
Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens

 ,
,  ,
, 
 , and
, and
Abstract
:1. Introduction
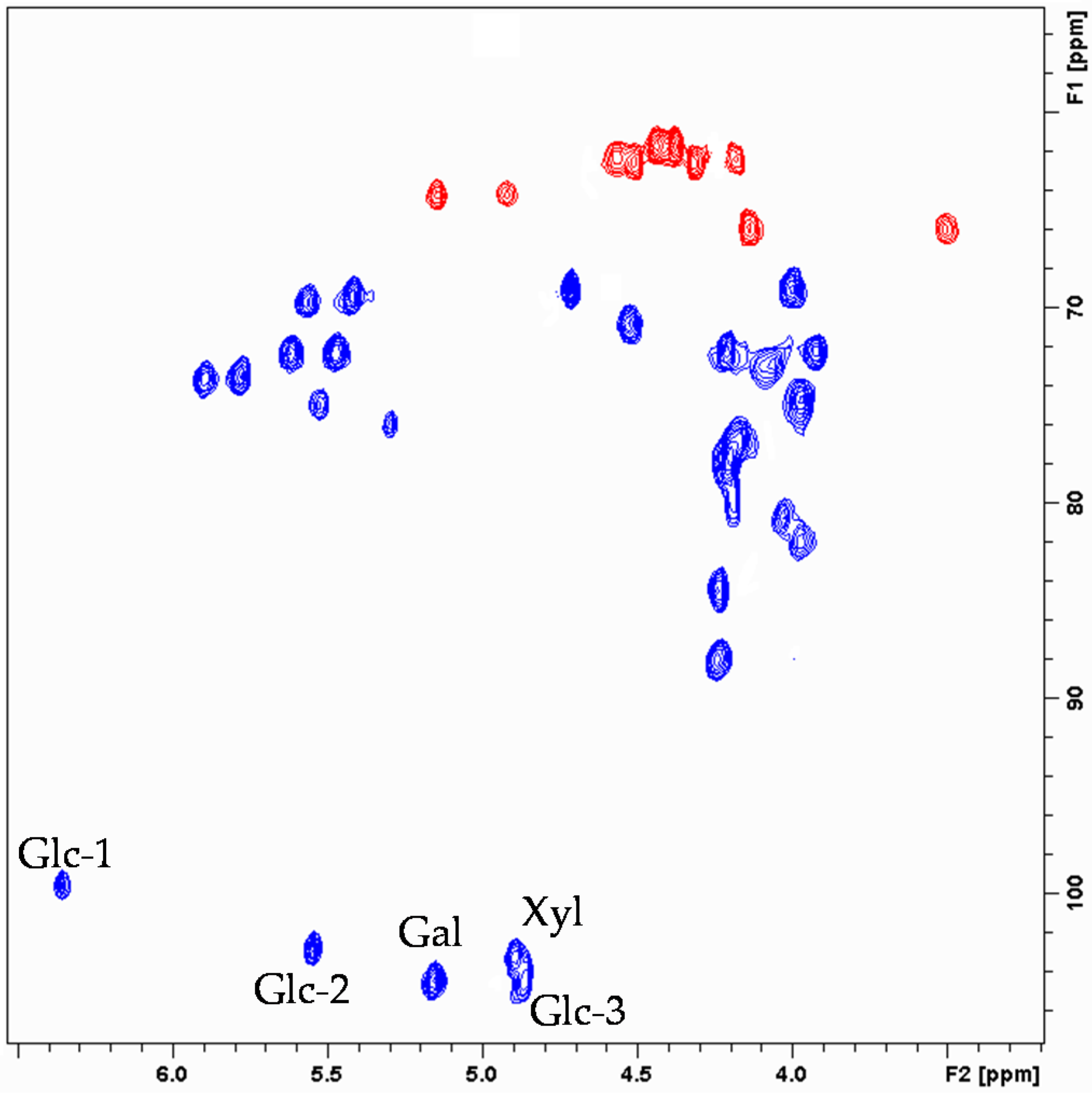
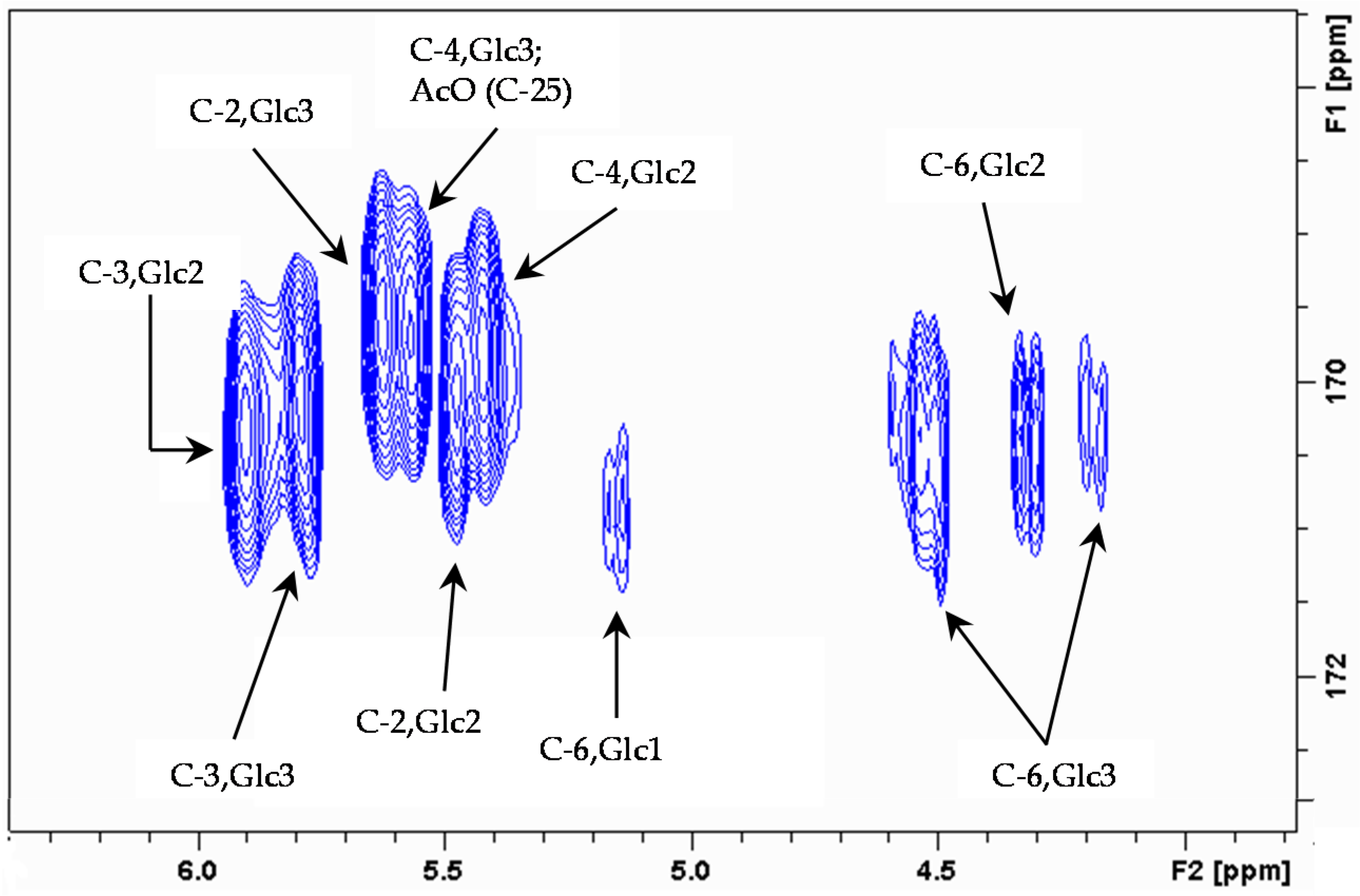
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation Procedures
3.4. Methanolysis of Crude Fraction of Glycolipids
3.5. Derivatization of Glycosides
3.6. Preparation of Monosaccharide Standards
3.7. Synthesis of the Acetonide of Compound 5
3.8. Bioassay Description (GPSD2 Screening Application) [15]
3.9. COS-7 Cells Bioassay [15]
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chester, M.A. IUPAC-IUB joint commission on biochemical nomenclature (JCBN) nomenclature of glycolipids—Recommendations 1997. Eur. J. Biochem. 1998, 257, 293–298. [Google Scholar] [PubMed]
- Barnathan, G.; Couzinet-Mossion, A.; Wielgosz-Collin, G. Glycolipids from Marine Invertebrates. In Outstanding Marine Molecules; Barre, S.L., Kornprobst, J.-M., Eds.; Wiley-Blackwell: Weinheim, Germany, 2014. [Google Scholar]
