
Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt against Ethanol-Induced Liver Injury in Rats


Abstract
:1. Introduction
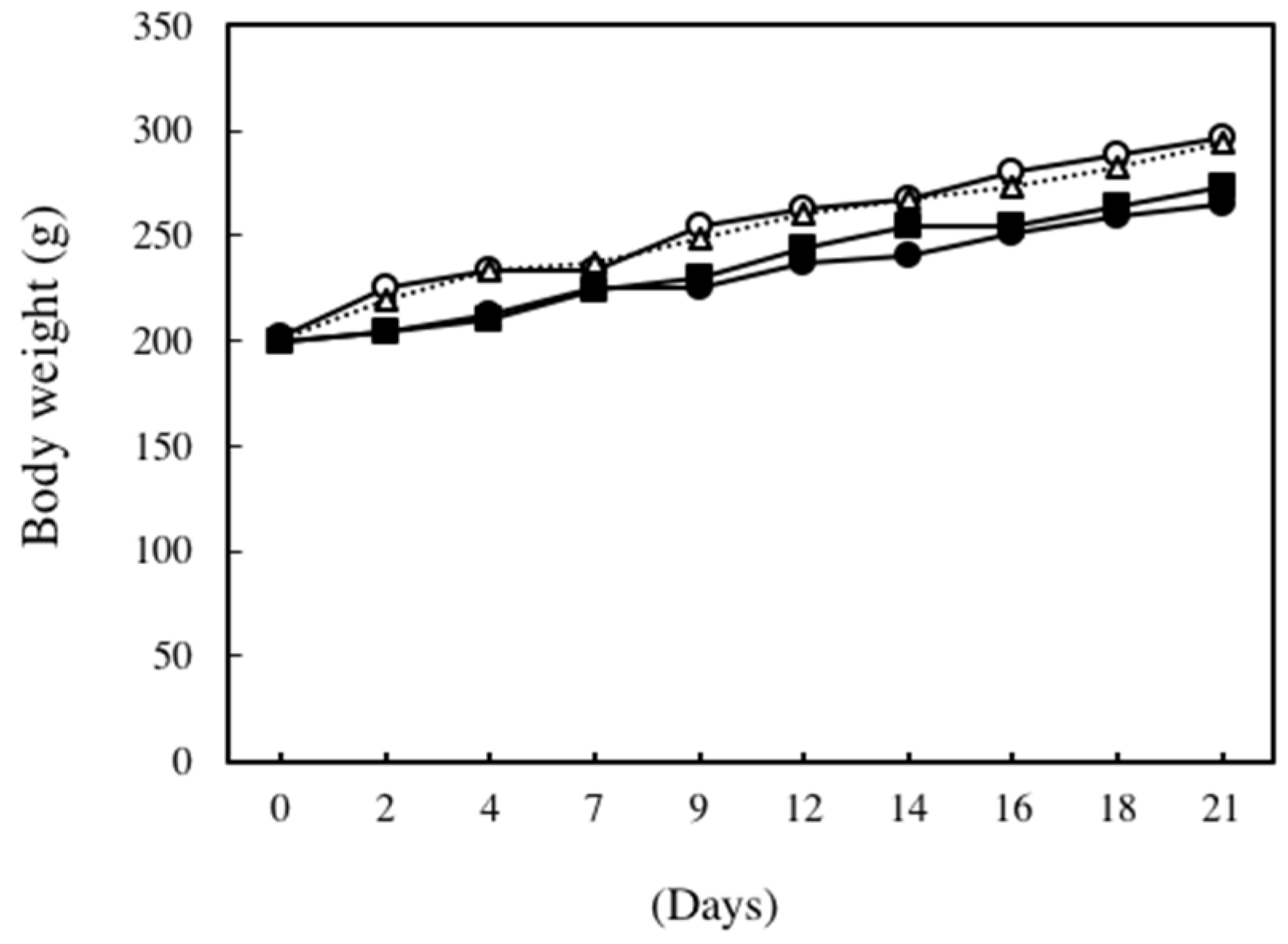
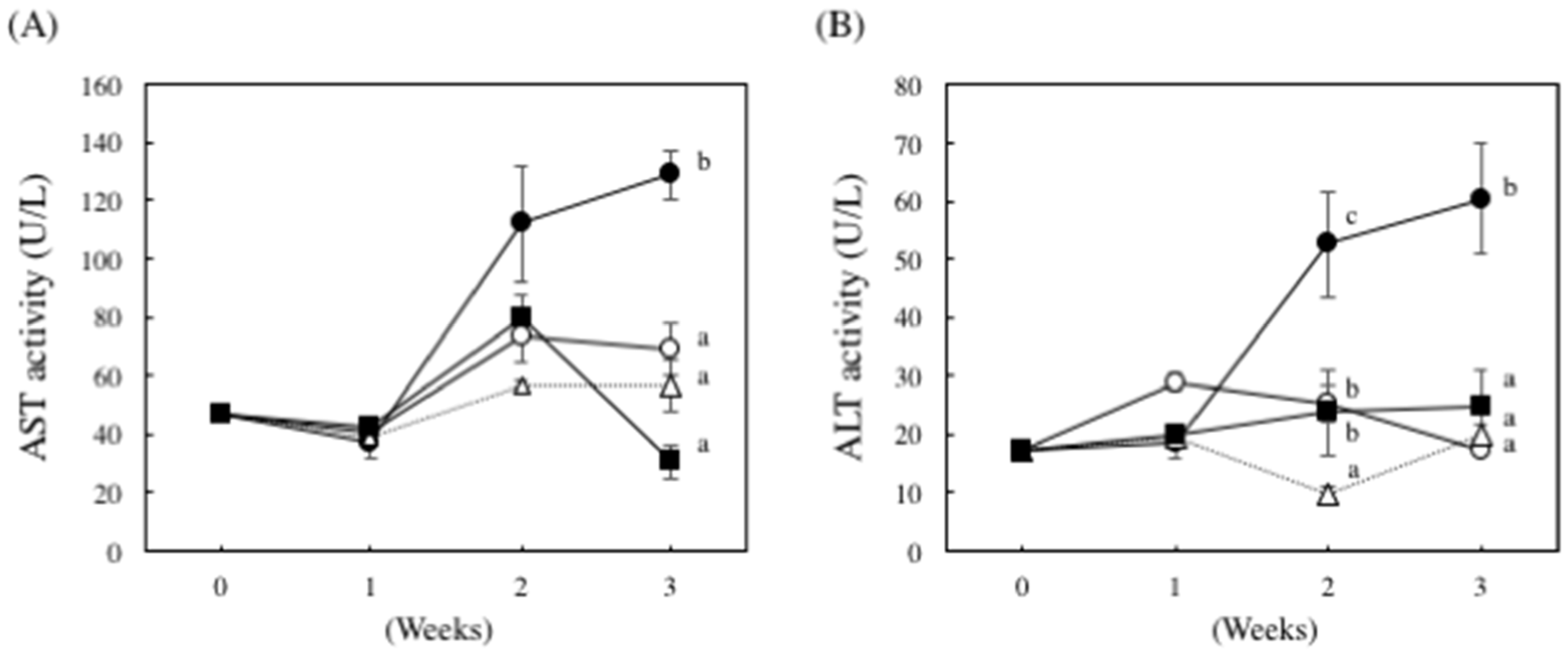
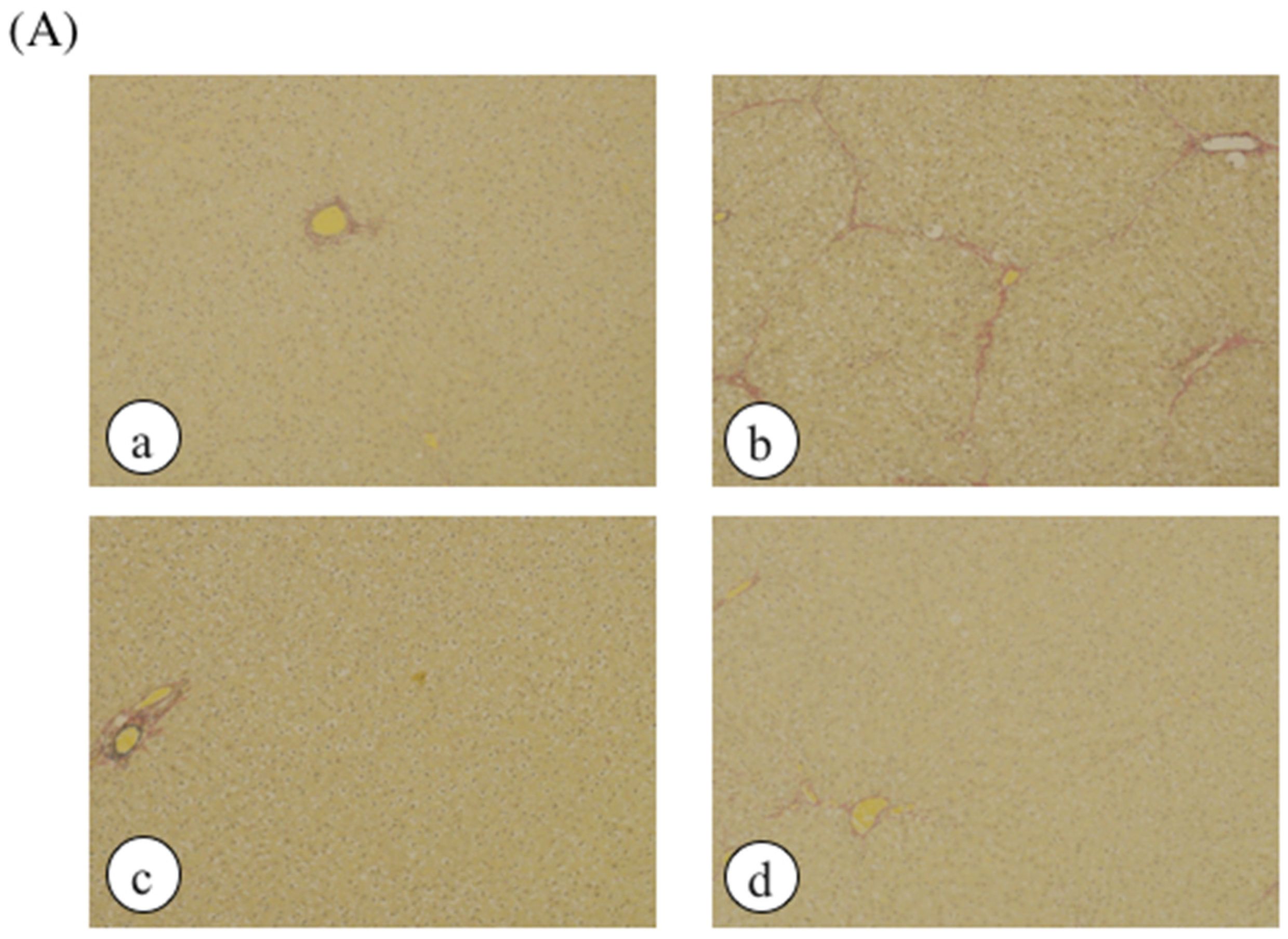
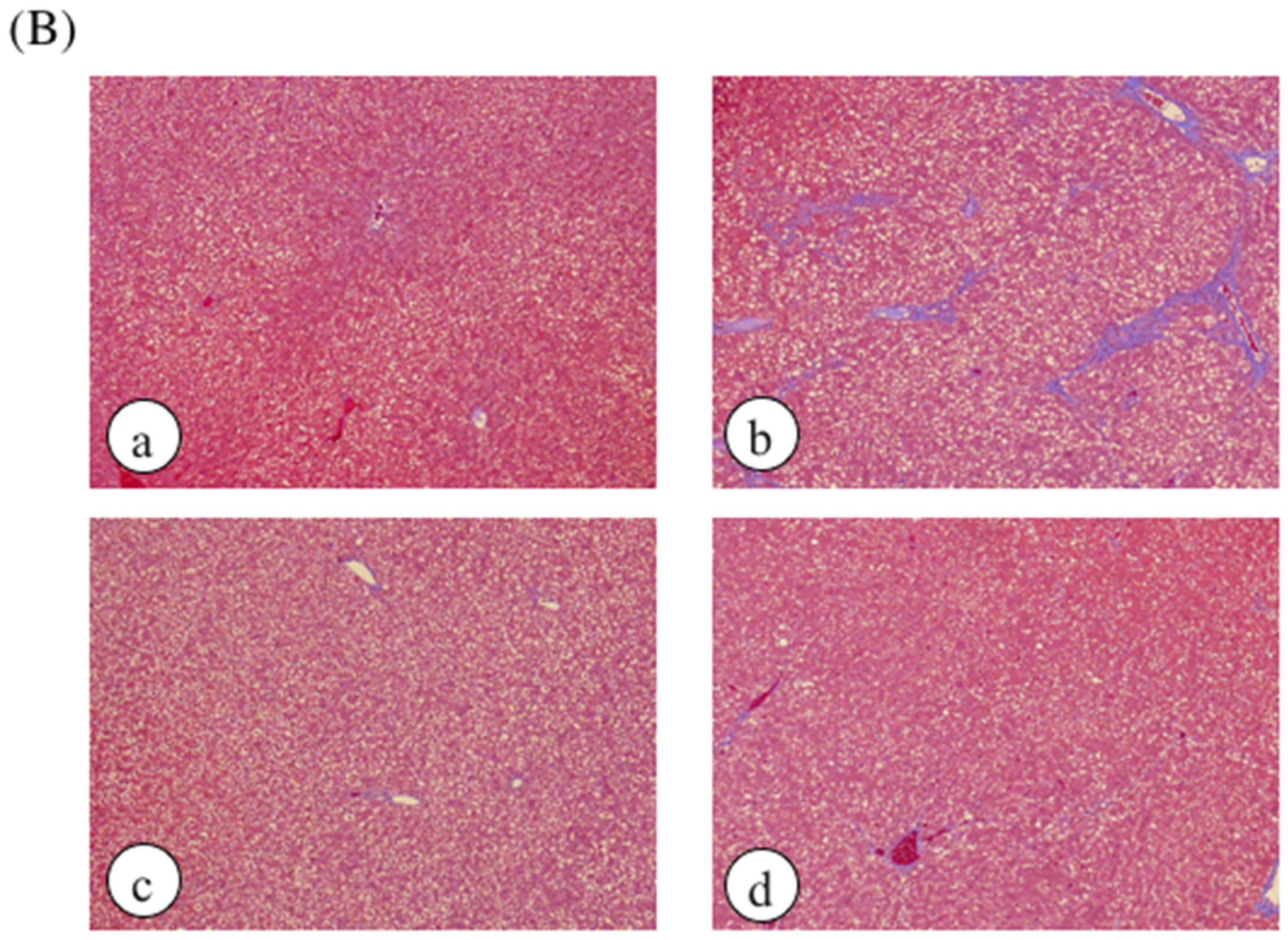
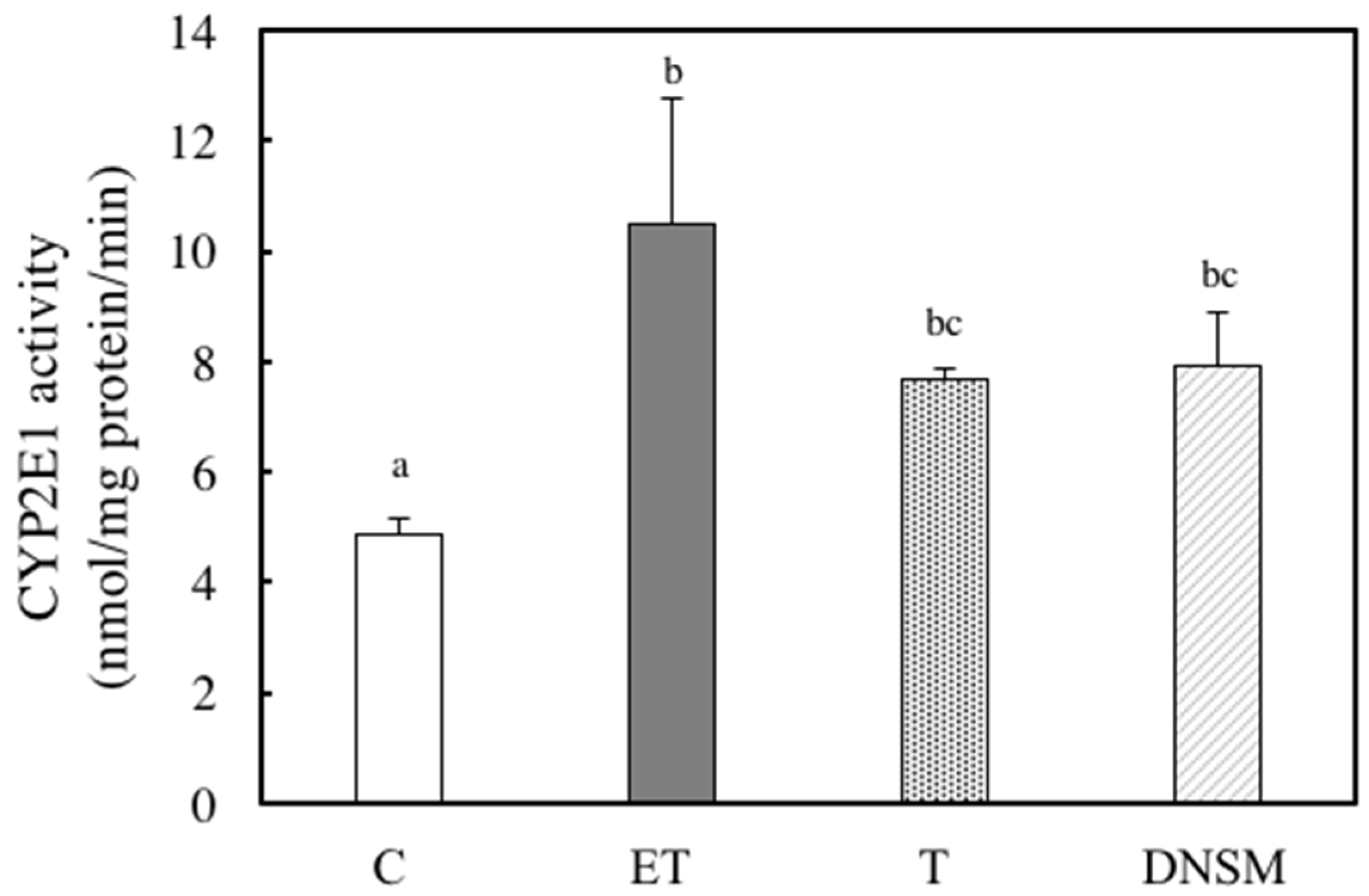
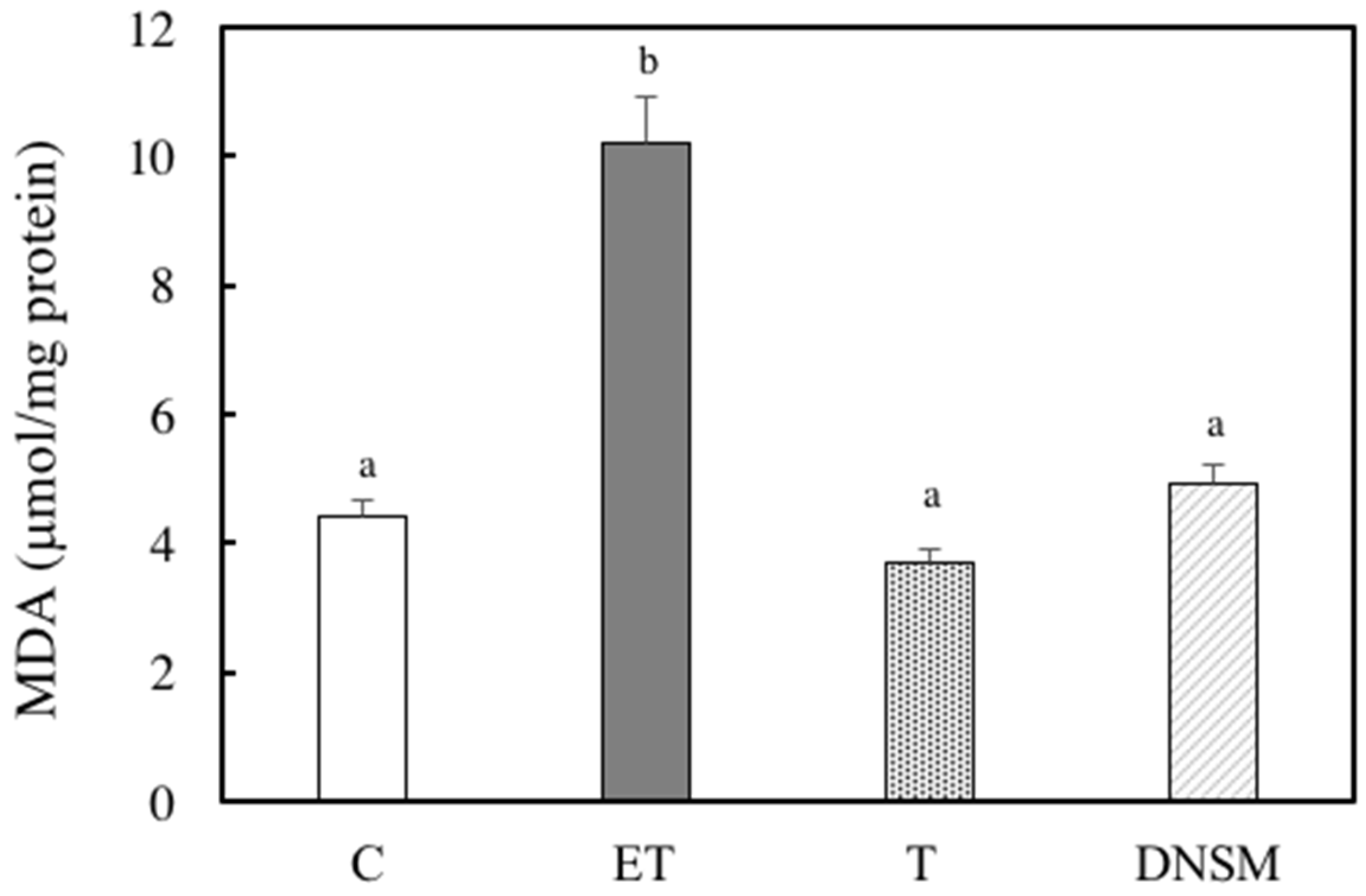
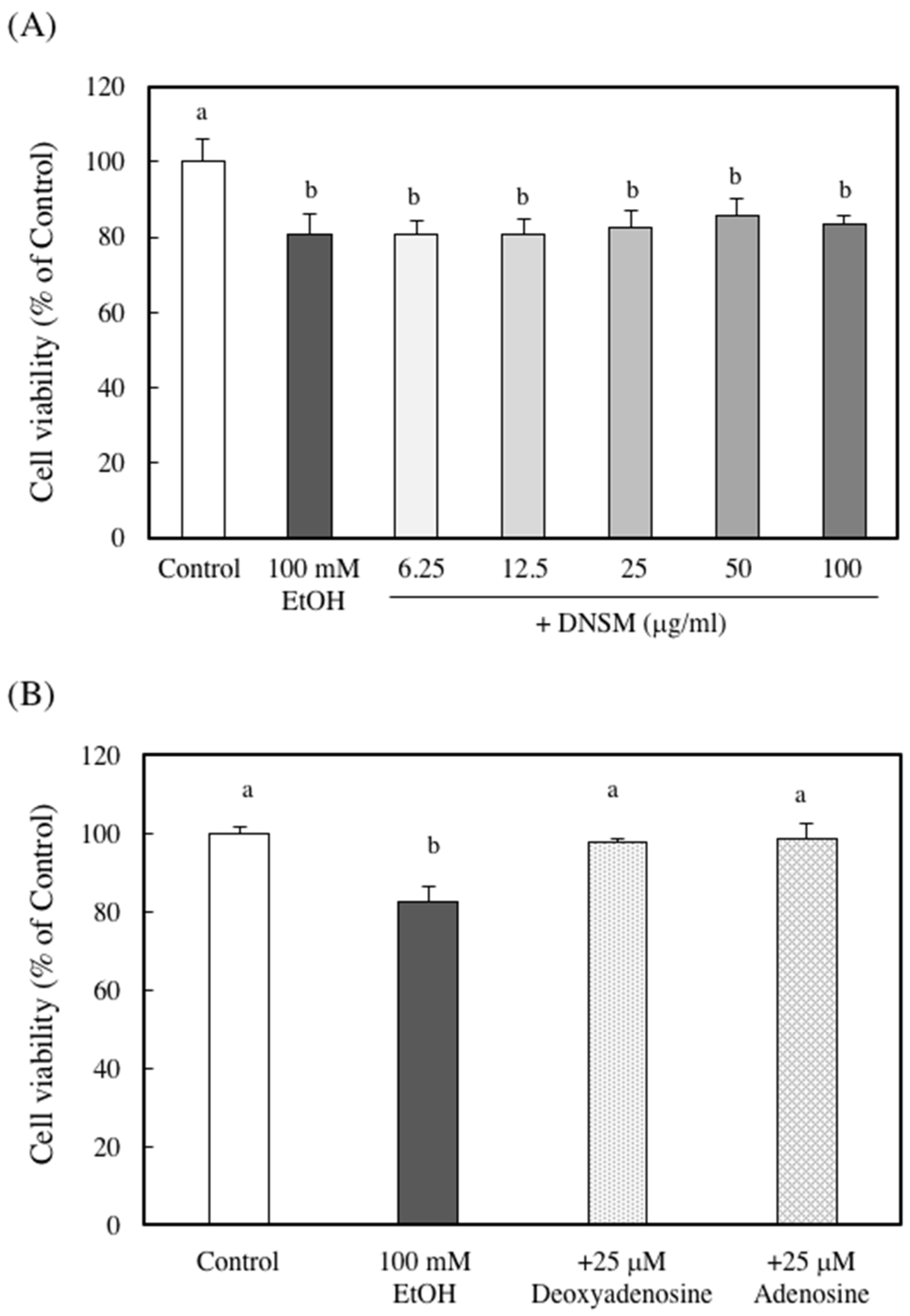
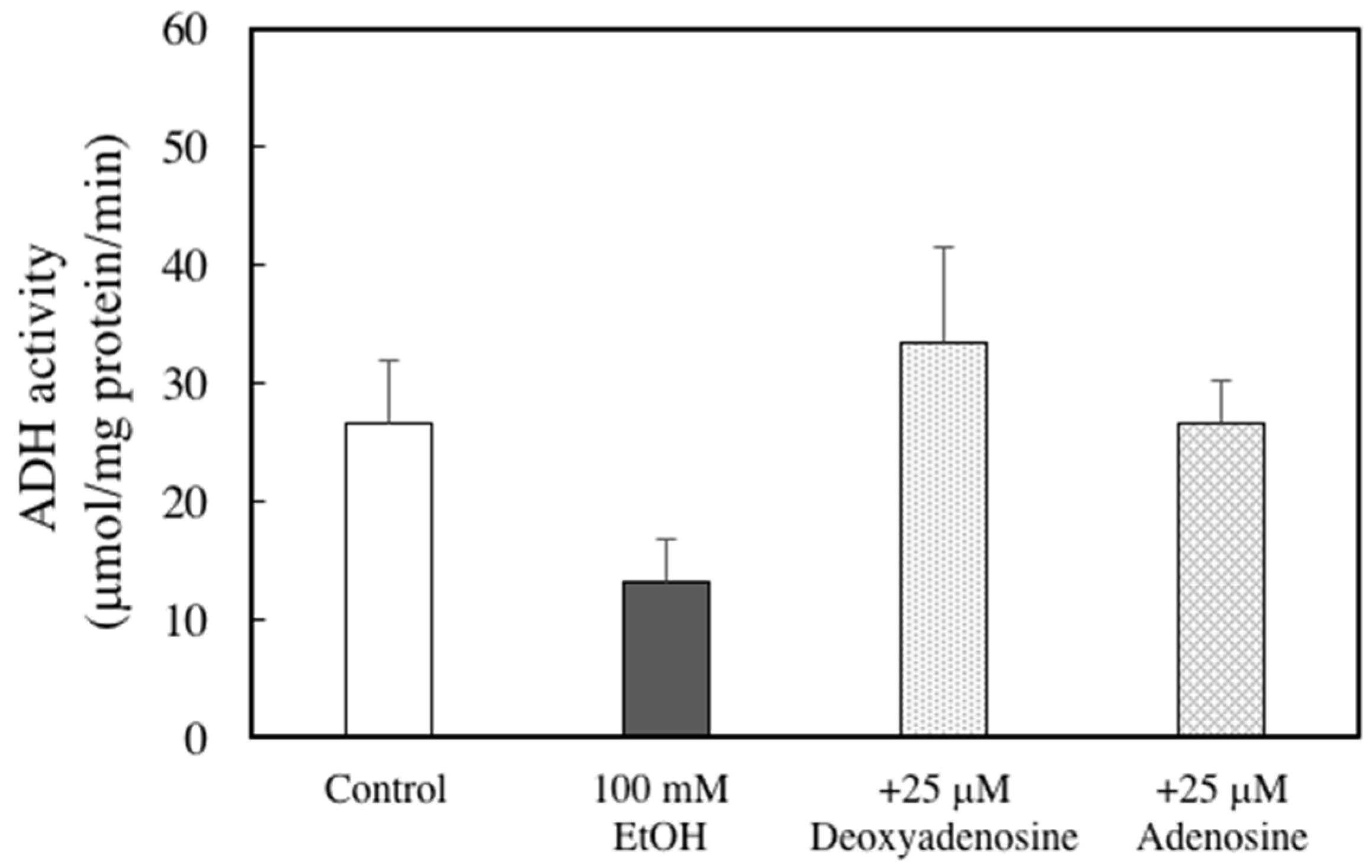
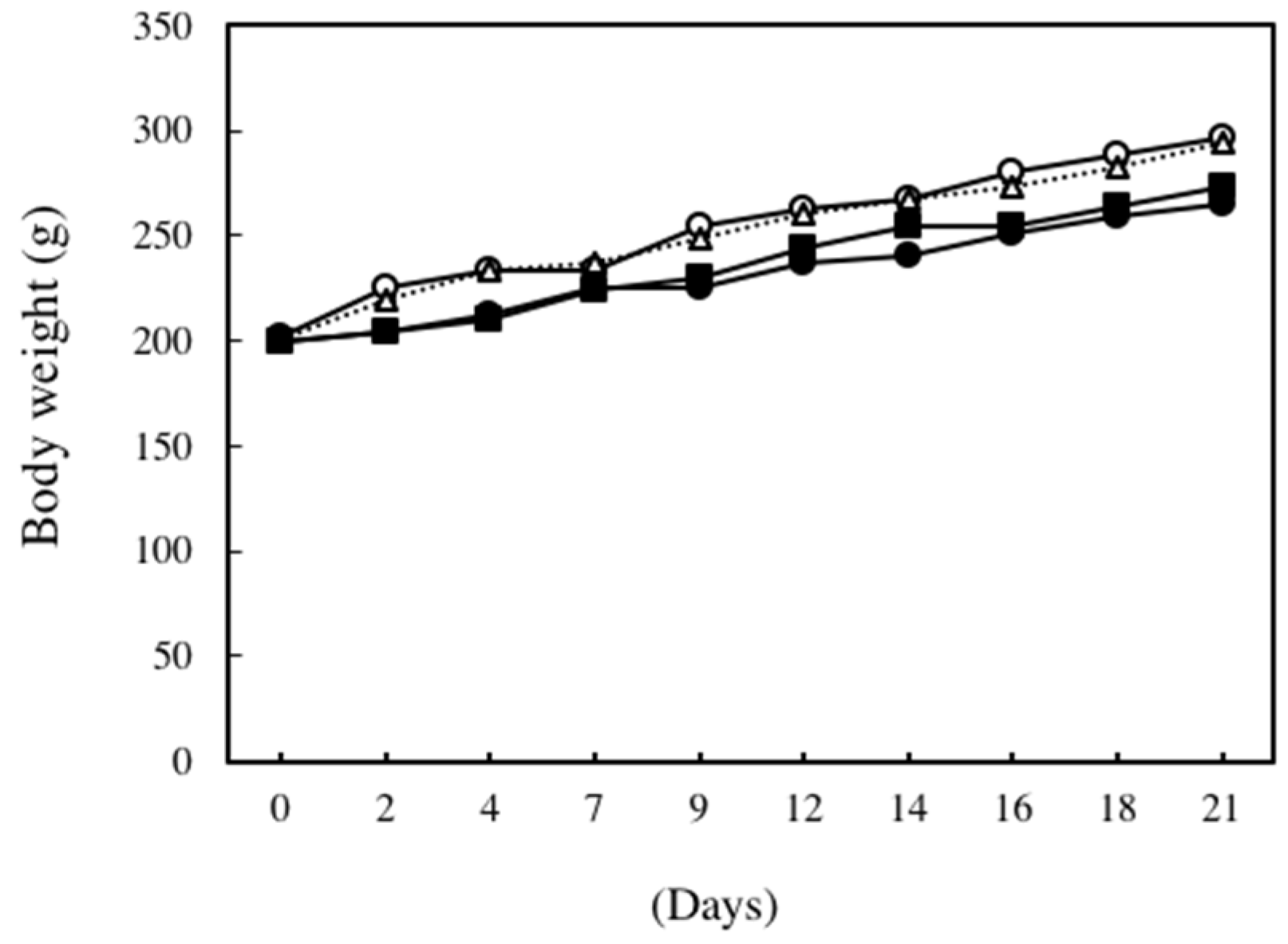
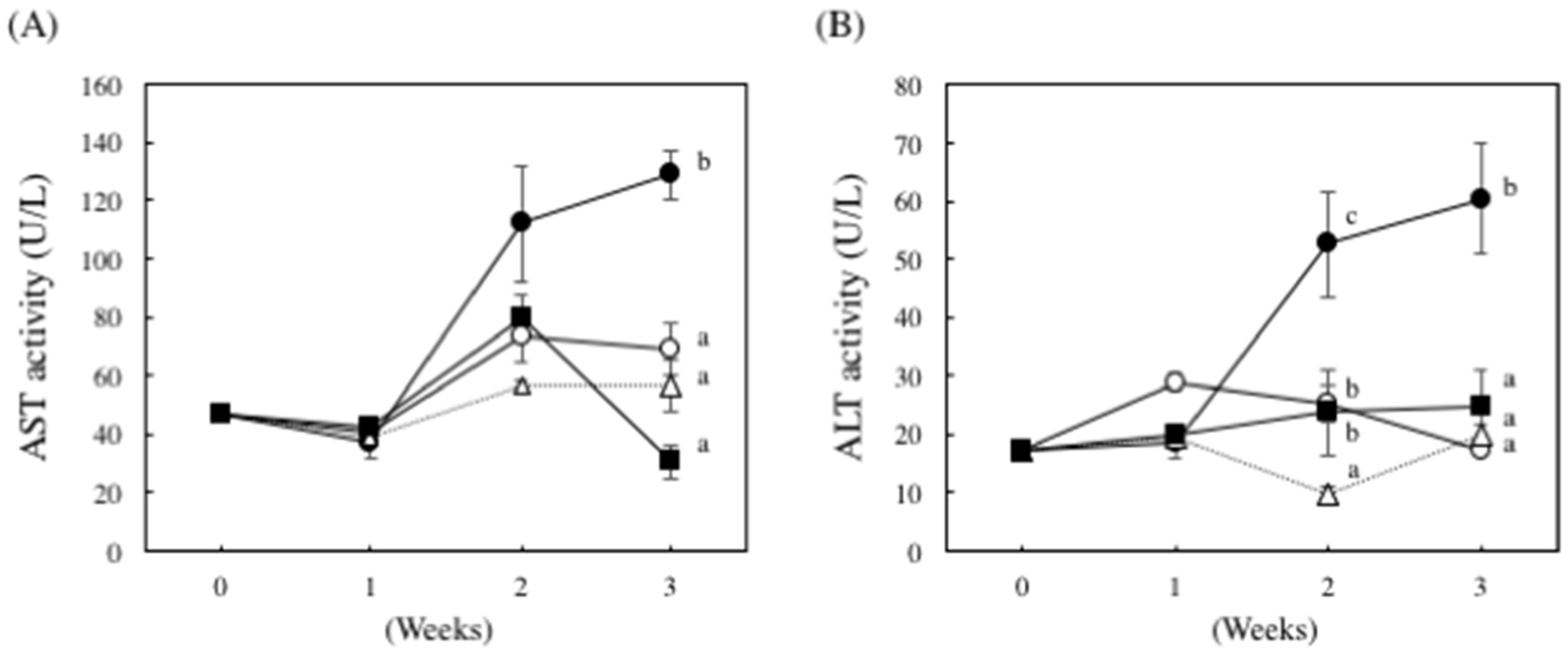
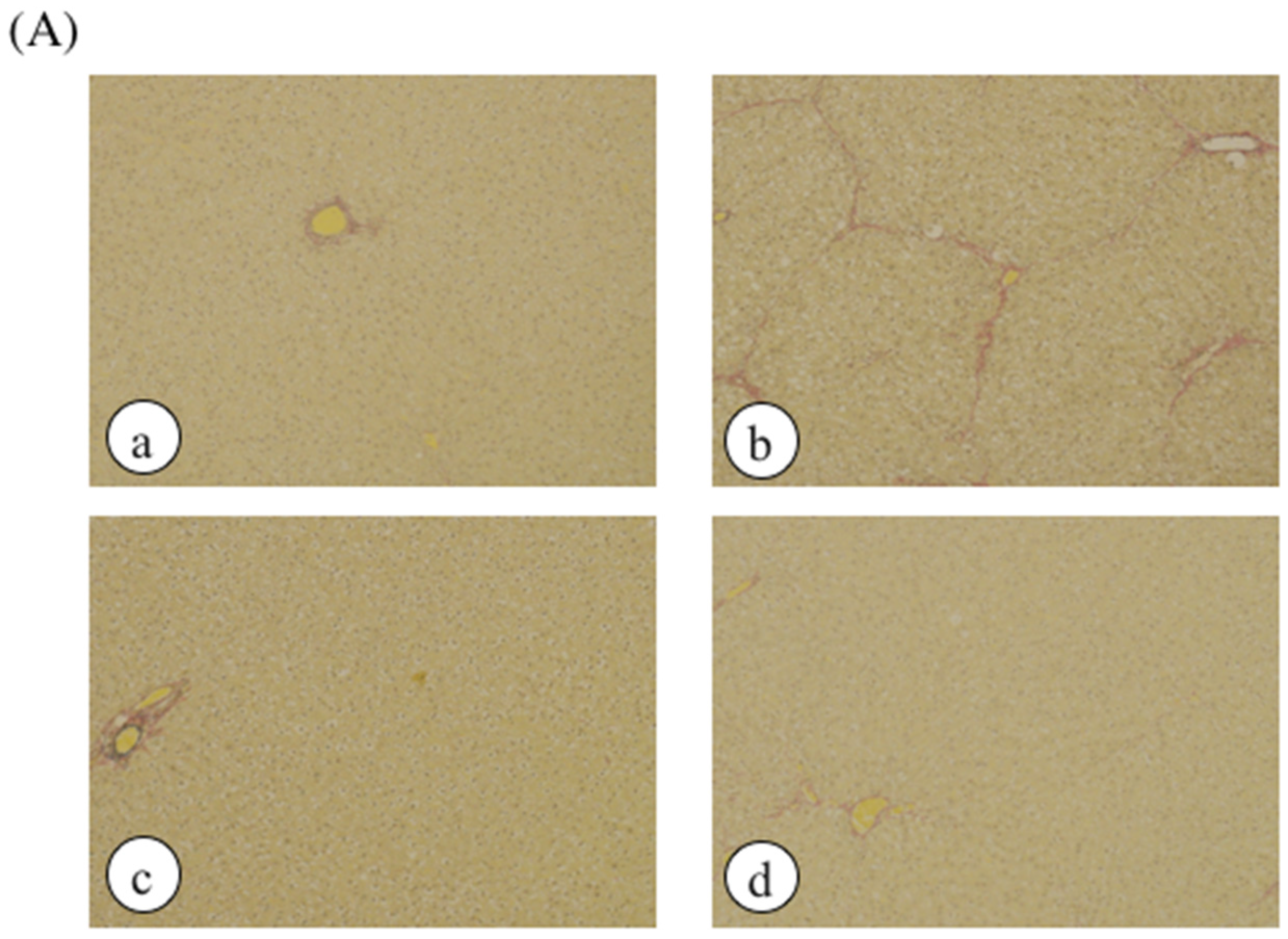
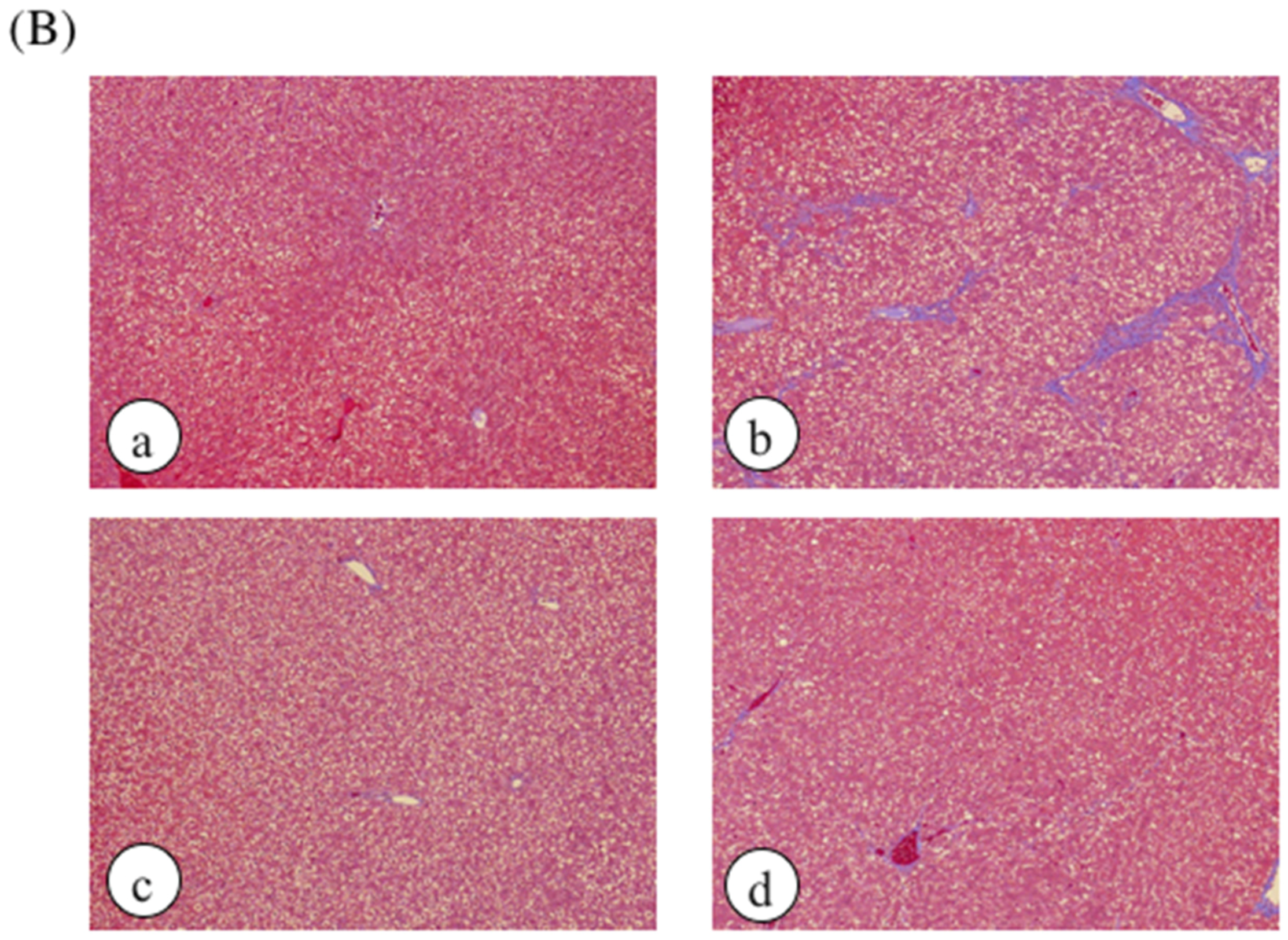
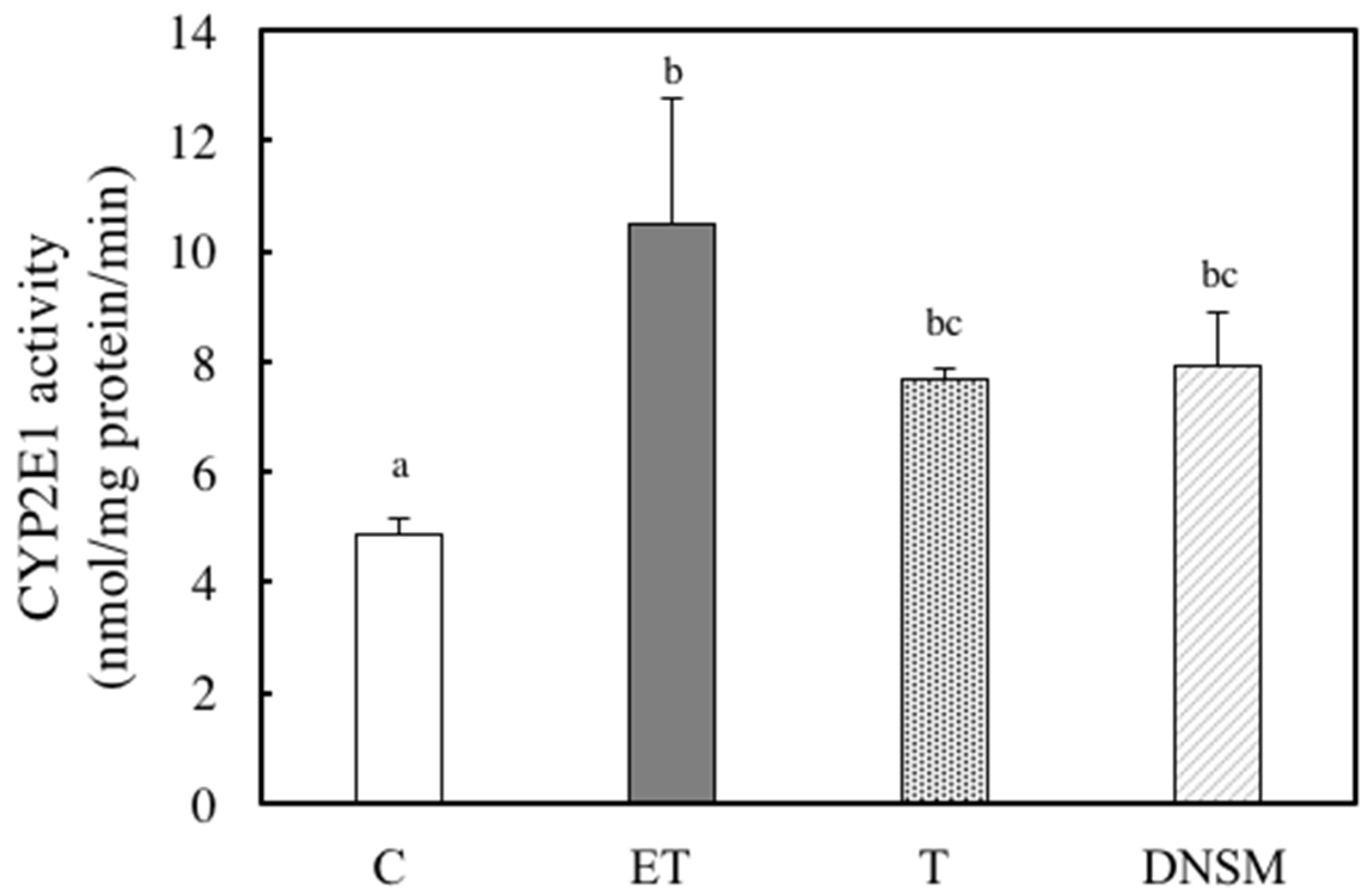
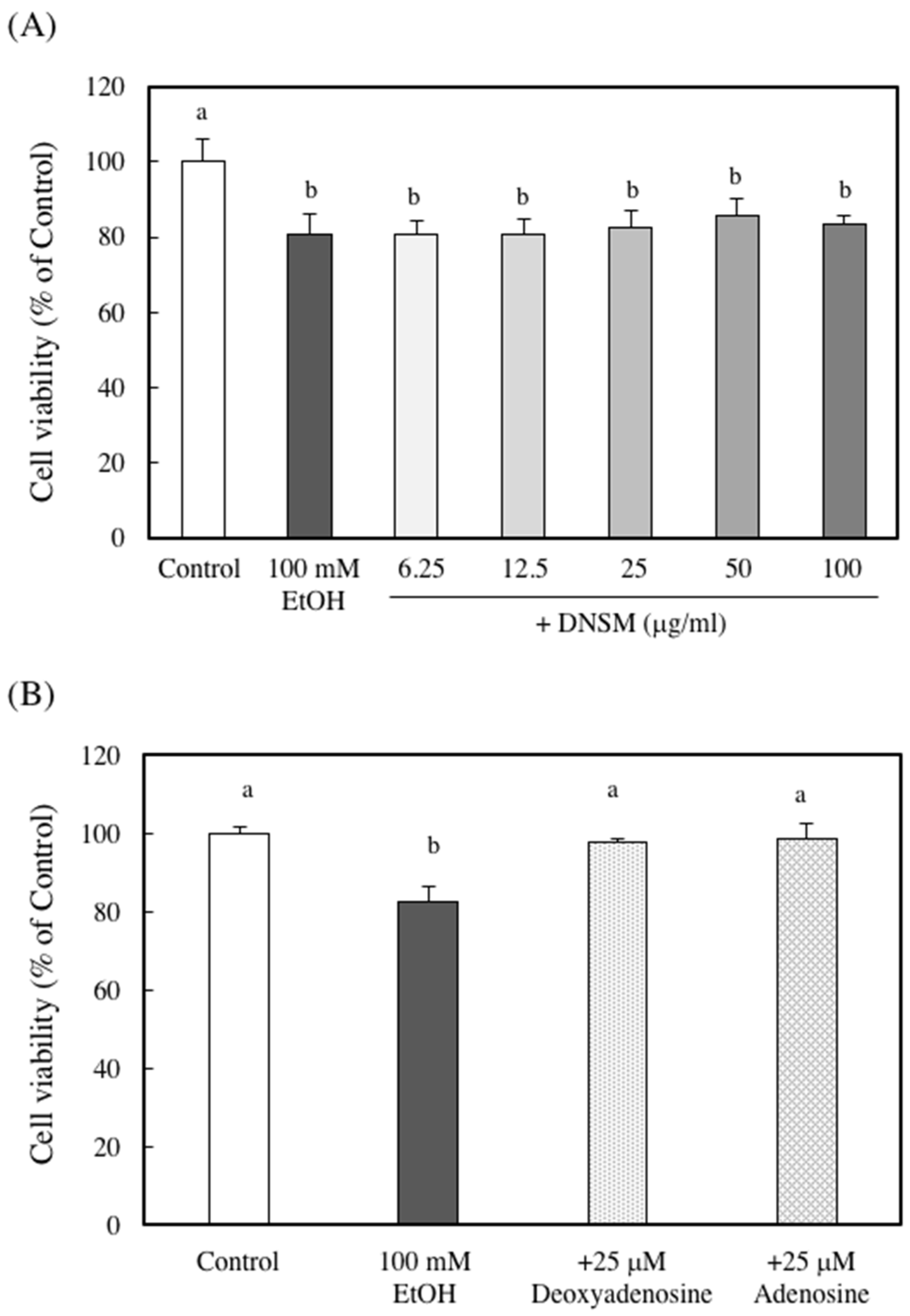
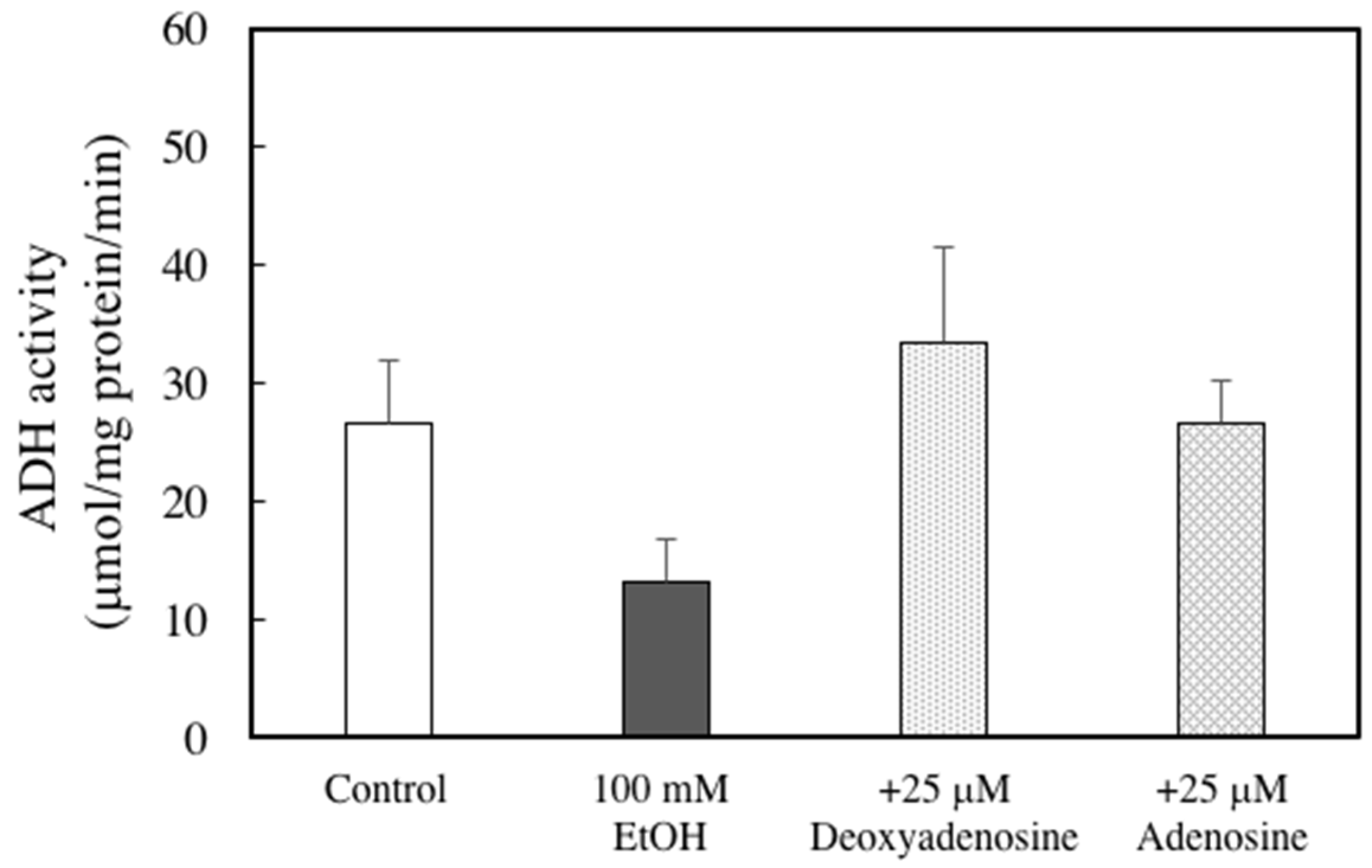
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Animal Experiments
4.4. Histological Analysis
4.5. Liver Damage Biomarkers
4.6. CYP2E1 Activity Analysis
4.7. Measurement of Lipid Peroxidation
4.8. Hepatocyte Preparation and Culture
4.9. Cell Viability Assay
4.10. Assay of ADH Activities
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta 1997, 257, 59–84. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Lu, S.C. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001, 15, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.R.; Mathurin, P.; Morgan, T.R. Alcoholic hepatitis. N. Engl. J. Med. 2009, 360, 2758–2769. [Google Scholar] [CrossRef] [PubMed]
