
Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics

Abstract
:
1. Introduction
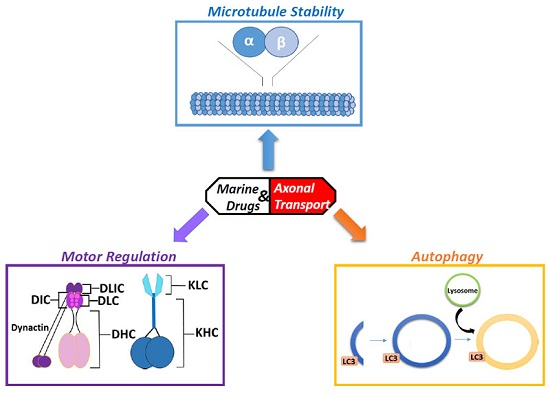
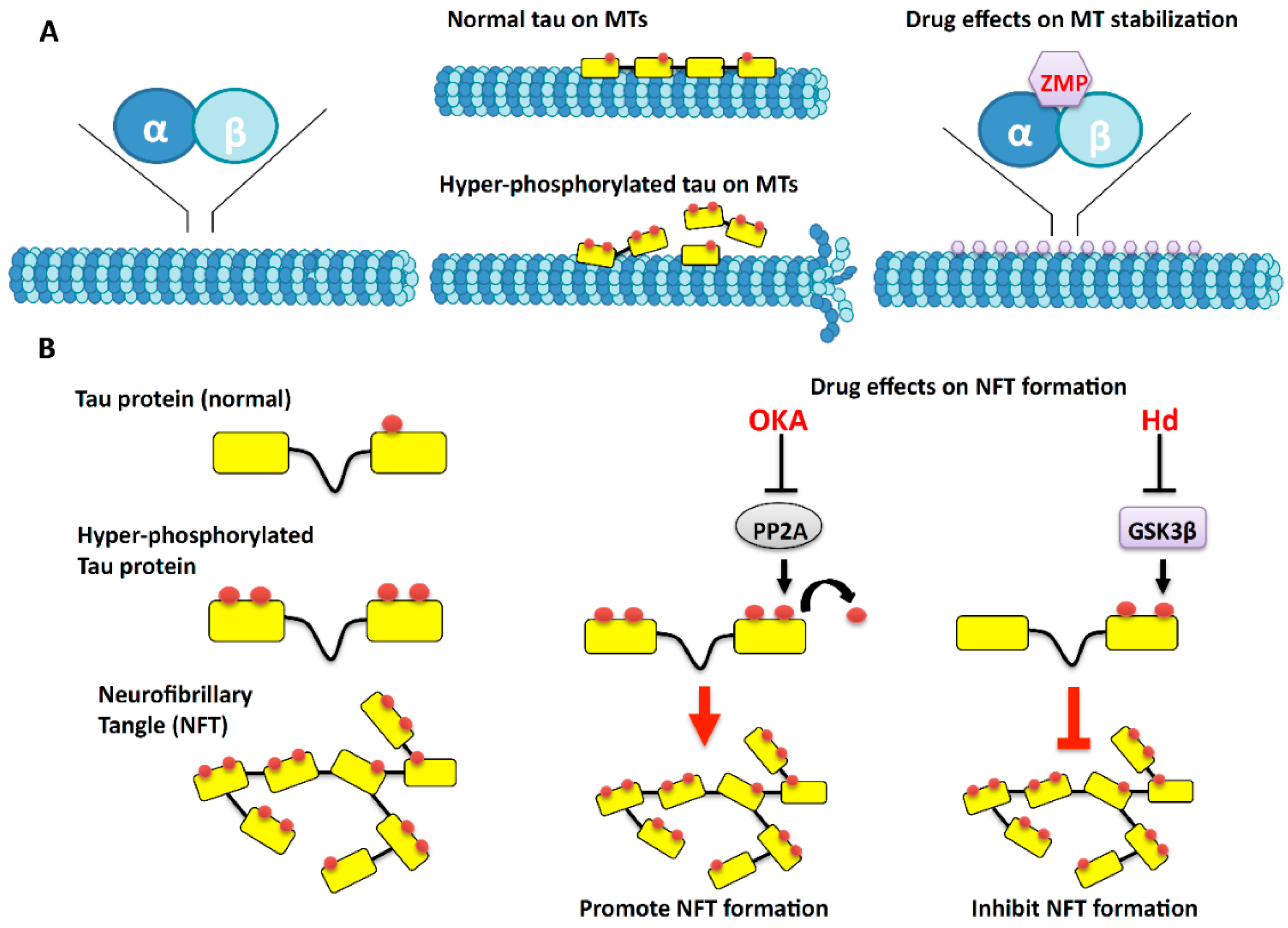
2. Microtubules: The Tracks of the Axonal Highway
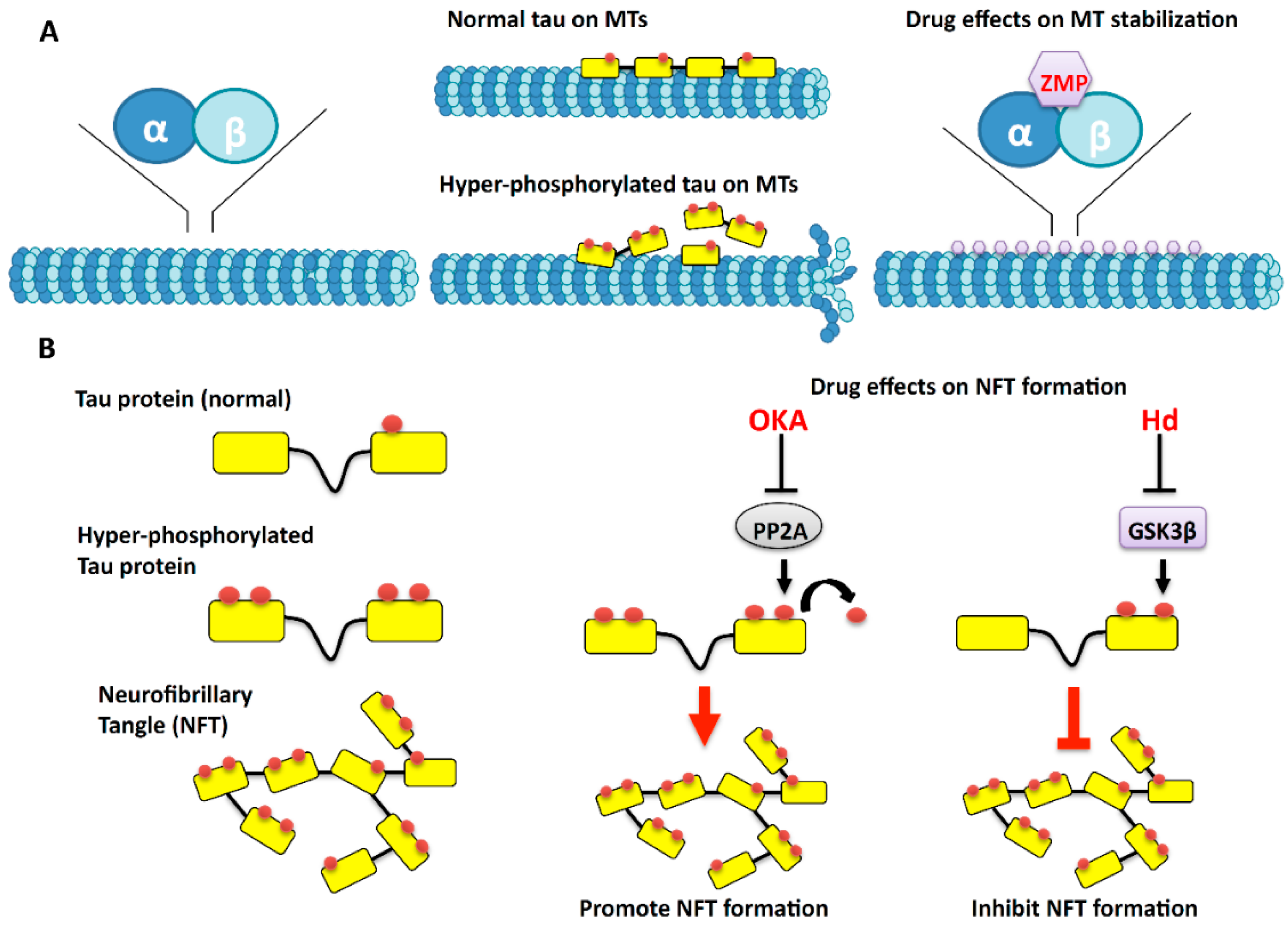
3. The Effects of Marine Drugs on Microtubules
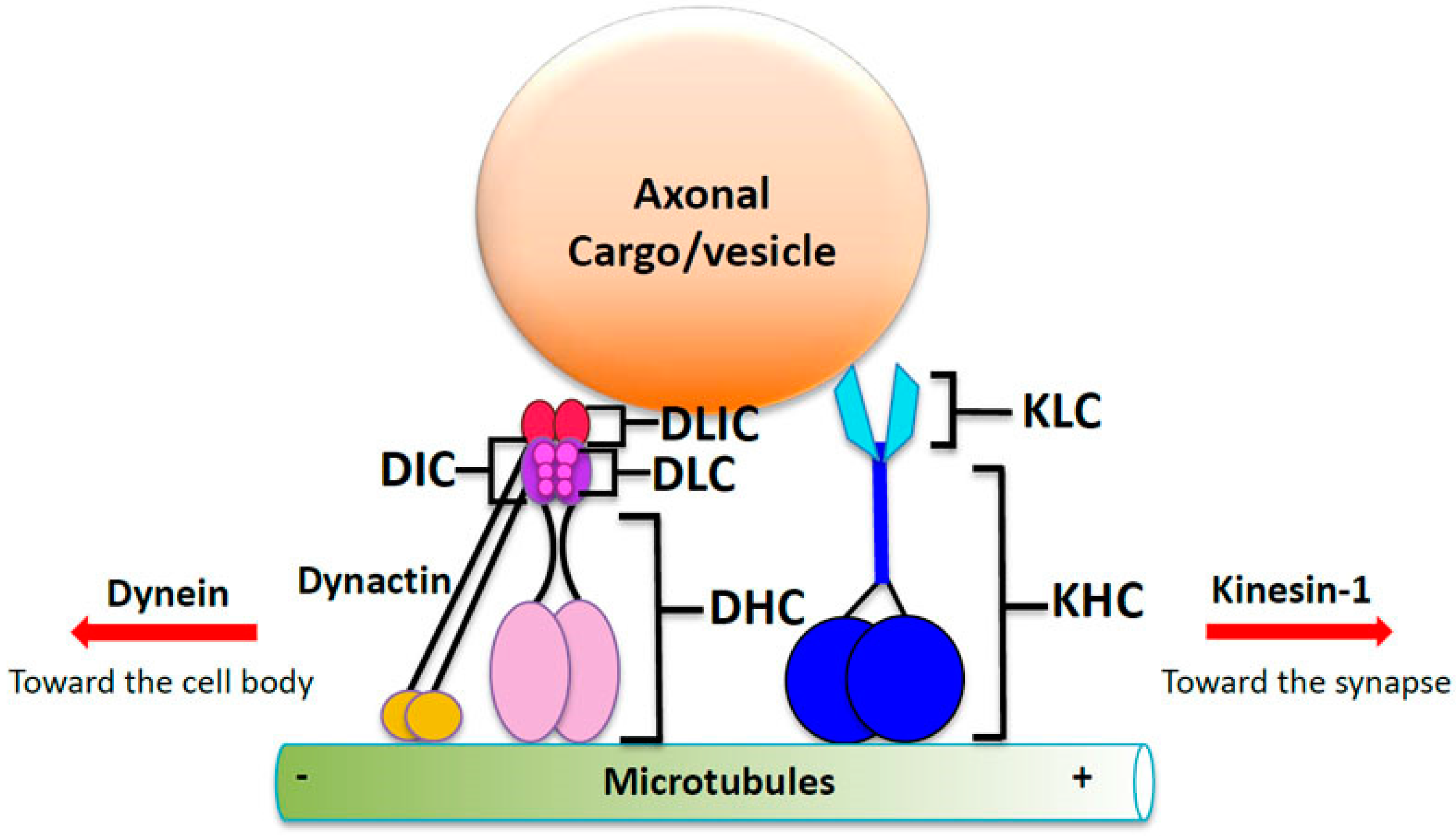
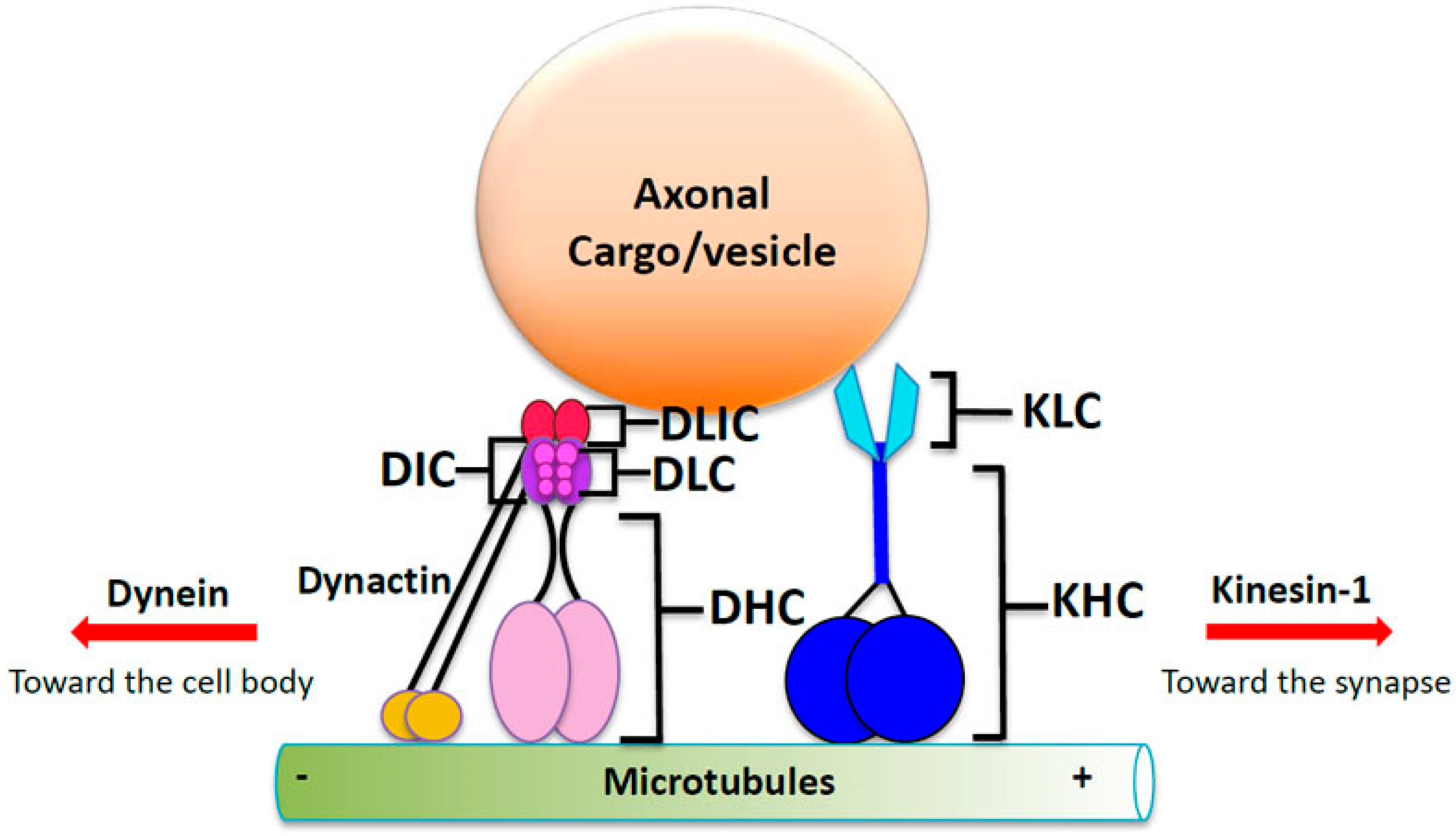
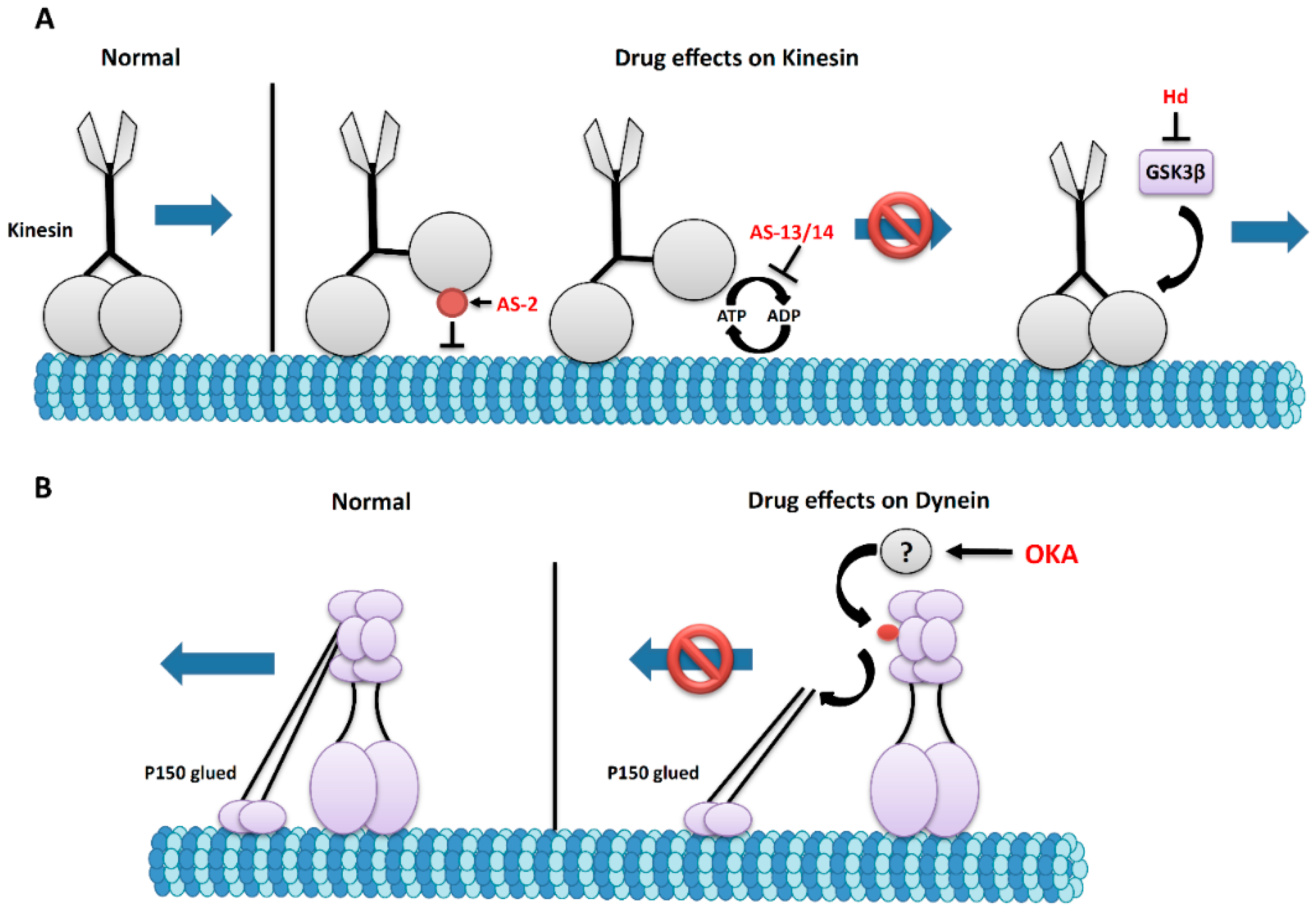
4. Molecular Motor Proteins Kinesin and Dynein: The Engines of Microtubule Transport
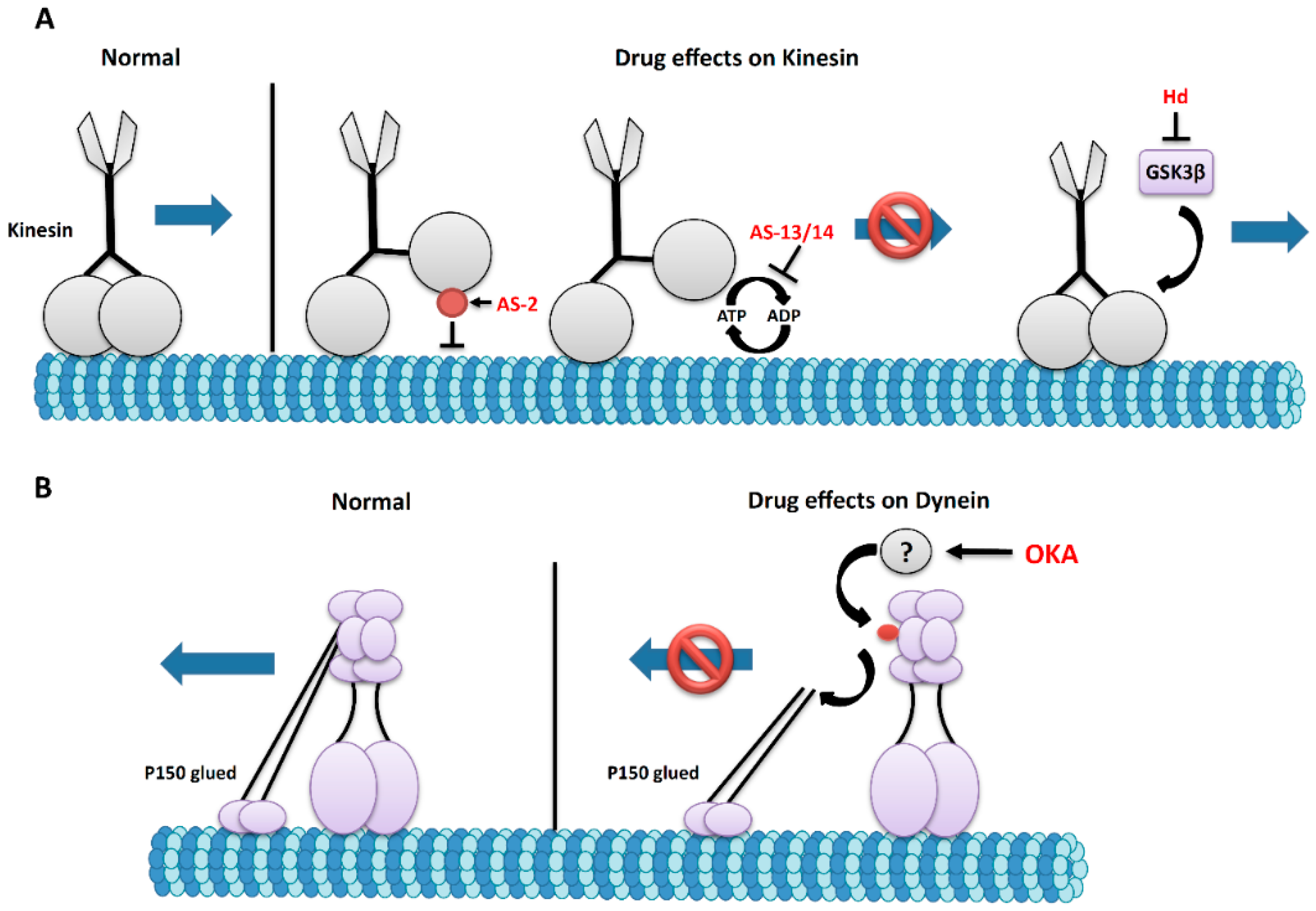
5. The Effects of Marine Drugs on Molecular Motors
6. The Autophagy-Lysosome Degradation Pathway: A Key to Neuronal Homeostasis and Survival
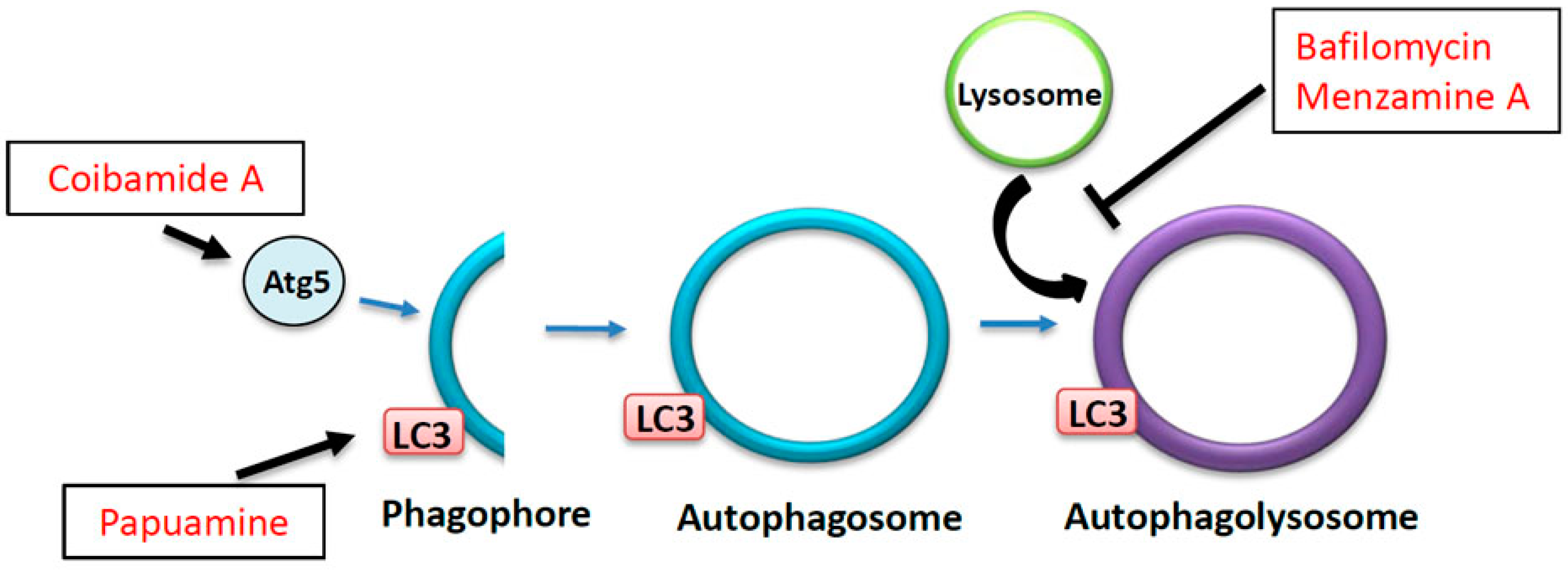
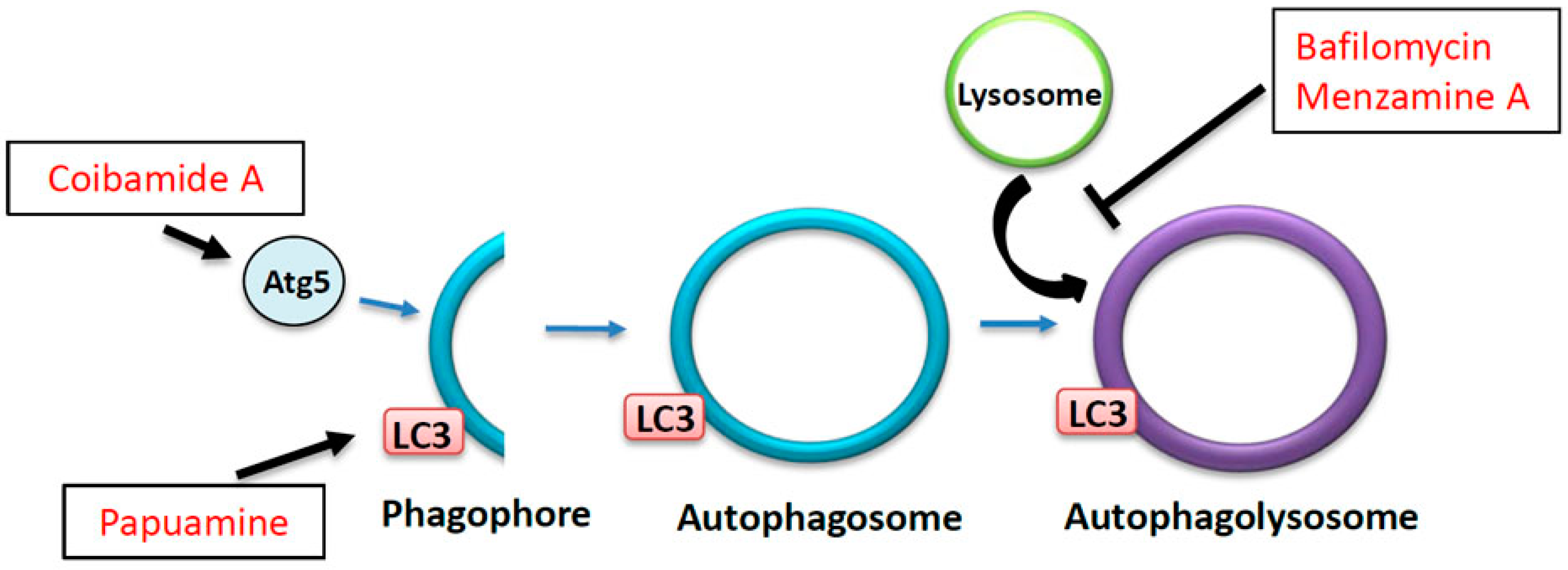
7. The Effects of Marine Drugs on the Autophagy-Lysosome Pathway
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Stokin, G.B.; Lillo, B.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Goldstein, L.S. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 2001, 32, 389–401. [Google Scholar] [CrossRef]
- Wirths, O.; Weis, J.; Szczygielski, J.; Multhaup, G.; Bayer, T.A. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol. 2006, 111, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, S.H.; Yu, Z.X.; Shelbourne, P.; Li, X.J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [PubMed]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2004, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Dolma, K.; Iacobucci, G.J.; Zheng, K.H.; Shandilya, J.; Toska, E.; White, J.; Spina, E.; Gunawardena, S. Presenilin influences glycogen synthase kinase-3 β (GSK-3β) for kinesin-1 and dynein function during axonal transport. Hum. Mol. Genet. 2014, 23, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Hansen, T.J.; Mickiewicz, M.; Kaczynski, T.J.; Fye, S.; Gunawardena, S. Disruption of axonal transport perturbs bone morphogenetic protein (BMP)—Signaling and contributes to synaptic abnormalities in two neurodegenerative diseases. PLoS ONE 2014, 9, e104617. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.F.; Yang, G.; Szpankowski, L.; Weaver, C.; Shah, S.B.; Robinson, J.T.; Hays, T.S.; Danuser, G.; Goldstein, L.S. Molecular motor function in axonal transport in vivo probed by genetic and computational analysis in Drosophila. Mol. Biol. Cell 2012, 23, 1700–1714. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Yang, G.; Goldstein, L.S. Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum. Mol. Genet. 2013, 22, 3828–3843. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; White, J.A.; Gunawardena, S. Axonal transport and neurodegenerative disease. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 29–47. [Google Scholar]
