
First Total Synthesis and Biological Screening of a Proline-Rich Cyclopeptide from a Caribbean Marine Sponge


Abstract
:1. Introduction
2. Results
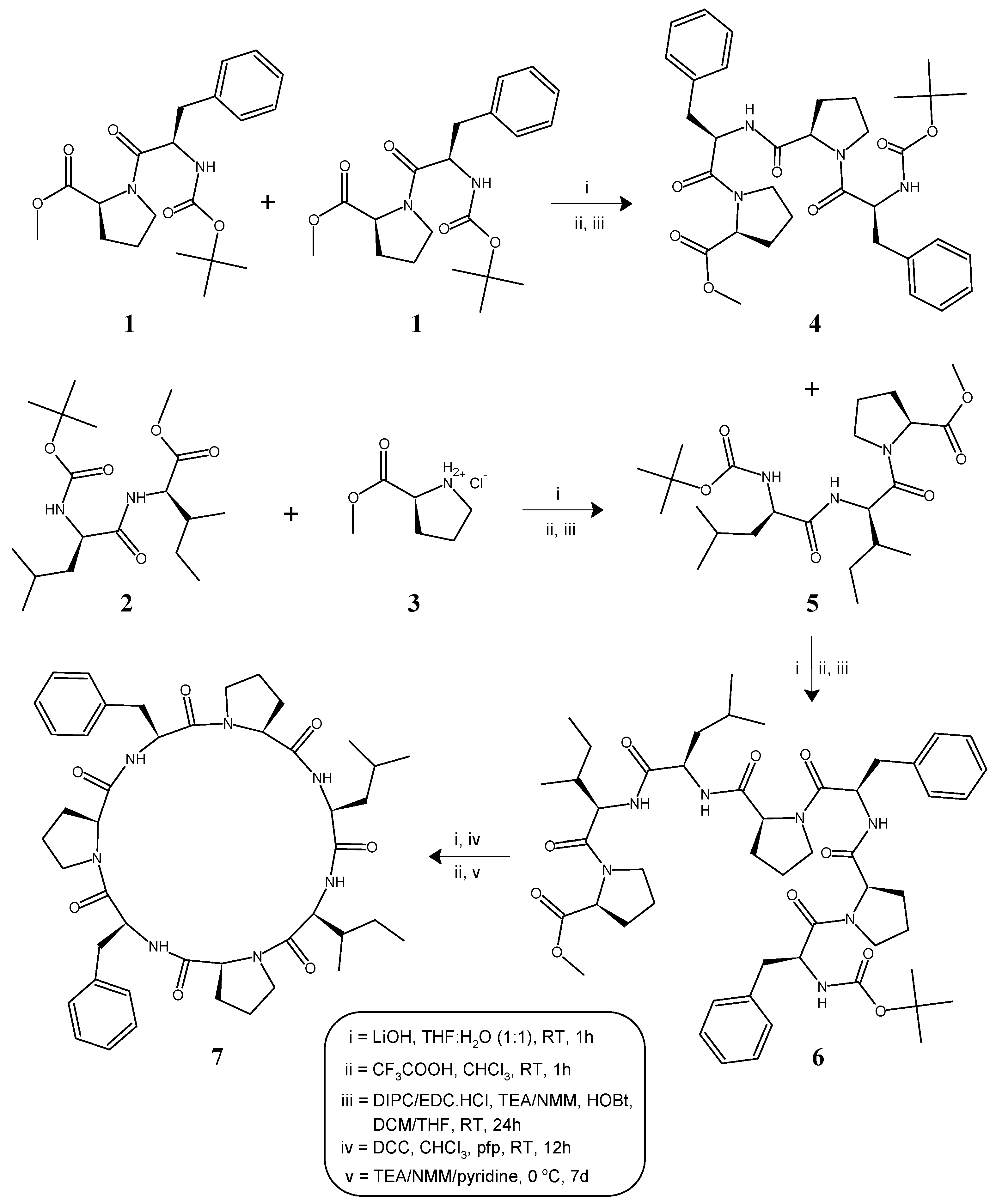
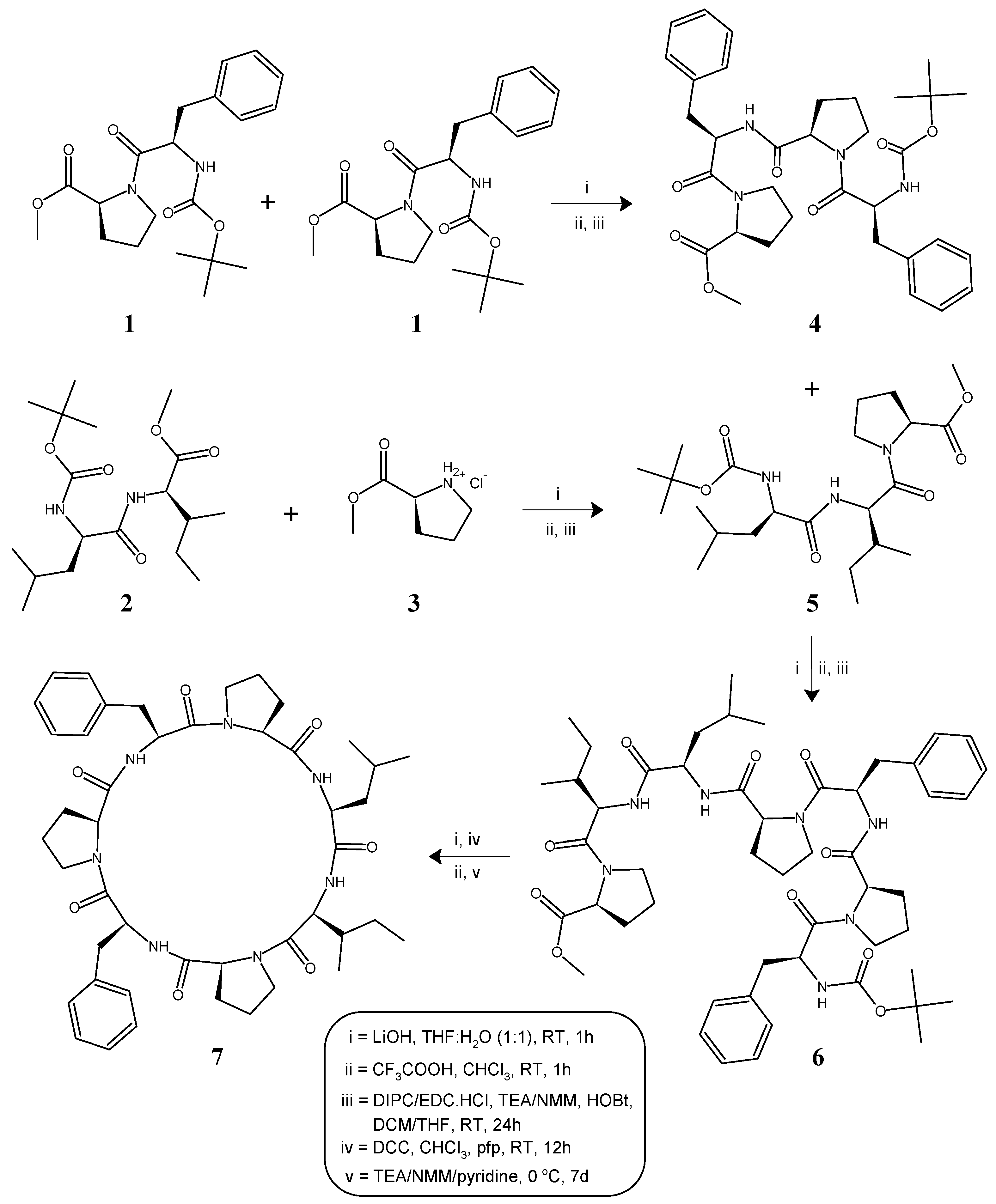
2.1. Synthesis
2.2. Pharmacology
3. Discussion
4. Materials and Methods
4.1. General Procedure for the Synthesis of Linear Tetra/Tripeptide Segments (4, 5)
4.2. Deprotection of the Tetrapeptide Unit (4) at the Amino Terminal
4.3. Deprotection of the Tripeptide Unit (5) at the Carboxyl Terminal
4.4. Procedure for the Synthesis of Linear Heptapeptide Unit and Its Cyclized form (6, 7)
4.5. Biological Evaluation
4.5.1. Anthelmintic Screening
4.5.2. Antibacterial Screening
4.5.3. Antifungal Screening
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Noro, J.C.; Kalaitzis, J.A.; Neilan, B.A. Bioactive natural products from Papua New Guinea marine sponges. Chem. Biodivers. 2012, 9, 2077–2095. [Google Scholar] [CrossRef] [PubMed]
- Proksch, P.; Ebel, R.; Edrada, R.A.; Wray, V.; Steube, K. Bioactive natural products from marine invertebrates and associated fungi. Prog. Mol. Subcell. Biol. 2003, 37, 117–142. [Google Scholar] [PubMed]
- Andavan, G.S.; Lemmens-Gruber, R. Cyclodepsipeptides from marine sponges: Natural agents for drug research. Mar. Drugs 2010, 8, 810–834. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Dahiya, R. Cyclic peptides as novel antineoplastic agents: A review. J. Sci. Pharm. 2003, 4, 125–131. [Google Scholar]
- Dahiya, R.; Pathak, D.; Himaja, M.; Bhatt, S. First total synthesis and biological screening of hymenamide E. Acta Pharm. 2006, 56, 399–415. [Google Scholar] [PubMed]
- Dahiya, R.; Gautam, H. Synthesis and pharmacological studies on a cyclooligopeptide from marine bacteria. Chin. J. Chem. 2011, 29, 1911–1916. [Google Scholar]
- Daletos, G.; Kalscheuer, R.; Koliwer-Brandl, H.; Hartmann, R.; de Voogd, N.J.; Wray, V.; Lin, W.; Proksch, P. Callyaerins from the marine sponge Callyspongia aerizusa: Cyclic peptides with antitubercular activity. J. Nat. Prod. 2015, 78, 1910–1925. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Gustafson, K.R.; Boswell, J.L.; Boyd, M.R. Haligramides A and B, two new cytotoxic hexapeptides from the marine sponge Haliclona nigra. J. Nat. Prod. 2000, 63, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; De Marino, S.; Sepe, V.; D’Auria, M.V.; Bifulco, G.; Débitus, C.; Bucci, M.; Vellecco, V.; Zampella, A. Solomonamides A and B, new anti-inflammatory peptides from Theonella swinhoei. Org. Lett. 2011, 13, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, A.; Bifulco, G.; Giannini, C.; Bucci, M.; Debitus, C.; Cirino, G.; Gomez-Paloma, L. Halipeptins A and B: Two novel potent anti-inflammatory cyclic depsipeptides from the Vanuatu marine sponge Haliclona species. J. Am. Chem. Soc. 2001, 123, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Gustafson, K.R.; Cartner, L.K.; Shigematsu, N.; Pannell, L.K.; Boyd, M.R. Microspinosamide, a new HIV-inhibitory cyclic depsipeptide from the marine sponge Sidonops microspinosa. J. Nat. Prod. 2001, 64, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Gustafson, K.R.; Cartner, L.K.; Wilson, J.A.; Shigematsu, N.; Hess, S.; Pannell, L.K.; Boyd, M.R.; McMahon, J.B. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod. 2004, 67, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, K.; Takada, K.; Okada, S.; Matsunaga, S. Nazumazoles D-F, cyclic pentapeptides that inhibit chymotrypsin, from the marine sponge Theonella swinhoei. J. Nat. Prod. 2016, 79, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Pathak, D. Cyclic peptides: New hope for antifungal therapy. Egypt. Pharm. J. (NRC) 2006, 5, 189–199. [Google Scholar]
- Schmidt, E.W.; Bewley, C.A.; Faulkner, D.J. Theopalauamide, a bicyclic glycopeptide from filamentous bacterial symbionts of the lithistid sponge Theonella swinhoei from Palau and Mozambique. J. Org. Chem. 1998, 63, 1254–1258. [Google Scholar] [CrossRef]
- Bose, U.; Hodson, M.P.; Shaw, P.N.; Fuerst, J.A.; Hewavitharana, A.K. Two peptides, cycloaspeptide A and nazumamide A from a sponge associated marine actinobacterium Salinispora sp. Nat. Prod. Commun. 2014, 9, 545–546. [Google Scholar] [PubMed]
- Mitova, M.; Popov, S.; De Rosa, S. Cyclic peptides from a Ruegeria strain of bacteria associated with the sponge Suberites domuncula. J. Nat. Prod. 2004, 67, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.; Fay, P. Nitrogen fixation in coral reef sponges with symbiotic cyanobacteria. Nature 1979, 79, 527–529. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Morinaka, B.I.; Molinski, T.F. Structures and solution conformational dynamics of stylissamides G and H from the Bahamian sponge Stylissa caribica. J. Nat. Prod. 2014, 77, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Min, C.C.; Teuscher, F.; Ebel, R.; Kakoschke, C.; Lin, W.; Wray, V.; Edrada-Ebel, R.; Proksch, P. Callyaerins A-F and H, new cytotoxic cyclic peptides from the Indonesian marine sponge Callyspongia aerizusa. Bioorg. Med. Chem. 2010, 18, 4947–4956. [Google Scholar] [CrossRef] [PubMed]