- Warabi, K.; Zimmerman, W.T.; Shen, J.K.; Gauthier, A.; Robertson, M.; Finlay, B.B.; van Soest, R.; Andersen, R.J. Pachymoside A—A novel glycolipid isolated from the marine sponge Pachymatisma johnstonia. Can. J. Chem. 2004, 82, 102–112. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Negron, V. Identification and Characterization of 2 New Methylicosadienoic Acids from Erylus-Formosus. J. Nat. Prod. 1991, 54, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, N.M.; Oyola, D.; Vicente, J.; Rodriguez, A.D. Identification of novel α-methoxylated phospholipid fatty acids in the caribbean sponge Erylus goffrilleri. Lipids 2007, 42, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Sata, N.; Asai, N.; Matsunaga, S.; Fusetani, N. Erylusamines, IL-6 Receptor Antagonists, from the Marine Sponge, Erylus placenta. Tetrahedron 1994, 50, 1105–1110. [Google Scholar] [CrossRef]
- Fusetani, N.; Sata, N.; Asai, N.; Matsunaga, S. Isolation and Structure Elucidation of Erylusamine-B, a New Class of Marine Natural-Products, Which Blocked an IL-6 Receptor, from the Marine Sponge Erylus placenta Thiele. Tetrahedron Lett. 1993, 34, 4067–4070. [Google Scholar] [CrossRef]