- Cosker, K.E.; Courchesne, S.L.; Segal, R.A. Action in the axon: Generation and transport of signaling endosomes. Curr. Opin. Neurobiol. 2008, 18, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Takita, J.; Tanaka, Y.; Setou, M.; Nakagawa, T.; Takeda, S.; Yang, H.W.; Terada, S.; Nakata, T.; Takei, Y. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bβ. Cell 2001, 105, 587–597. [Google Scholar] [CrossRef]
- Reid, E.; Kloos, M.; Ashley-Koch, A.; Hughes, L.; Bevan, S.; Svenson, I.K.; Graham, F.L.; Gaskell, P.C.; Dearlove, A.; Pericak-Vance, M.A.; et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 2002, 71, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot–Marie–Tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dachsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009, 41, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.H.; Goldstein, L.S. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000, 28, 449–459. [Google Scholar] [CrossRef]
- Satpute-Krishnan, P.; DeGiorgis, J.A.; Conley, M.P.; Jang, M.; Bearer, E.L. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2006, 103, 16532–16537. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.M.; Holzbaur, E.L. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol. 2013, 202, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Zala, D.; Hinckelmann, M.V.; Yu, H.; Lyra da Cunha, M.M.; Liot, G.; Cordelieres, F.P. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 2013, 152, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Anderson, E.; Zimmerman, K.; Zheng, K.H.; Rouhani, R.; Gunawardena, S. Huntingtin differentially regulates the axonal transport of a sub-set of Rab-containing vesicles in vivo. Hum. Mol. Genet. 2015, 24, 7182–7195. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; You, Y.M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Björkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.R.; Hill, J.; Utton, M.A.; Asuni, A.A.; Ackerley, S.; Grierson, A.J.; Miller, C.C.; Davies, A.M.; Buchman, V.L.; Anderton, B.H.; et al. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J. Cell Sci. 2004, 117, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.V.; Nixon, R.A. Defective neurofilament transport in mouse models of amyotrophic lateral sclerosis: A review. Neurochem. Res. 2003, 28, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2004, 21, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C.; Borisy, G.G.; Taylor, E.W. The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 1968, 7, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.; Mandelkow, E. Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 1995, 7, 72–81. [Google Scholar] [CrossRef]
- Evans, L.; Mitchison, T.; Kirschner, M. Influence of the centrosome on the structure of nucleated microtubules. J. Cell Biol. 1985, 100, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Borisy, G.G. Structural polarity and directional growth of microtubules of Chlamydomonas flagella. J. Mol. Biol. 1974, 90, 3812–3840. [Google Scholar] [CrossRef]