- Whitson, E.L.; Ratnayake, A.S.; Bugni, T.S.; Harper, M.K.; Treland, C.M. Isolation, structure elucidation and synthesis of eudistomides A and B, lipopeptides from a fijian ascidian Eudistoma sp. J. Org. Chem. 2009, 74, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Goeger, D.; Maier, C.S.; Gerwick, W.H. The Wewakpeptins, cyclic depsipeptides from a papua new guinea collection of the marine cyanobacterium Lyngbya semiplena. J. Org. Chem. 2005, 70, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R. Cyclopolypeptides with antifungal interest. Coll. Pharm. Commun. 2013, 1, 1–15. [Google Scholar]
- Fang, W.Y.; Dahiya, R.; Qin, H.L.; Mourya, R.; Maharaj, S. Natural proline-rich cyclopolypeptides from marine organisms: Chemistry, synthetic methodologies and biological status. Mar. Drugs 2016, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Benelkebir, H. Synthesis of Cyclic Peptide Natural Products and Inhibitors of Histone Modifying Enzymes. Ph.D. Thesis, School of Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Southampton, UK, 2011; pp. 57–60. [Google Scholar]
- Arai, M.; Yamano, Y.; Fujita, M.; Setiawan, A.; Kobayashi, M. Stylissamide X, a new proline-rich cyclic octapeptide as an inhibitor of cell migration, from an Indonesian marine sponge of Stylissa sp. Bioorg. Med. Chem. Lett. 2012, 22, 1818–1821. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Pathak, D. Synthetic studies on a natural cyclic tetrapeptide—Halolitoralin C. J. Pharm. Res. 2006, 5, 69–73. [Google Scholar]
- Dahiya, R.; Pathak, D. Synthesis, characterization and biological evaluation of halolitoralin B—A natural cyclic peptide. Asian J. Chem. 2007, 19, 1499–1505. [Google Scholar]
- Dahiya, R.; Pathak, D. First total synthesis and biological evaluation of halolitoralin A. J. Serb. Chem. Soc. 2007, 72, 101–107. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis, characterization and biological evaluation of a glycine-rich peptide—Cherimolacyclopeptide E. J. Chil. Chem. Soc. 2007, 52, 1224–1229. [Google Scholar] [CrossRef]
