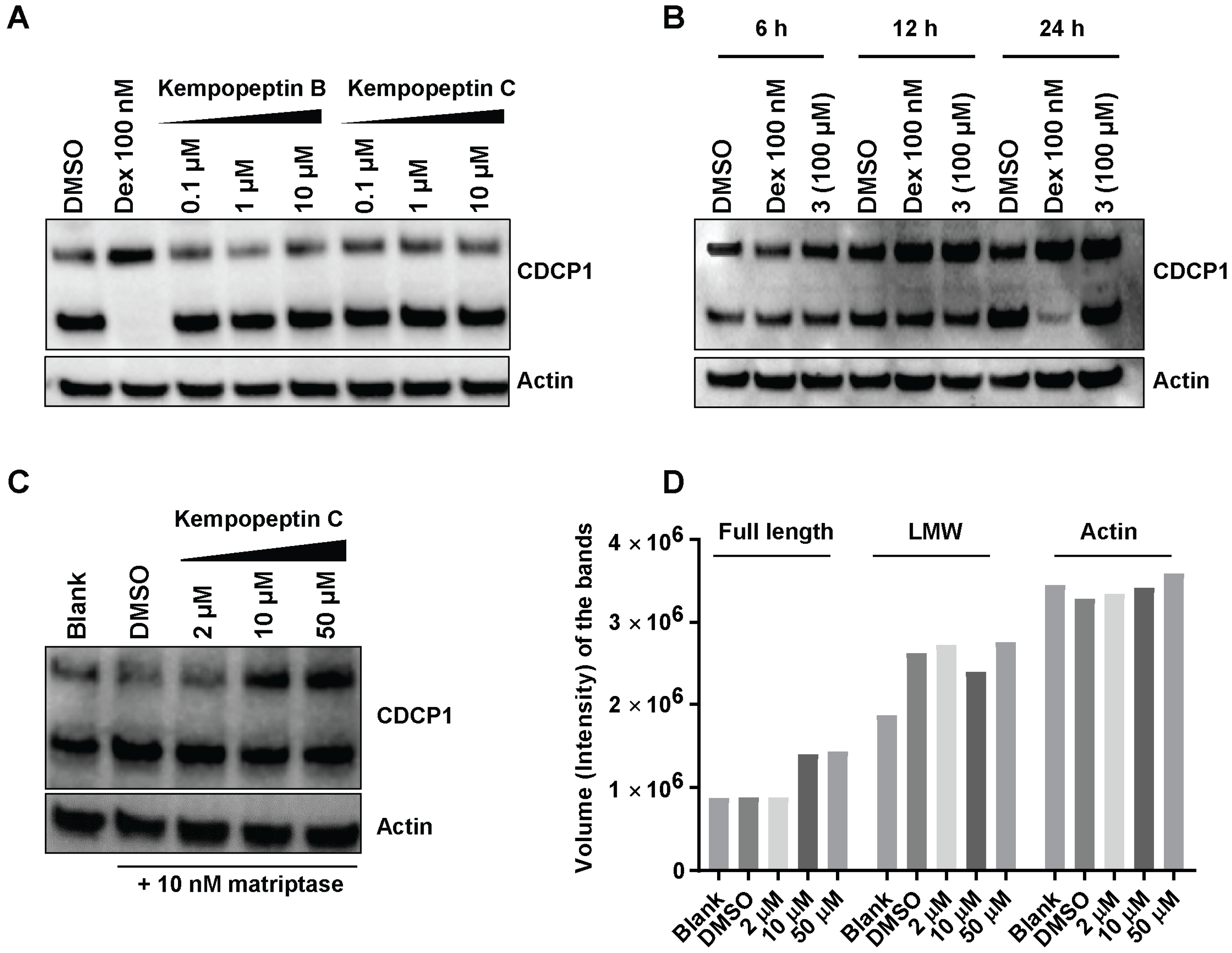
2.8. Effects of Kempopeptins B and C on the CDCP1 Cleavage in MDA-MB-231
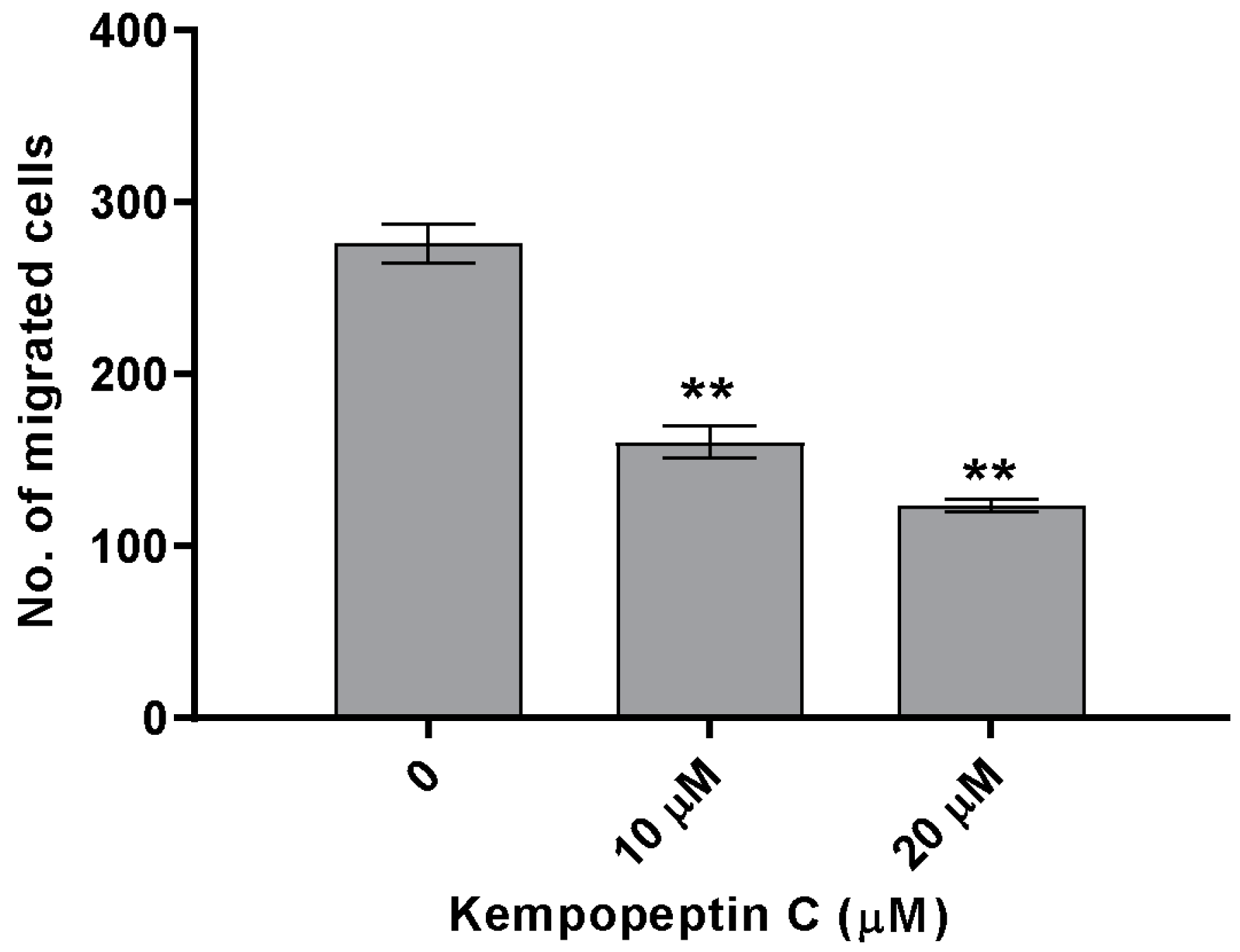
2.9. Effects of KempopeptinC on the Migration of MDA-MB-231
3. Experimental Section
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. In Vitro Protease Inhibition Assays
3.5. Molecular Docking
3.6. In Vitro Cleavage of Desmoglein 2 and CDCP1 by Matriptase
3.7. Cell Viability Assay
3.8. Expression Levels of Dsg-2 and CDCP1 in Breast Cancer Cell Lines
3.9. Cleavage of CDCP1 in the MDA-MB-231 Cell Line
3.10. Transwell Migration Assays
3.11. Proteasome Activity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Netzel-arnett, S.; Hooper, J.D.; Szabo, R.; Madison, E.L.; Quigley, J.P.; Bugge, H.; Antalis, T.M. Membrane anchored serine proteases: A rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003, 22, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.L.; Sanders, A.J.; Mason, M.D.; Jiang, W.G. Type II transmembrane serine protease (TTSP) deregulation in cancer. Front. Biosci. 2011, 16, 539–552. [Google Scholar] [CrossRef]
- Shi, Y.E.; Torri, J.; Yieh, L.; Wellstein, A.; Lippman, M.E.; Dickson, R.B. Identification and characterization of a novel matrix-degrading protease from hormone-dependent human breast cancer cells. Cancer Res. 1993, 53, 1409–1415. [Google Scholar] [PubMed]
- Bhatt, A.S.; Erdjument-Bromage, H.; Tempst, P.; Craik, C.S.; Moasser, M.M. Adhesion signaling by a novel mitotic substrate of src kinases. Oncogene 2005, 24, 5333–5343. [Google Scholar] [CrossRef] [PubMed]
- Uhland, K. Matriptase and its putative role in cancer. Cell. Mol. Life Sci. 2006, 63, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- List, K. Matriptase: A culprit in cancer? Future Oncol. 2009, 5, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ustach, C.V.; Huang, W.; Conley-LaComb, M.K.; Lin, C.Y.; Che, M.; Abrams, J.; Kim, H.R.C. A novel signaling axis of matriptase/PDGF-D/β-PDGFR in human prostate cancer. Cancer Res. 2010, 70, 9631–9640. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Dickson, R.B.; Lin, C.Y. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J. Biol. Chem. 2000, 275, 36720–36725. [Google Scholar] [CrossRef] [PubMed]