- Wegner, A. Head to tail polymerization of actin. J. Mol. Biol. 1976, 108, 139–150. [Google Scholar] [CrossRef]
- Kirschner, M.W.; Mitchison, T. Beyond self-assembly: From microtubules to morphogenesis. Cell 1986, 45, 329–342. [Google Scholar] [CrossRef]
- Sudo, H.; Baas, P.W. Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum. Mol. Genet. 2011, 20, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Dong, S.; Gu, F.; Hu, Y.; Zhao, Z. Advances in the pathogenesis of Alzheimer’s disease: Focusing on tau-mediated neurodegeneration. Transl. Neurodegener. 2012, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Lee, V.M.; Trojanowski, J.Q. Therapeutic strategies for tau mediated neurodegeneration. J. Neurol. Neurosurg. Psychiatry 2013, 84, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Naini, S.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxidative Med. Cell. Longev. 2015, 2015, 17. [Google Scholar]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Nath, C. Okadaic acid: A tool to study regulatory mechanisms for neurodegeneration and regeneration in Alzheimer’s disease. Neural Regen. Res. 2015, 10, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [PubMed]
- Sengottuvel, V.; Leibinger, M.; Pfreimer, M.; Andreadaki, A.; Fischer, D. Taxol facilitates axon regeneration in the mature CNS. J. Neurosci. 2011, 31, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Higa, T. Zampanolide, a New Cytotoxic Macrolide from a Marine Sponge. Tetrahedron Lett. 1996, 37, 5535–5538. [Google Scholar] [CrossRef]
- Field, J.J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodríguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S.J.; et al. Zampanolide, a potent new microtubule-stabilizing agent, covalently reacts with the taxane luminal site in tubulin α,β-heterodimers and microtubules. Chem. Biol. 2012, 19, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Bruno, I.; Bifulco, G.; Casapullo, A.; Debitus, C.; Gomez-Paloma, L.; Riccio, R. Dactylolide, a New Cytotoxic Macrolide from the Vanuatu Sponge Dactylospongia sp. Eur. J. Org. Chem. 2001, 2001, 775–778. [Google Scholar] [CrossRef]
- Kitagawa, I.; Kobayashi, M.; Kitanaka, K.; Kido, M.; Kyogoku, Y. Marine natural products. XII. On the chemical constituents of the Okinawan marine sponge Hymeniacidon aldis. Chem. Pharm. Bull. 1983, 31, 2321–2328. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- West, L.M.; Northcote, P.T.; Battershill, C.N. Peloruside A: A potent cytotoxic macrolide isolated from the New Zealand marine sponge Mycale sp. J. Org. Chem. 2000, 65, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.H.; Singh, A.J.; Northcote, P.T. Microtubule-stabilizing drugs from marine sponges: Focus on peloruside A and zampanolide. Mar. Drugs 2010, 8, 1059–1079. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissolute. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Canales, A.; Rodríguez-salarichs, J.; Trigili, C.; Nieto, L.; Coderch, C.; Andreu, J.M.; Paterson, I.; Jiménez-Barbero, J.; Díaz, J.F. Insights into the interaction of discodermolide and docetaxel with tubulin. Mapping the binding sites of microtubule-stabilizing agents by using an integrated NMR and computational approach. ACS Chem. Biol. 2011, 6, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Freeze, B.S. (+)-Discodermolide: Total Synthesis, Construction of Novel Analogues, and Biological Evaluation. Tetrahedron 2007, 64, 261–298. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Das, V.; Miller, J.H. Microtubule stabilization by peloruside A and paclitaxel rescues degenerating neurons from okadaic acid-induced tau phosphorylation. Eur. J. Neurosci. 2012, 35, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Pettit, G.R.; Hamel, E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [PubMed]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal: Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Beckwith, M.; Urba, W.J.; Longo, D.L. Growth inhibition of human lymphoma cell lines by the marine products, dolastatins 10 and 15. J. Natl. Cancer Inst. 1993, 85, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.; Jackson, W.H.; Pettit, G.R.; Wells, A.; Kraft, A.S. Treatment of human prostate cancer cells with dolastatin 10, a peptide isolated from a marine shell-less mollusc. Prostate 1998, 34, 175–181. [Google Scholar] [CrossRef]
- Jordan, M.A.; Thrower, D.; Wilson, L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991, 51, 2212–2222. [Google Scholar] [PubMed]
- Mooberry, S.L.; Leal, R.M.; Tinley, T.L.; Luesch, H.; Moore, R.E.; Corbett, T.H. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int. J. Cancer 2003, 104, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Bai, R.L.; Paull, K.D.; Herald, C.L.; Malspeis, L.; Pettit, G.R.; Hamel, E. Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data. J. Biol. Chem. 1991, 266, 15882–15889. [Google Scholar] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Zhang, X. Mechanism of action cryptophycin. Interaction with the Vinca alkaloid domain of tubulin. J. Biol. Chem. 1996, 271, 6192–6198. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.A.; Vaisberg, E.A.; Shanina, N.A.; Magretova, N.N.; Chernyak, V.Y.; Gelfand, V.I. The quaternary structure of bovine brain kinesin. EMBO J. 1988, 7, 353–356. [Google Scholar] [PubMed]
- Bloom, G.S.; Wagner, M.C.; Pfister, K.K.; Brady, S.T. Native structure and physical properties of bovine brain kinesin and identifcation of the ATP-binding subunit polypeptide. Biochemistry 1988, 27, 3409–3416. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.; Mandelkow, E.M. Kinesin motors and disease. Trends Cell Biol. 2002, 12, 585–591. [Google Scholar] [CrossRef]