- Dahiya, R.; Kaur, K. Synthetic and biological studies on natural cyclic heptapeptide: Segetalin E. Arch. Pharm. Res. 2007, 30, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R. Synthesis of a phenylalanine-rich peptide as potential anthelmintic and cytotoxic agent. Acta Pol. Pharm. 2007, 64, 509–516. [Google Scholar] [PubMed]
- Dahiya, R. Synthetic and pharmacological studies on longicalycinin A. Pak. J. Pharm. Sci. 2007, 20, 317–323. [Google Scholar] [PubMed]
- Dahiya, R.; Kumar, A. Synthesis and biological activity of a potent analog of natural cyclopeptide. Int. J. Nat. Appl. Sci. 2007, 3, 433–440. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis, spectroscopic and biological investigation of cyclic octapeptide: Cherimolacyclopeptide G. Turk. J. Chem. 2008, 32, 205–215. [Google Scholar]
- Dahiya, R. Total synthesis and biological potential of psammosilenin A. Arch. Pharm. Chem. Life Sci. 2008, 341, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R. Synthetic studies on a cyclic hexapeptide from Dianthus superbus. Chem. Pap. 2008, 62, 527–535. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis and in vitro cytotoxic activity of a natural peptide of plant origin. J. Iran. Chem. Soc. 2008, 5, 445–452. [Google Scholar] [CrossRef]
- Dahiya, R.; Sharma, R.D. Synthesis and bioactivity of a novel cyclic hexapeptide from Stellaria delavayi. Eur. J. Sci. Res. 2008, 21, 277–287. [Google Scholar]
- Dahiya, R.; Kumar, A. Synthetic and biological studies on a cyclopolypeptide of plant origin. J. Zhejiang Univ. Sci. B 2008, 9, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Maheshwari, M.; Kumar, A. Toward the synthesis and biological evaluation of hirsutide. Monatsh. Chem. 2009, 140, 121–127. [Google Scholar] [CrossRef]
- Dahiya, R.; Kumar, A.; Gupta, R. Synthesis, cytotoxic and antimicrobial screening of a proline-rich cyclopolypeptide. Chem. Pharm. Bull. 2009, 57, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Maheshwari, M.; Yadav, R. Synthetic, cytotoxic and antimicrobial activity studies on annomuricatin B. Z. Naturforsch. B 2009, 64, 237–244. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Total synthesis and antimicrobial activity of a natural cycloheptapeptide of marine-origin. Mar. Drugs 2010, 8, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Gautam, H. Synthetic and pharmacological studies on a natural cyclopeptide from Gypsophila arabica. J. Med. Plants Res. 2010, 4, 1960–1966. [Google Scholar]