- Benaud, C.M.; Oberst, M.; Dickson, R.B.; Lin, C.Y. Deregulated activation of matriptase in breast cancer cells. Clin. Exp. Metastasis 2002, 19, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Welman, A.; Sproul, D.; Mullen, P.; Muir, M.; Kinnaird, A.R.; Harrison, D.J.; Faratian, D.; Brunton, V.G.; Frame, M.C. Diversity of matriptase expression level and function in breast cancer. PLoS ONE 2012, 7, e34182. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.S.; Cheng, T.F.; Tsai, W.C.; Sheu, L.F.; Chiang, H.; Yu, C.P. Expression of the serine protease, matriptase, in breast ductal carcinoma of Chinese women: Correlation with clinicopathological parameters. Histol. Histopathol. 2007, 22, 305–309. [Google Scholar] [PubMed]
- Wadhawan, V.; Kolhe, Y.A.; Sangith, N.; Gautam, A.K.S.; Venkatraman, P. From prediction to experimental validation: Desmoglein 2 is a functionally relevant substrate of matriptase in epithelial cells and their reciprocal relationship is important for cell adhesion. Biochem. J. 2012, 447, 61–70. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wortmann, A.; Burke, L.J.; Reid, J.C.; Adams, M.N.; Abdul-Jabbar, I.; Quigley, J.P.; Leduc, R.; Kirchhofer, D.; Hooper, J.D. Proteolysis-induced N-terminal ectodomain shedding of the integral membrane glycoprotein CUB domain-containing protein 1 (CDCP1) is accompanied by tyrosine phosphorylation of its C-terminal domain and recruitment of Src and PKC? J. Biol. Chem. 2010, 285, 26162–26173. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Gaudry, C.A. Are desmosomes more than tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 2000, 1, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.P.; Bornslaeger, E.A.; Norvell, S.M.; Palka, H.L.; Green, K.J. Desmosomes: Intercellular adhesive junctions specialized for attachment of intermediate filaments. Int. Rev. Cytol. 1999, 185, 237–302. [Google Scholar] [PubMed]
- Law, M.E.; Corsino, P.E.; Jahn, S.C.; Davis, B.J.; Chen, S.; Patel, B.; Pham, K.; Lu, J.; Sheppard, B.; Nørgaard, P.; et al. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013, 32, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; Duncan, D.; Moreno, O.; Madison, E.L.; Agus, D.B. CVS-3983, a selective matriptase inhibitor, suppresses the growth of androgen independent prostate tumor xenografts. Prostate 2004, 61, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jiang, S.; Lee, S.L.; Lin, C.Y.; Johnson, M.D.; Dickson, R.B.; Michejda, C.J.; Roller, P.P. Design and synthesis of novel and potent inhibitors of the type II transmembrane serine protease, matriptase, based upon the sunflower trypsin inhibitor-1. J. Med. Chem. 2007, 50, 5976–5983. [Google Scholar] [CrossRef] [PubMed]