- Yang, J.T.; Laymon, R.A.; Goldstein, L.S. A three-domain structure of kinesin heavy chain revealed by DNA sequence and microtubule binding analysis. Cell 1989, 56, 879–889. [Google Scholar] [CrossRef]
- Hirokawa, N.; Pfister, K.K.; Yorifuji, H.; Wagner, M.C.; Brady, S.T.; Bloom, G.S. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell 1989, 56, 867–878. [Google Scholar] [CrossRef]
- Scholey, J.M.; Heuser, J.; Yang, J.T.; Goldstein, L.S. Identification of globular mechanochemical heads of kinesin. Nature 1989, 338, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.J.; Allan, V.J. Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 2006, 25, 5457–5468. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins, KIFs: Structure, function, and dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Holzbaur, E.L. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr. Opin. Cell Biol. 1999, 11, 45–53. [Google Scholar] [CrossRef]
- Pfister, K.K.; Fisher, E.M.; Gibbons, I.R.; Hays, T.S.; Holzbaur, E.L.; McIntosh, J.R.; Porter, M.E.; Schroer, T.A.; Vaughan, K.T.; Witman, G.B.; et al. Cytoplasmic dynein nomenclature. J. Cell Biol. 2005, 171, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Holzbaur, E.L. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem. 1995, 270, 28806–28811. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Maciejewski, M.W.; Takebe, S.; King, S.M. Solution structure of the Tctex1 dimer reveals a mechanism for dynein-cargo interactions. Structure 2005, 13, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; Karki, S.; Tokito, M.; Holzbaur, E.L. Dynein binds to beta-catenin and may tether microtubules at adherens junctions. Nat. Cell Biol. 2001, 3, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Lee, F.S.; Kong, H.; Chuang, J.; Arevalo, J.; Perez, P.; Sung, C.; Chao, M.V. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J. Neurosci. 2001, 21, RC125. [Google Scholar] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein–dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Vaughan, K.T.; Vallee, R.B. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chain and p150Glued. J. Cell Biol. 1995, 131, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhapparova, O.N.; Bryantseva, S.A.; Dergunova, L.V.; Raevskaya, N.M.; Burakov, A.V.; Bantysh, O.B.; Shanina, N.A.; Nadezhdina, E.S. Dynactin subunit p150Glued isoforms notable for differential interaction with microtubules. Traffic 2009, 10, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef] [PubMed]
- Holleran, E.A.; Karki, S.; Holzbaur, E.L. The role of the dynactin complex in intracellular motility. Int. Rev. Cytol. 1998, 182, 69–109. [Google Scholar] [PubMed]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, P.S.; Leszyk, J.D.; Vaughan, K.T. Cytoplasmic dynein intermediate chain phosphorylation regulates binding to dynactin. J. Biol. Chem. 2001, 276, 26171–26179. [Google Scholar] [CrossRef] [PubMed]