- Goobes, R.; Rudi, A.; Kashman, Y.; Ilan, M.; Loya, Y. Three new glycolipids from a Red Sea sponge of the genus Erylus. Tetrahedron 1996, 52, 7921–7928. [Google Scholar] [CrossRef]
- Van Altena, I.; van Soest, R.; Roberge, M.; Andersen, R.J. Trisphaerolide A, a novel polyketide from the Dominican sponge Erylus trisphaerus. J. Nat. Prod. 2003, 66, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Malachowski, W.P.; Winters, M.; DuHadaway, J.B.; Lewis-Ballester, A.; Badir, S.; Wai, J.; Rahman, M.; Sheikh, E.; LaLonde, J.M.; Yeh, S.R.; et al. O-alkylhydroxylamines as rationally-designed mechanism-based inhibitors of indoleamine 2,3-dioxygenase-1. Eur. J. Med. Chem. 2016, 108, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Ueng, S.H.; Tseng, C.T.; Hung, M.S.; Song, J.S.; Wu, J.S.; Liao, F.Y.; Fan, Y.S.; Wu, M.H.; Hsiao, W.C.; et al. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, U.F.; Majjigapu, S.R.; Vogel, P.; Zoete, V.; Michiein, O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J. Med. Chem. 2015, 58, 9421–9437. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: http://www.cancer.gov/about-cancer/treatment/clinical-trials/search/results?protocolsearchid=14884282 (accessed on 15 April 2016).