- Zima, T.; Fialova, L.; Mestek, O.; Janebova, M.; Crikovska, J.; Malbonan, I.; Stipek, S.; Milkulikova, L.; Popov, P. Oxidative stress, metabolism of ethanol and alcohol-related disease. J. Biomed. Sci. 2001, 8, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes-adaptive increase after ethanol feeding. Science 1968, 162, 912–918. [Google Scholar] [CrossRef]
- Lieber, C.S. CYP2E1: From ASH to NASH. Hepatol. Res. 2004, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guengrich, F.P. Oxidative cleavage of carboxylic esters by cytochrome-P-450. J. Biol. Chem. 1987, 262, 8459–8462. [Google Scholar]
- Porter, T.D.; Coon, M.J. Cytochrome P-450: Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J. Biol. Chem. 1991, 266, 13469–13472. [Google Scholar] [PubMed]
- Robertson, G.; Leclercq, I.; Farrell, G.C. Nonalcoholic p-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [PubMed]
- Lieber, C.A.; De Carli, L.M.; Sorrel, M.F. Experimental methods of ethanol administration. Hepatology 1989, 10, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Horne, W.; Kamimura, S.; Niemela, O.; Parkkla, S.; Ylaherttula, S.; Brinttenham, G.M. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Investig. 1995, 96, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Tipoe, G.L.; Liong, E.C.; Casey, C.A.; Donohue, T.M., Jr.; Eagon, P.K.; So, H.; Leung, Y.M.; Fogt, F.; Nanji, A.A. A voluntary oral ethanol-feeding rat model associated with necroinflammatory liver injury. Alcohol