- Encalada, S.E.; Szpankowski, L.; Goldstein, L.S.B. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell 2011, 144, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Hancock, W.O. Bidirectional cargo transport: Moving beyond tug of war. Nat. Rev. Mol. Cell Biol. 2014, 15, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Kilaru, S.; Fink, G.; Collemare, J.; Roger, Y.; Steinberg, G. Kinesin-3 and dynein cooperate in long-range retrograde endosome motility along a nonuniform microtubule array. Mol. Biol. Cell 2011, 22, 3645–3657. [Google Scholar] [CrossRef] [PubMed]
- Ally, S.; Larson, A.; Barlan, K.; Rice, S.; Gelfand, V.J. Opposite-polarity motors activate one another to trigger cargo transport in live cells. Cell Biol. 2009, 187, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie–Tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Füger, P.; Sreekumar, V.; Schüle, R.; Kern, J.V.; Stanchev, D.T.; Schneider, C.D.; Karle, K.N.; Daub, K.J.; Siegert, V.K.; Flötenmeyer, M.; et al. Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet. 2012, 8, e1003066. [Google Scholar] [CrossRef] [PubMed]
- Dhaenens, C.M.; Van Brussel, E.; Schraen-Maschke, S.; Pasquier, F.; Delacourte, A.; Sablonnière, B. Association study of three polymorphisms of kinesin light-chain 1 gene with Alzheimer’s disease. Neurosci. Lett. 2004, 368, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- LaMonte, B.H.; Wallace, K.E.; Holloway, B.A.; Shelly, S.S.; Ascaño, J.; Tokito, M.; Van Winkle, T.; Howland, D.S.; Holzbaur, E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 2002, 34, 715–727. [Google Scholar] [CrossRef]
- Lai, C.; Lin, X.; Chandran, J.; Shim, H.; Yang, W.J.; Cai, H. The G59S mutation in p150 (glued) causes dysfunction of dynactin in mice. J. Neurosci. 2007, 27, 13982–13990. [Google Scholar] [CrossRef] [PubMed]
- Laird, F.M.; Farah, M.H.; Ackerley, S.; Hoke, A.; Maragakis, N.; Rothstein, J.D.; Griffin, J.; Price, D.L.; Martin, L.J.; Wong, P.C. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 2008, 28, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Rosenbohm, A.; Sperfeld, A.D.; Uttner, I.; Reske, S.; Krause, B.J.; Sedlmeier, R.; Meyer, T.; Hanemann, C.O.; Stumm, G.; et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 2005, 58, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.J.; Hebbar, S.; Gao, X.A.; Alexander, M.; Pandey, J.P.; Walla, M.D.; Cotham, W.E.; King, S.J.; Smith, D.S. GSK-3β Phosphorylation of Cytoplasmic Dynein Reduces Ndel1 Binding to Intermediate Chains and Alters Dynein Motility. Traffic 2015, 16, 941–961. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine Natural Products. Nat. Prod. Rep. 1997, 14, 259. [Google Scholar] [CrossRef]