- Austin, C.J.; Kahlert, J.; Issa, F.; Reed, J.H.; Smith, J.R.; Ioppolo, J.A.; Ong, J.A.; Jamie, J.F.; Hibbs, D.; Rendina, L.M. The first indoleamine-2,3-dioxygenase-1 (IDO1) inhibitors containing carborane. Dalton Trans. 2014, 43, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Cerejo, M.; Andrade, G.; Roca, C.; Sousa, J.; Rodrigues, C.; Pinheiro, R.; Chatterjee, S.; Vieira, H.; Calado, P. A Powerful Yeast-Based Screening Assay for the Identification of Inhibitors of Indoleamine 2,3-Dioxygenase. J. Biomol. Screen. 2012, 17, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Doco, T.; O’Neill, M.A.; Pellerin, P. Determination of the neutral and acidic glycosyl-residue compositions of plant polysaccharides by GC-EI-MS analysis of the trimethylsilyl methyl glycoside derivatives. Carbohyd. Polym. 2001, 46, 249–259. [Google Scholar] [CrossRef]
- Clayden, J.; Johnson, P.; Pink, J.H.; Helliwell, M. Atropisomeric amides as chiral ligands: Using (−)-sparteine-directed enantioselective silylation to control the conformation of a stereogenic axis. J. Org. Chem. 2000, 65, 7033–7040. [Google Scholar] [CrossRef] [PubMed]