Clin. Exp. Res. 2008, 32, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Siegers, C.P.; Pauli, V.; Korb, G.; Younes, M. Hepatoprotection by malotilate against carbon tetrachloride-alcohol-induced liver fibrosis. Agents Actions 1986, 18, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Satake, N.; Yamashita, H.; Tamura, A.; Sasaki, M.; Matsui-Yuasa, I.; Tabuchi, M.; Akahoshi, Y.; Terada, M.; Kojima-Yuasa, A. Ecklonia-cava polyphenol protects the liver against ethanol-induced injury in rats. Biochim. Biophysica. Acta 2012, 1820, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Sasaki, M.; Yamashita, H.; Matsui-Yuasa, I.; Saku, T.; Hikima, T.; Tabuchi, M.; Munakata, H.; Kojima-Yuasa, A. Yerba-mate (Ilex paraguarienesis) extract prevents ethanol-induced liver injury in rats. J. Funct. Food. 2013, 5, 1714–1723. [Google Scholar] [CrossRef]
- Carver, J.D. Dietary nucleotides: Effects on the immune and gastrointestinal systems. Acta Paediatr. Suppl. 1999, 88, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Sun, S.; Abiru, T.; Winship, T.; Kuchan, M.J. Dietary nucleotides modulate antigen-specific type 1 and type 2 T-cell responses in young c57bl/6 mice. Nutrition 2000, 16, 442–446. [Google Scholar] [CrossRef]
- Sudo, N.; Aiba, Y.; Oyama, N.; Yu, X.N.; Matsunaga, M.; Koga, Y.; Kudo, C. Dietary nucleic acid and intestinal microbiota synergistically promote a shift in the Th1/Th2 balance toward Th1-dominant immunity. Clin. Exp. Allergy 2004, 135, 132–135. [Google Scholar]