- Dahiya, R.; Gautam, H. Toward the first total synthesis of gypsin D: A natural cyclopolypeptide from Gypsophila arabica. Am. J. Sci. Res. 2010, 11, 150–158. [Google Scholar]
- Dahiya, R.; Gautam, H. Solution phase synthesis and bioevaluation of cordyheptapeptide B. Bull. Pharm. Res. 2011, 1, 1–10. [Google Scholar]
- Dahiya, R.; Gautam, H. Synthesis, characterization and biological evaluation of cyclomontanin D. Afr. J. Pharm. Pharmacol. 2011, 5, 447–453. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Toward the synthesis and biological screening of a cyclotetrapeptide from marine bacteria. Mar. Drugs 2011, 9, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S. First total synthesis and biological potential of a heptacyclopeptide of plant origin. Chin. J. Chem. 2016, 34, 1158–1164. [Google Scholar] [CrossRef]
- Dahiya, R.; Singh, S. Synthesis, characterization and biological screening of diandrine A. Acta Pol. Pharm. 2017, in press. [Google Scholar]
- Dahiya, R.; Singh, S. Synthesis, characterization, and biological activity studies on fanlizhicyclopeptide A. Iran. J. Pharm. Res. 2017, in press. [Google Scholar]
- Huang, T.; Zou, Y.; Wu, M.C.; Zhao, Q.J.; Hu, H.G. Total synthesis of proline-rich cyclic octapeptide stylissamide X. Chem. Nat. Compd. 2015, 51, 523–526. [Google Scholar] [CrossRef]
- Akindele, T.; Gise, B.; Sunaba, T.; Kita, M.; Kigoshi, H. Total synthesis of stylissatin A, a cyclic peptide that inhibits nitric oxide production. Bull. Chem. Soc. Jpn. 2015, 88, 600–609. [Google Scholar] [CrossRef]
- Bodanszky, M.; Bodanszky, A. The Practice of Peptide Synthesis; Springer: New York, NY, USA, 1984; pp. 78–143. [Google Scholar]
- Garg, L.C.; Atal, C.K. Anthelmintic activity of Myrsine Africana. Indian J. Pharm. Sci. 1963, 59, 240–245. [Google Scholar]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1996, 45, 493–496. [Google Scholar]
- Schmidt, G.; Grube, A.; Kock, M. Stylissamides A–D—New proline-containing cyclic heptapeptides from the marine sponge Stylissa caribica. Eur. J. Org. Chem. 2007, 24, 4103–4110. [Google Scholar] [CrossRef]