- Farady, C.J.; Sun, J.; Darragh, M.R.; Miller, S.M.; Craik, C.S. The mechanism of inhibition of antibody-based inhibitors of membrane-type serine protease 1 (MT-SP1). J. Mol. Biol. 2007, 369, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Taori, K.; Paul, V.J.; Luesch, H. Lyngbyastatins 8–10, elastase inhibitors with cyclic depsipeptide scaffolds isolated from the marine cyanobacterium Lyngbya semiplena. Mar. Drugs 2009, 7, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Matthew, S.; Rocca, J.R.; Paul, V.J.; Luesch, H. Lyngbyastatins 5–7, potent elastase inhibitors from Floridian marine cyanobacteria, Lyngbya spp. J. Nat. Prod. 2007, 70, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Kempopeptins A and B, serine protease inhibitors with different selectivity profiles from a marine cyanobacterium, Lyngbya sp. J. Nat. Prod. 2008, 71, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Taori, K.; Biggs, J.S.; Jakoncic, J.; Ostrov, D.A.; Paul, V.J.; Luesch, H. Potent elastase inhibitors from cyanobacteria: Structural basis and mechanisms mediating cytoprotective and anti-inflammatory effects in bronchial epithelial cells. J. Med. Chem. 2013, 56, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Smitka, T.A.; Bonjouklian, R. Atomic structure of the trypsin-A90720A complex: A unified approach to structure and function. Chem. Biol. 1994, 1, 113–117. [Google Scholar] [CrossRef]
- Gademann, K.; Portmann, C.; Blom, J.F.; Zeder, M.; Jüttner, F. Multiple toxin production in the cyanobacterium microcystis: Isolation of the toxic protease inhibitor cyanopeptolin 1020. J. Nat. Prod. 2010, 73, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Bonjouklian, R.; Smitka, T.A.; Hunt, A.H.; Occolowitz, J.L.; Perun, T.J.; Doolin, L.; Stevenson, S.; Knauss, L.; Wijayaratne, R.; Szewczyk, S.; et al. A90720A, a serine protease inhibitor isolated from a terrestrial blue-green alga Microchaete loktakensis. Tetrahedron 1996, 52, 395–404. [Google Scholar] [CrossRef]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine Cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Ploutno, A.; Shoshan, M.; Carmeli, S. Three Novel Protease Inhibitors from a Natural Bloom of the Cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 2002, 65, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Reshef, V.; Carmeli, S. Protease inhibitors from a water bloom of the cyanobacterium Microcystis aeruginosa. Tetrahedron 2001, 57, 2885–2894. [Google Scholar] [CrossRef]