- He, Y.; Sanderson, I.R.; Walker, W.A. Uptake, transport and metabolism of exogenous nucleosides in intestinal epithelial cell culture. J. Nutr. 1994, 124, 1942–1949. [Google Scholar] [PubMed]
- Tanaka, M.; Lee, K.; Martinez-Augustin, O.; He, Y.; Sanderson, L.R.; Walker, W.A. Exogenous nucleotides after the proliferation, differentiation and apoptosis of human small intestinal epitherium. J. Nutr. 1996, 126, 424–433. [Google Scholar] [PubMed]
- Sakai, T.; Taki, T.; Nakamoto, A.; Tazaki, S.; Arakawa, M.; Nakamoto, M.; Tsutsumi, R.; Shuto, E. Dietary ribonucleic acid suppresses inflammation of adipose Tissue and improves glucose intolerance that is medicated by immune cells in C57BL/6 Mice fed a high-fat diet. J. Nutr. Sci. Vitaminol. 2015, 61, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Goto, M.; Matsui-Yuasa, I.; Kojima-Yuasa, A. Ecklonia cava polyphenol has a protective effect against ethanol-induced liver injury in a cyclic AMP-dependent manner. Mar. Drugs 2015, 13, 3877–3891. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Geró, D.; Stangl, R.; Rosero, O.; Szijártó, A.; Lotz, G.; Mohácsik, P.; Szoleczky, P.; Coletta, C.; Szabó, C. Adenisine and inosine exert cytoprotective effects in an in vitro model of liver ischemia-reperfusion injury. Int. J. Mol. Med. 2013, 31, 437–446. [Google Scholar] [PubMed]