- Cychon, C.; Kock, M. Stylissamides E and F, cyclic heptapeptides from the Caribbean sponge Stylissa caribica. J. Nat. Prod. 2010, 73, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Clewlow, P.J.; Dufrense, C.; Doubek, D.L.; Cerny, R.L.; Rutzler, K. Antineoplastic agents. 193. Isolation and structure of the cyclic peptide hymenistatin 1. Can. J. Chem. 1990, 68, 708–711. [Google Scholar] [CrossRef]
- Vera, B.; Vicente, J.; Rodriguez, A.D. Isolation and structural elucidation of euryjanicins B-D, proline-containing cycloheptapeptides from the Caribbean marine sponge Prosuberites laughlini. J. Nat. Prod. 2009, 72, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Tabudravu, J.; Morris, L.A.; Kettenes-van den Bosch, J.J.; Jaspars, M. Wainunuamide, a histidine-containing proline-rich cyclic heptapeptide isolated from the Fijian marine sponge Stylotella aurantium. Tetrahedron Lett. 2001, 42, 9273–9276. [Google Scholar] [CrossRef]
- Williams, D.E.; Patrick, B.O.; Behrisch, H.W.; Van Soest, R.; Roberge, M.; Andersen, R.J. Dominicin, a cyclic octapeptide, and laughine, a bromopyrrole alkaloid, isolated from the Caribbean marine sponge Eurypon laughlini. J. Nat. Prod. 2005, 68, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, A.; Piaz, F.D.; Orrù, S.; Debitus, C.; Roussakis, C.; Pucci, P.; Gomez-Paloma, L. Axinellins A and B: New proline-containing antiproliferative cyclopeptides from the Vanuatu sponge Axinella carteri. Eur. J. Org. Chem. 1998, 11, 2659–2665. [Google Scholar] [CrossRef]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Leng, Q.; Begum, M.D.; Woodle, M.C.; Scaria, P.; Chou, S.-T.; Mixson, A.J. Peptide-based antifungal therapies against emerging infections. Drugs Future 2010, 35, 197. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Mourya, R. Synthetic studies on novel nitroquinazolinone analogs with antimicrobial potential. Bull. Pharm. Res. 2013, 3, 51–57. [Google Scholar]
- Dahiya, R.; Kaur, K. Synthesis and pharmacological investigation of segetalin C as a novel antifungal and cytotoxic agent. Arzneim. Forsch. 2008, 58, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Pathak, D. Synthetic studies on novel benzimidazolopeptides with antimicrobial, cytotoxic and anthelmintic potential. Eur. J. Med. Chem. 2007, 42, 772–798. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Mourya, R. Synthesis and antimicrobial screening of some novel halogenated phenoxyacetyl amino acid and peptide analogs. Bull. Pharm. Res. 2012, 2, 56–65. [Google Scholar]