- Shih, F.Y.; Chen, T.H.; Lu, M.C.; Chen, W.F.; Wen, Z.H.; Kuo, Y.H.; Sung, P.J. Cladieunicellins K and L, New Eunicellin-Based Diterpenoids from an Octocoral Cladiella sp. Int. J. Mol. Sci. 2013, 14, 21781–21789. [Google Scholar] [CrossRef] [PubMed]
- Wojnar, J.M.; Northcote, P.T. The Agminosides: Naturally Acetylated Glycolipids from the New Zealand Marine Sponge Raspailia agminata. J. Nat. Prod. 2011, 74, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Peddie, V.; Takada, K.; Okuda, S.; Ise, Y.; Morii, Y.; Yamawaki, N.; Takatani, T.; Arakawa, O.; Okada, S.; Matsunaga, S. Cytotoxic Glycosylated Fatty Acid Amides from a Stelletta sp. Marine Sponge. J. Nat. Prod. 2015, 78, 2808–2813. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Morinaka, B.I. Integrated approaches to the configurational assignment of marine natural products. Tetrahedron 2012, 68, 9307–9343. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Nuzzo, G.; D’Angelo, D.; Borbone, E.; Fusco, A.; Fontana, A. Mycalol: A natural lipid with promising cytotoxic properties against human anaplastic thyroid carcinoma cells. Angew. Chem. 2013, 52, 9256–9260. [Google Scholar] [CrossRef] [PubMed]