- Hasemi, M.; Karami-Tehrani, F.; Ghavami, S.; Maddika, S. Adenosine and deoxyadenosine induced apoptosis in oestrogen receptor-positive and -negative human breast cancer cells via the intrinsic pathway. Cell Prolif. 2005, 38, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Adenosine protects Sprague Dawley rats from high-fat diet and repeated acute restraint stress-induced intestinal inflammation and altered expression of nutrient transporters. J. Anim. Physiol. Anim. Nutr. 2015, 99, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.J.; MacDougalg, O.A.; Mezey, E. Regulation of rat alcohol dehydrogenase by cyclic AMP in primary hepatocyte culture. Arch. Biochem. Biophys. 1995, 321, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Oesch-Bartlomowicz, B.; Padma, P.R.; Becker, R.; Richter, B.; Hengstler, J.G.; Freeman, J.E.; Wolf, C.R.; Oesch, F. Differential modulation of CYP2E1 activity by cAMP-dependent protein kinase upon Ser129 replacement. Exp. Cell Res. 1998, 242, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Charest, R.; Blckmore, P.F.; Exton, J.H. Characterization of responses of isolated rat hepatocytes to ATP and ADP. J. Biol. Chem. 1985, 260, 15789–15794. [Google Scholar] [PubMed]
- Okajima, F.; Tokumitsu, Y.; Kondo, Y.; Ui, M. P2-Purinergic receptors are coupled to two signal transduction systems leading to inhibition of cAMP generation and to production of inositol trisphosphate in rat hepatocytes. J. Biol. Chem. 1987, 262, 13483–13490. [Google Scholar] [PubMed]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem. Pharmacol. 