- Dahiya, R.; Kumar, A.; Yadav, R. Synthesis and biological activity of peptide derivatives of iodoquinazolinones/nitroimidazoles. Molecules 2008, 13, 958–976. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Compound | Earthworm Species | |||||
|---|---|---|---|---|---|---|
| M. konk. | P. core. | E. euge. | ||||
| Mean Paralyzing Time (min) ‡ | Mean Death Time (min) ‡ | Mean Paralyzing Time (min) | Mean Death Time (min) | Mean Paralyzing Time (min) | Mean Death Time (min) | |
| 6 | 13.50 ± 0.11 | 21.52 ± 0.40 | 17.21 ± 0.22 | 27.55 ± 0.26 | 12.48 ± 0.44 | 23.28 ± 0.17 |
| 7 | 09.13 ± 0.31 | 15.48 ± 0.52 | 12.55 ± 0.37 | 21.27 ± 0.19 | 09.25 ± 0.35 | 18.09 ± 0.22 |
| Control # | – | – | – | – | – | – |
| Mebendazole | 13.63 ± 0.30 | 22.43 ± 0.21 | 17.56 ± 0.43 | 29.49 ± 0.17 | 13.50 ± 0.34 | 24.07 ± 0.49 |
| Compound | Diameter of Zone of Inhibition (mm) | |||||||
|---|---|---|---|---|---|---|---|---|
| Bacterial Strains | Fungal Strains | |||||||
| B. sub. | S. aur. | P. aeru. | K. pneu. | C. alb. | M. audo. | A. niger | T. menta. | |
| 6 | – | – | 14(6) | 19(6) | 17(6) | 19(6) | – | 18(6) |
| 7 | 10(25) | 11(12.5) | 18(6) | 22(6) | 22(6) | 23(6) | – | 22(6) |
| Control * | – | – | – | – | – | – | – | – |
| Gatifloxacin | 18(12.5) † | 27(6) | 23(6) | 25(6) | – | – | – | – |
| Griseofulvin | – | – | – | – | 20(6) | 18(6) | 20(12.5) | 19(6) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahiya, R.; Singh, S.; Sharma, A.; Chennupati, S.V.; Maharaj, S. First Total Synthesis and Biological Screening of a Proline-Rich Cyclopeptide from a Caribbean Marine Sponge. Mar. Drugs 2016, 14, 228. https://doi.org/10.3390/md14120228
Dahiya R, Singh S, Sharma A, Chennupati SV, Maharaj S. First Total Synthesis and Biological Screening of a Proline-Rich Cyclopeptide from a Caribbean Marine Sponge. Marine Drugs. 2016; 14(12):228. https://doi.org/10.3390/md14120228
Chicago/Turabian StyleDahiya, Rajiv, Sunil Singh, Ajay Sharma, Suresh V. Chennupati, and Sandeep Maharaj. 2016. "First Total Synthesis and Biological Screening of a Proline-Rich Cyclopeptide from a Caribbean Marine Sponge" Marine Drugs 14, no. 12: 228. https://doi.org/10.3390/md14120228
APA StyleDahiya, R., Singh, S., Sharma, A., Chennupati, S. V., & Maharaj, S. (2016). First Total Synthesis and Biological Screening of a Proline-Rich Cyclopeptide from a Caribbean Marine Sponge. Marine Drugs, 14(12), 228. https://doi.org/10.3390/md14120228