- Sakowicz, R.; Berdelis, M.S.; Ray, K.; Blackburn, C.L.; Hopmann, C.; Faulkner, D.J.; Goldstein, L.S. A marine natural product inhibitor of kinesin motors. Science 1998, 280, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Brier, S.; Carletti, E.; DeBonis, S.; Hewat, E.; Lemaire, D.; Kozielski, F. The marine natural product adociasulfate-2 as a tool to identify the MT-binding region of kinesins. Biochemistry 2006, 45, 15644–15653. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.E.; Hong, W.; Zachariah, M.M.; Harper, M.K.; Matainaho, T.K.; Van Wagoner, R.M.; Ireland, C.M.; Vershinin, M. Single-molecule inhibition of human kinesin by adociasulfate-13 and -14 from the sponge Cladocroce aculeata. Proc. Natl. Acad. Sci. USA 2013, 110, 18880–18885. [Google Scholar] [CrossRef] [PubMed]
- Runnegar, M.T.; Wei, X.; Hamm-Alvarez, S.F. Increased protein phosphorylation of cytoplasmic dynein results in impaired motor function. Biochem. J. 1999, 342, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kopec, K.; Chambers, J.P. Effect of Alzheimer’s brain extracts on dynein immunoreactivity in PC12 cells. Exp. Biol. Med. 1997, 216, 429–437. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Choi, J.E.; Choi, J.M.; Kim, D.H. Dynein cleavage and microtubule accumulation in okadaic acid-treated neurons. Neurosci. Lett. 2008, 437, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.J.; Baas, P.W. Microtubules released from the neuronal centrosome are transported into the axon. J. Cell Sci. 1995, 108, 2761–2769. [Google Scholar] [PubMed]
- Scheinfeld, M.H.; Roncarati, R.; Vito, P.; Lopez, P.A.; Abdallah, M.; D’adamio, L. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer’s beta-amyloid precursor protein (APP). J. Biol. Chem. 2002, 277, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Araseki, M.; Nozawa, K.; Furukori, K.; Araki, Y.; Matsushima, T.; Nakaya, T.; Hata, S.; Saito, Y.; Uchida, S.; et al. Quantitative analysis of APP axonal transport in neurons: Role of JIP1 in enhanced APP anterograde transport. Mol. Biol. Cell 2014, 25, 3569–3580. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Gonzalez-Santiago, L.; Zarich, N.; Davalos, A.; Aranda, J.F.; Alonso, M.A.; Lasuncion, M.A.; Rojas, J.M.; Munoz, A. Plitidepsin cellular binding and Rac1/JNK pathway activation depend on membrane cholesterol content. Mol. Pharmacol. 2006, 70, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- PharmaMar announces license agreement with STA for APLIDIN® (plitidepsin) in oncology. Available online: http://www.eurekalert.org/pub_releases/2015-08/p-pal082015.php (accessed on 3 February 2016).
- Noda, T.; Suzuki, K.; Ohsumi, Y. Yeast autophagosomes: De novo formation of a membrane structure. Trends Cell Biol. 2002, 12, 231–235. [Google Scholar] [CrossRef]