2013, 86, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Park, J.; Li, Y.; Min, K.N.; Kong, G.; Hur, G.M.; Kim, J.M.; Shong, M.; Jung, M.-S.; Park, J.K.; et al. β-Lapachone alleviates alcoholic fatty liver disease in rats. Cell. Signal. 2014, 26, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Su, B.; Fan, S.; Fei, H.; Zhao, W. Protective effect of oligomeric proanthocyanidins against alcohol-induced liver steatosis and injury in mice. Biochem. Biophys. Res. Commun. 2015, 458, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Stenesen, D.; Suh, J.M.; Seo, J.; Yu, K.; Lee, K.-S.; Kim, J.-S.; Min, K.-J.; Graff, J.M. Dietary adenine controls adult lifespan via adenosine nucleotide biosynthesis and AMPK, and regulates the longevity benefit of caloric restriction. Cell Metab. 2013, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; Affatato, A.; Canistro, D.; Broccoli, M.; Trespidi, S.; Pozzetti, L.; Biagi, G.L.; Cantelli-Forti, G.; Paoline, M. Induction and suppression of cytochrome P450 isoenzymes and generation of oxygen radicals by procymidone in liver, kidney and lung of CD1 mice. Mutat. Res. 2003, 527, 67–80. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohnishi, N.; Yagi, K. Assay for lipidperoxide in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Moldéus, P.; Högber, J.; Orrenius, S. Isolation and use of liver cells. Methods Enzymol. 1978, 52, 60–71. [Google Scholar] [PubMed]