- Scherl-Mostageer, M.; Sommergruber, W.; Abseher, R.; Hauptmann, R.; Ambros, P.; Schweifer, N. Identification of a novel gene, CDCP1, overexpressed in human colorectal cancer. Oncogene 2001, 20, 4402–4408. [Google Scholar] [CrossRef]
- Bühring, H.J.; Kuçi, S.; Conze, T.; Rathke, G.; Bartolović, K.; Grünebach, F.; Scherl-Mostageer, M.; Brümmendorf, T.H.; Schweifer, N.; Lammers, R. CDCP1 identifies a broad spectrum of normal and malignant stem/progenitor cell subsets of hematopoietic and nonhematopoietic origin. Stem Cells 2004, 22, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.; Morii, E.; Kimura, H.; Tomita, Y.; Takakuwa, T.; Hasegawa, J.; Kim, Y.K.; Miyoshi, Y.; Noguchi, S.; Nishida, T.; et al. Epigenetic regulation of the expression of the novel stem cell marker CDCP1 in cancer cells. J. Pathol. 2006, 210, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Awakura, Y.; Nakamura, E.; Takahashi, T.; Kotani, H.; Mikami, Y.; Kadowaki, T.; Myoumoto, A.; Akiyama, H.; Ito, N.; Kamoto, T.; et al. Microarray-based identification of CUB-domain containing protein 1 as a potential prognostic marker in conventional renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2008, 134, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.; Oda, T.; Inoue, M.; Uekita, T.; Sakai, R.; Okumura, M.; Aozasa, K.; Morii, E. Expression of CUB domain containing protein (CDCP1) is correlated with prognosis and survival of patients with adenocarcinoma of lung. Cancer Sci. 2009, 100, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Conn, E.M.; Wortmann, A.; Partridge, J.J.; Kupriyanova, T.A.; Ardi, V.C.; Hooper, J.D.; Quigley, J.P. Functional role of cell surface CUB domain-containing protein 1 in tumor cell dissemination. Mol. Cancer Res. 2009, 7, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Yang, T.M.; Zaitsevskaia, T.; Xia, Y.; Dunn, C.A.; Sigle, R.O.; Knudsen, B.; Carter, W.G. Adhesion or plasmin regulates tyrosine phosphorylation of a novel membrane glycoprotein p80/gp140/CUB domain-containing protein 1 in epithelia. J. Biol. Chem. 2004, 279, 14772–14783. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; Cochrane, R.; Hiscox, S.; Jiang, W.; Sweetland, H.; Mansel, R. The role of desmoglein 2 and E-cadherin in the invasion and motility of human breast cancer cells. Int. J. Oncol. 1997, 11, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Hennings, L.; Haun, R.S. Desmoglein 2 is a substrate of kallikrein 7 in pancreatic cancer. BMC Cancer 2008, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Mahoney, M.G. Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adhes. Migr. 2009, 3, 148–154. [Google Scholar] [CrossRef]
- Peitsch, W.K.; Doerflinger, Y.; Fischer-Colbrie, R.; Huck, V.; Bauer, A.T.; Utikal, J.; Goerdt, S.; Schneider, S.W. Desmoglein 2 depletion leads to increased migration and upregulation of the chemoattractant secretoneurin in melanoma cells. PLoS ONE 2014, 9, e89491. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Altendorf-Hofmann, A.; Jackstadt, R.; Horst, D.; Assmann, G.; Zhao, Y.; Bruns, C.; Kirchner, T.; Knösel, T. Desmogleins as prognostic biomarkers in resected pancreatic ductal adenocarcinoma. Br. J. Cancer 2015, 113, 1460–1466. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | C/H no. | δH (J in Hz) | δC, mult a | COSY | HMBC |
|---|---|---|---|---|---|
| Val-1 | 1 | 172.4, qC | |||
| 2 | 4.66, dd (9.2, 5) | 56.0, CH | NH, H-3 | 1, 5, 1 (N,O-diMe-Cl-Tyr) | |
| 3 | 2.06, m | 30.3, CH | H-2, H3-4, H3-5 | 5 | |
| 4 | 0.87, d (6.9) | 19.0, CH3 | H-3 | 3 | |
| 5 | 0.76, d (6.9) | 17.3, CH3 | H-3 | 2, 3 | |
| NH | 7.69, d (9.4) | H-2 | 1 (N,O-diMe-Cl-Tyr) | ||
| N,O-diMe-Cl-Tyr | 1 | 169.2, qC | |||
| 2 | 5.05, dd (11.7, 2.2) | 60.1, CH | H-3a, H-3b | ||
| 3a | 3.22, dd (−14, 2.2) | 32.6, CH2 | H-2, H-3b | 4, 9 | |
| 3b | 2.80, dd (−14, 11.7) | H-2, H-3a | 4, 9 | ||
| 4 | 130.7, qC | ||||
| 5 | 7.25 d (1.5) | 130.4, CH | 6, 7 | ||
| 6 | 121.4, qC | ||||
| 7 | 153.6, C | ||||
| 8 | 7.06, d (8.5) | 113.0, CH | H-9 | 3, 6, 7 | |
| 9 | 7.14, dd (8.6, 1.5) | 129.2, CH | H-8 | 3, 7 | |
| O-Me | 3.78, s | 56.0, CH3 | 7 | ||
| N-Me | 2.74, s | 29.9, CH3 | 2, 1 (Ile) | ||
| Ile | 1 | 169.7, qC | |||
| 2 | 4.38, d (11) | 53.9, CH | 1 | ||
| 3 | 1.82, m | 32.6, CH | H-6 | ||
| 4a | 1.11, m | 23.4, CH2 | H-4b, H-5 | 5 | |
| 4b | 0.65, m | H-4a | |||
| 5 | 0.62, t (6.5) | 9.86, CH3 | H-4a | 3, 4 | |
| 6 | −0.14, d (6.5) | 13.4, CH3 | H-3 | 2, 3, 4 | |