- Yen, W.L.; Klionsky, D.J. How to live long and prosper: Autophagy, mitochondria, and aging. Physiology (Bethesda) 2008, 23, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.A.; Reggiori, F.; Dunn, W.A.; Klionsky, D.J. Atg23 Is Essential for the Cytoplasm to Vacuole Targeting Pathway and Efficient Autophagy but Not Pexophagy. J. Biol. Chem. 2003, 278, 48445–48452. [Google Scholar] [CrossRef] [PubMed]
- Legakis, J.E.; Yen, W.; Klionsky, D.J. A cycling protein complex required for selective autophagy. Autophagy 2007, 3, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12–Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The Reversible Modification Regulates the Membrane-Binding State of Apg8/Aut7 Essential for Autophagy and the Cytoplasm to Vacuole Targeting Pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Okamoto, K.; Mizuno, Y.; Fujita, Y. Bunina bodies in amyotrophic lateral sclerosis. Neuropathology 2008, 28, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; van der Zee, J.; Bettens, K.; Engelborghs, S.; Vandenbulcke, M.; Robberecht, C.; Dillen, L.; Merlin, C.; Geerts, N.; Graff, C.; et al. Genetic variability in SQSTM1 and risk of early-onset Alzheimer dementia: A European early-onset dementia consortium study. Neurobiol. Aging 2015, 36, e15–e22. [Google Scholar] [CrossRef] [PubMed]
- Calderilla-Barbosa, L.; Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.T.; Moscat, J.; Yan, J.; Wooten, M.W.; Wooten, M.C. Interaction of SQSTM1 with the motor protein dynein—SQSTM1 is required for normal dynein function and trafficking. J. Cell Sci. 2014, 127, 4052–4063. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kallifatidis, G.; Hoepfner, D.; Jaeg, T.; Guzman, E.A.; Wright, A.E. The marine natural product manzamine A targets vacuolar ATPases and inhibits autophagy in pancreatic cancer cells. Mar. Drugs 2013, 11, 3500–3516. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Greenwood, J.A.; Löhr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.; Yomogida, S.; Tomizawa, A.; Yamazaki, H.; Ukai, K.; Mangindaan, R.E.; Namikoshi, M.; Ishikawa, M. Papuamine causes autophagy following the reduction of cell survival through mitochondrial damage and JNK activation in MCF-7 human breast cancer cells. Int. J. Oncol. 2013, 43, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, J.A.; Banerjee, R.; Gunawardena, S. Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics. Mar. Drugs 2016, 14, 102. https://doi.org/10.3390/md14050102
White JA, Banerjee R, Gunawardena S. Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics. Marine Drugs. 2016; 14(5):102. https://doi.org/10.3390/md14050102
Chicago/Turabian StyleWhite, Joseph A., Rupkatha Banerjee, and Shermali Gunawardena. 2016. "Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics" Marine Drugs 14, no. 5: 102. https://doi.org/10.3390/md14050102
APA StyleWhite, J. A., Banerjee, R., & Gunawardena, S. (2016). Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics. Marine Drugs, 14(5), 102. https://doi.org/10.3390/md14050102