- Zhang, S.Z.; Lipsky, M.M.; Trump, B.F.; Hsu, I.C. Neutral red (NR) assay for cell viability and xenobiotic-induced cytotoxicity in primary culture of human and rat hepatocytes. Cell Biol. Toxicol. 1990, 6, 219–234. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Organ Weight (g) | ||||
|---|---|---|---|---|---|
| Liver | Kidney | Spleen | Visceral Fat | Epididymal Fat | |
| C | 10.82 ± 0.38 | 1.60 ± 0.03 | 0.76 ± 0.04 | 6.52 ± 0.55 | 7.49 ± 0.61 |
| ET | 10.54 ± 0.46 | 1.58 ± 0.05 | 0.75 ± 0.03 | 4.54 ± 0.48 | 4.74 ± 0.37 |
| T | 12.35 ± 0.47 | 1.70 ± 0.08 | 0.72 ± 0.02 | 6.22 ± 0.52 | 6.02 ± 0.20 |
| DNSM | 10.76 ± 0.34 | 1.66 ± 0.02 | 0.70 ± 0.03 | 4.79 ± 0.52 | 5.44 ± 0.52 |
| Nucleotides * | Amount (g/100 g) |
|---|---|
| 5′-dCMP | 6.01 |
| 5′-dAMP | 9.15 |
| 5′-dTMP | 9.26 |
| 5′-dGMP | 6.93 |
| Total | 31.35 |
| Amino Acids | Amount (g/100 g) |
| Arg | 17.80 |
| Lys | 2.66 |
| His | 0.65 |
| Phe | 0.89 |
| Tyr | 0.88 |
| Leu | 1.95 |
| Ile | 1.25 |
| Met | 0.60 |
| Val | 2.12 |
| Ala | 1.95 |
| Gly | 4.11 |
| Pro | 2.62 |
| Glu | 3.48 |
| Ser | 2.49 |
| Thr | 1.27 |
| Asp | 2.24 |
| Trp | 0.20 |
| Cys | 0.25 |
| Total | 47.41 |
| Components (g) | Control | 0.12% DNSM |
|---|---|---|
| Casein | 200 | 200 |
| l-Cystine | 3 | 3 |
| Cornstarch | 397.486 | 396.286 |
| α-Cornstarch | 132 | 132 |
| Sucrose | 100 | 100 |
| Soybean oil | 70 | 70 |