| Ahp | 2 | 169.8, qC | |||
| 3 | 4.45, m | 48.6, CH | NH, H-4a, H-4b | 2, 6 | |
| 4a | 2.58, m | 21.4, CH2 | H-3, H-4b | ||
| 4b | 1.74, m | H-3, H-4a | |||
| 5 | 1.76, m | 29.4, CH2 | H-6 | ||
| 6 | 4.93, br s | 73.6, CH | H-5, 6-OH | ||
| 6-OH | 6.17, d (2.7) | H-6 | |||
| NH | 7.35, d (9.4) | H-3 | 1 (Lys) | ||
| Lys | 1 | 170.3, qC | |||
| 2 | 4.28, br | 51.5, CH | NH, H-3b | ||
| 3a | 2.03, m | 29.0, CH2 | H-3b, H-5 | ||
| 3b | 1.42, m | H-2, H-3a | |||
| 4 | 1.26, m | 21.9, CH2 | H-3a | ||
| 5 | 1.49, m | 25.9, CH2 | H-6 | ||
| 6 | 2.73, m | 38.4, CH2 | H-5, NH2 | ||
| NH | 8.45, d (8.5) | H-2 | 1 (Thr) | ||
| NH2 | 7.63, br s | H-6 | |||
| Thr | 1 | 169.2, qC | |||
| 2 | 4.64, d (9.2) | 54.5, CH | NH, H-3 | 1, 1 (Val-1) | |
| 3 | 5.51, q (6.9) | 71.5, CH | H-2, H3-4 | 4, 1 (Val-1) | |
| 4 | 1.22, d (6.5) | 17.3, CH3 | H-3 | 3 | |
| NH | 7.83, d (7.7) | H-2 | 1 (Val-2) | ||
| Val-2 | 1 | 172.4, qC | |||
| 2 | 4.34, d (8.0) | 57.3, CH | NH, H-3 | 1, 4, 5 | |
| 3 | 2.03, m | 29.8, CH | H-2, H-5 | ||
| 4 | 0.87, d (6.8) | 19.0, CH3 | |||
| 5 | 0.85, d (6.8) | 17.8, CH3 | H-3 | ||
| NH | 7.84, d (8.1) | H-2 | 1, 1 (Ba) | ||
| Ba | 1 | 172.3, qC | |||
| 2 | 2.17, m | 36.7, CH2 | H-3 | 1, 4 | |
| 3 | 1.52, m | 18.5, CH2 | H-2, H3-4 | 1, 4 | |
| 4 | 0.86, t (7.5) | 13.2, CH3 | H-3 |
| Compound | Trypsin | Plasmin | Matriptase |
|---|---|---|---|
| Kempopeptin B (2) | 1.49 ± 0.18 µM | 1.05 ± 0.19 µM | 1.83 ± 0.19 µM |
| Kempopeptin C (3) | 0.19 ± 0.05 µM | 0.36 ± 0.09 µM | 0.28 ± 0.03 µM |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Awadhi, F.H.; Salvador, L.A.; Law, B.K.; Paul, V.J.; Luesch, H. Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer. Mar. Drugs 2017, 15, 290. https://doi.org/10.3390/md15090290
Al-Awadhi FH, Salvador LA, Law BK, Paul VJ, Luesch H. Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer. Marine Drugs. 2017; 15(9):290. https://doi.org/10.3390/md15090290
Chicago/Turabian StyleAl-Awadhi, Fatma H., Lilibeth A. Salvador, Brian K. Law, Valerie J. Paul, and Hendrik Luesch. 2017. "Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer" Marine Drugs 15, no. 9: 290. https://doi.org/10.3390/md15090290
APA StyleAl-Awadhi, F. H., Salvador, L. A., Law, B. K., Paul, V. J., & Luesch, H. (2017). Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer. Marine Drugs, 15(9), 290. https://doi.org/10.3390/md15090290