| Cellulose powder | 50 | 50 |
| Mineral mix (AIN-93G-MX) 1 | 35 | 35 |
| Vitamin mix (AIN-93VX) 2 | 10 | 10 |
| Choline hydrogen tartrate | 2.5 | 2.5 |
| t-Butylhydroquinone | 0.014 | 0.014 |
| DNSM | 0 | 1.2 |
| Total | 1000 | 1000 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojima-Yuasa, A.; Goto, M.; Yoshikawa, E.; Morita, Y.; Sekiguchi, H.; Sutoh, K.; Usumi, K.; Matsui-Yuasa, I. Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt against Ethanol-Induced Liver Injury in Rats. Mar. Drugs 2016, 14, 232. https://doi.org/10.3390/md14120232
Kojima-Yuasa A, Goto M, Yoshikawa E, Morita Y, Sekiguchi H, Sutoh K, Usumi K, Matsui-Yuasa I. Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt against Ethanol-Induced Liver Injury in Rats. Marine Drugs. 2016; 14(12):232. https://doi.org/10.3390/md14120232
Chicago/Turabian StyleKojima-Yuasa, Akiko, Mayu Goto, Eri Yoshikawa, Yuri Morita, Hirotaka Sekiguchi, Keita Sutoh, Koji Usumi, and Isao Matsui-Yuasa. 2016. "Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt against Ethanol-Induced Liver Injury in Rats" Marine Drugs 14, no. 12: 232. https://doi.org/10.3390/md14120232
APA StyleKojima-Yuasa, A., Goto, M., Yoshikawa, E., Morita, Y., Sekiguchi, H., Sutoh, K., Usumi, K., & Matsui-Yuasa, I. (2016). Protective Effects of Hydrolyzed Nucleoproteins from Salmon Milt against Ethanol-Induced Liver Injury in Rats. Marine Drugs, 14(12), 232. https://doi.org/10.3390/md14120232