- Solladié, G.; Hanquet, G.; Rolland, C. Stereoselective Sulfoxide Directed Reduction of 1,2-Diketo-Derivatives to Enantiomerically Pure Syn and Anti 1,2-Diols. Tetrahedron Lett. 1997, 38, 5847–5850. [Google Scholar] [CrossRef]
- Bubb, W.A. NMR spectroscopy in the study of carbohydrates: Characterizing the structural complexity. Concepts Magn. Reson. A 2003, 19, 1–19. [Google Scholar] [CrossRef]
- Beier, R.C.; Mundy, B.P.; Strobel, G.A. Assignment of Anomeric Configuration and Identification of Carbohydrate Residues by C-13 Nmr. 1. Galactopyranosides and Glucopyranosides and Furanosides. Can. J. Chem. 1980, 58, 2800–2804. [Google Scholar] [CrossRef]
- Shin, J.; Lee, H.S.; Woo, L.; Rho, J.R.; Seo, Y.; Cho, K.W.; Sim, C.J. New triterpenoid saponins from the sponge Erylus nobilis. J. Nat. Prod. 2001, 64, 767–771. [Google Scholar] [CrossRef] [PubMed]
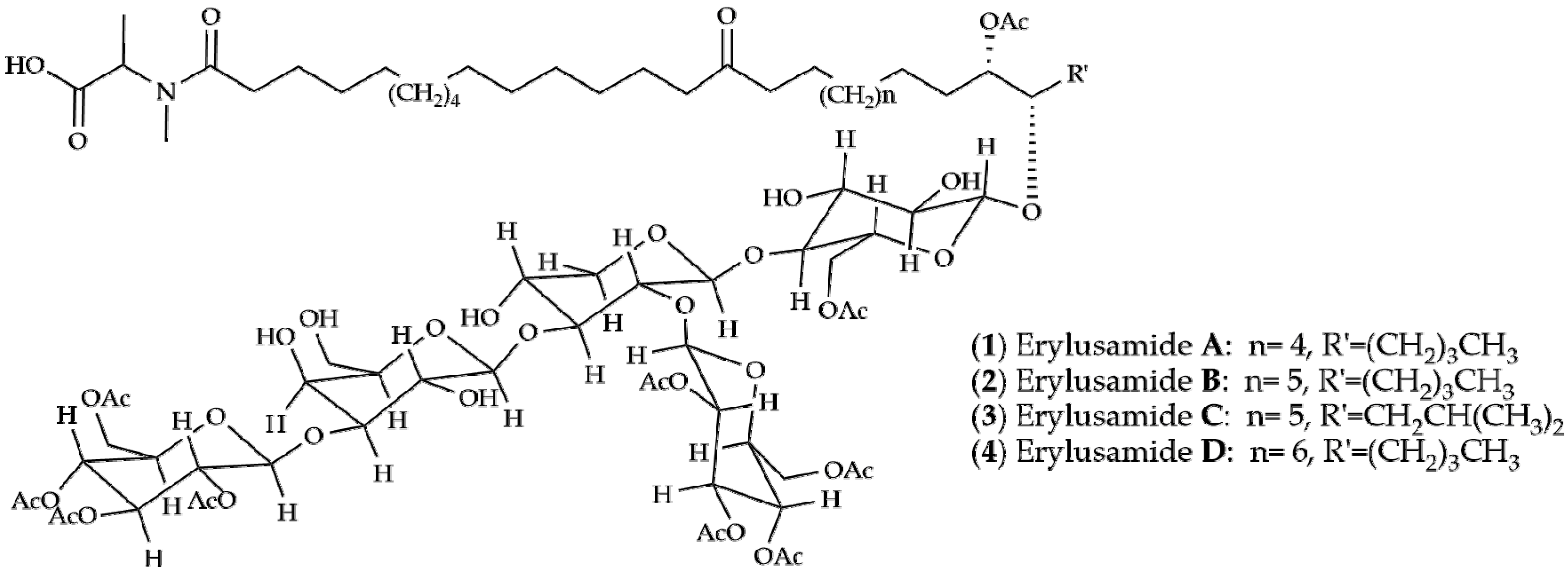
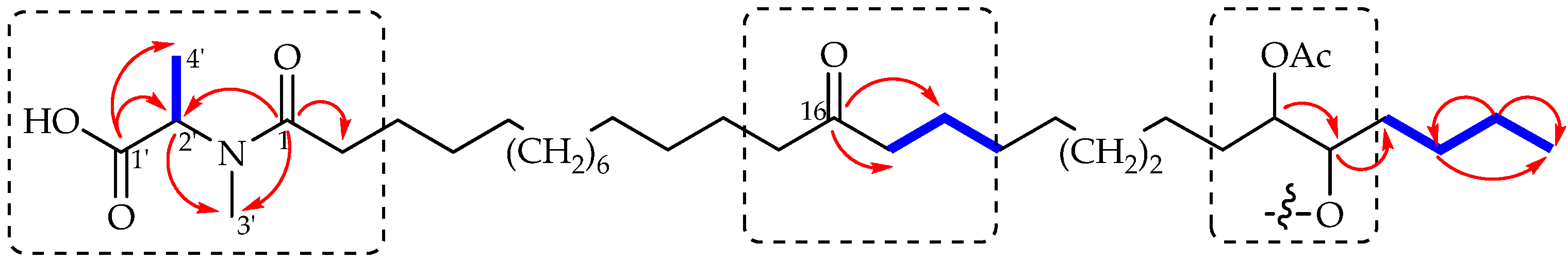
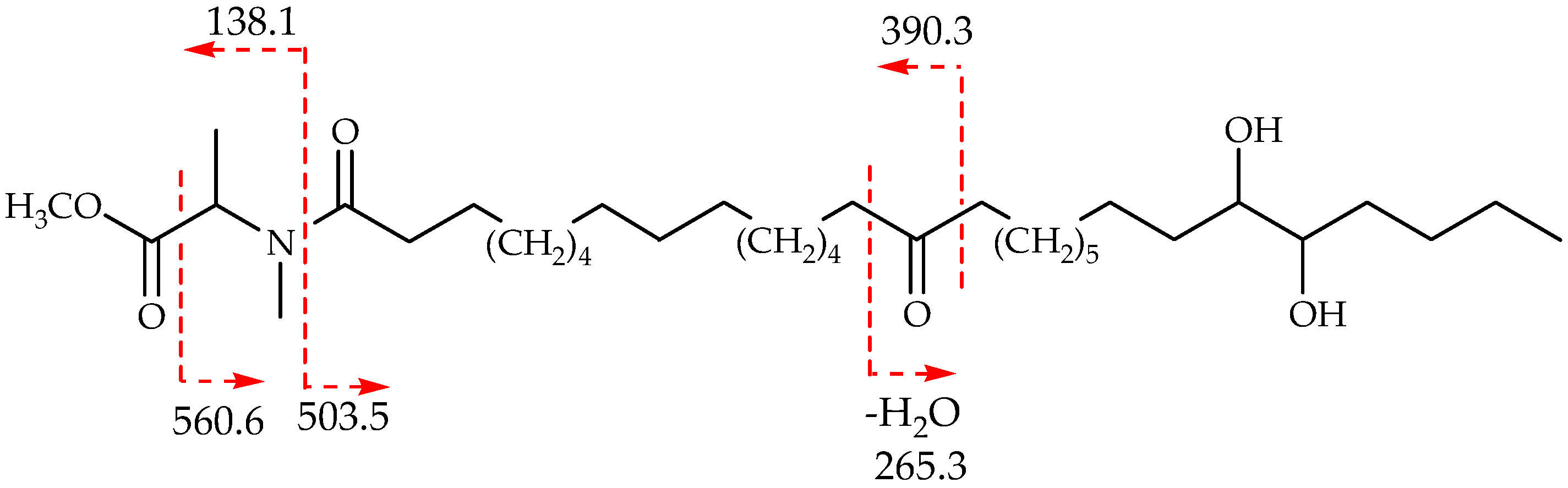
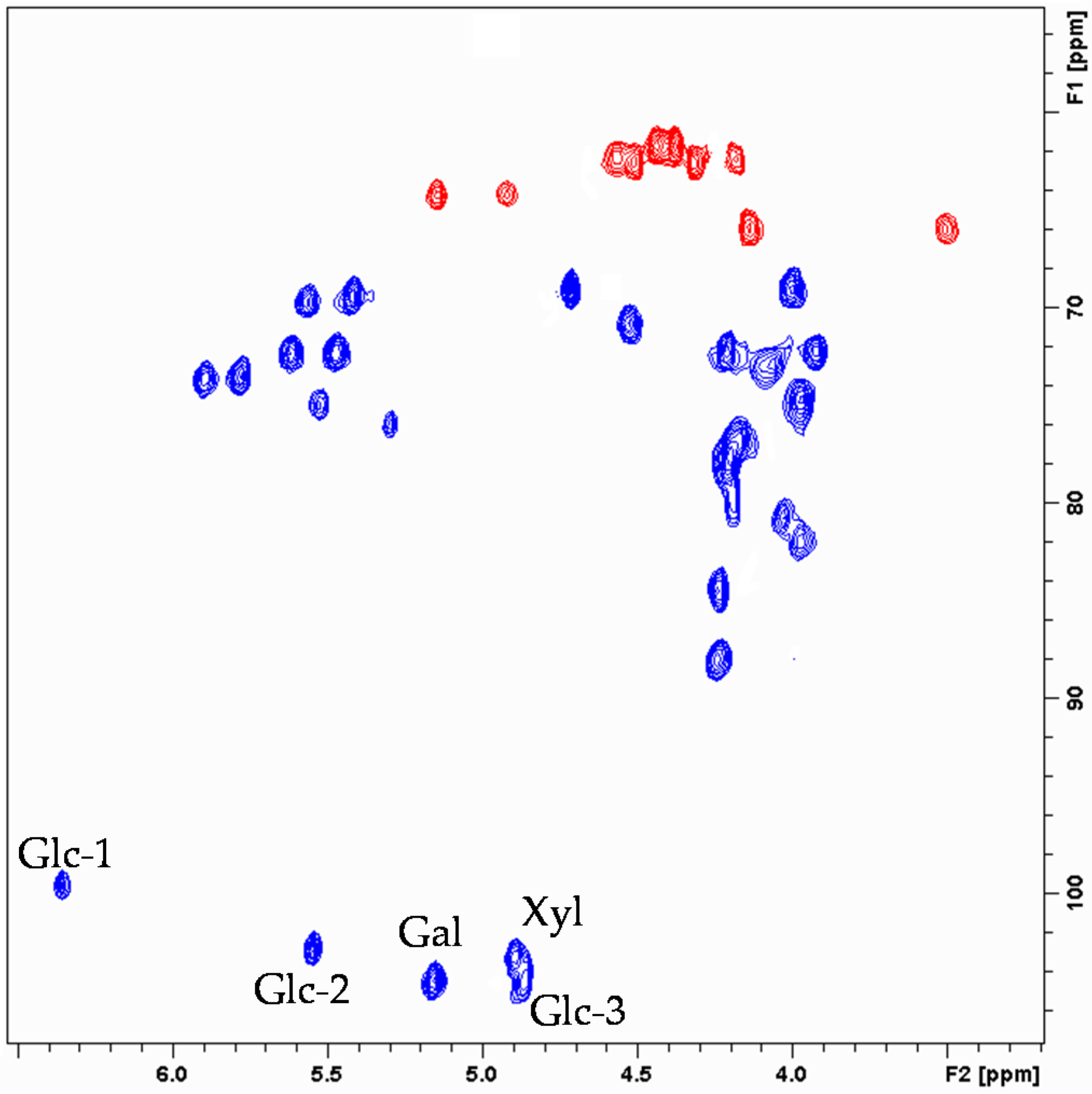
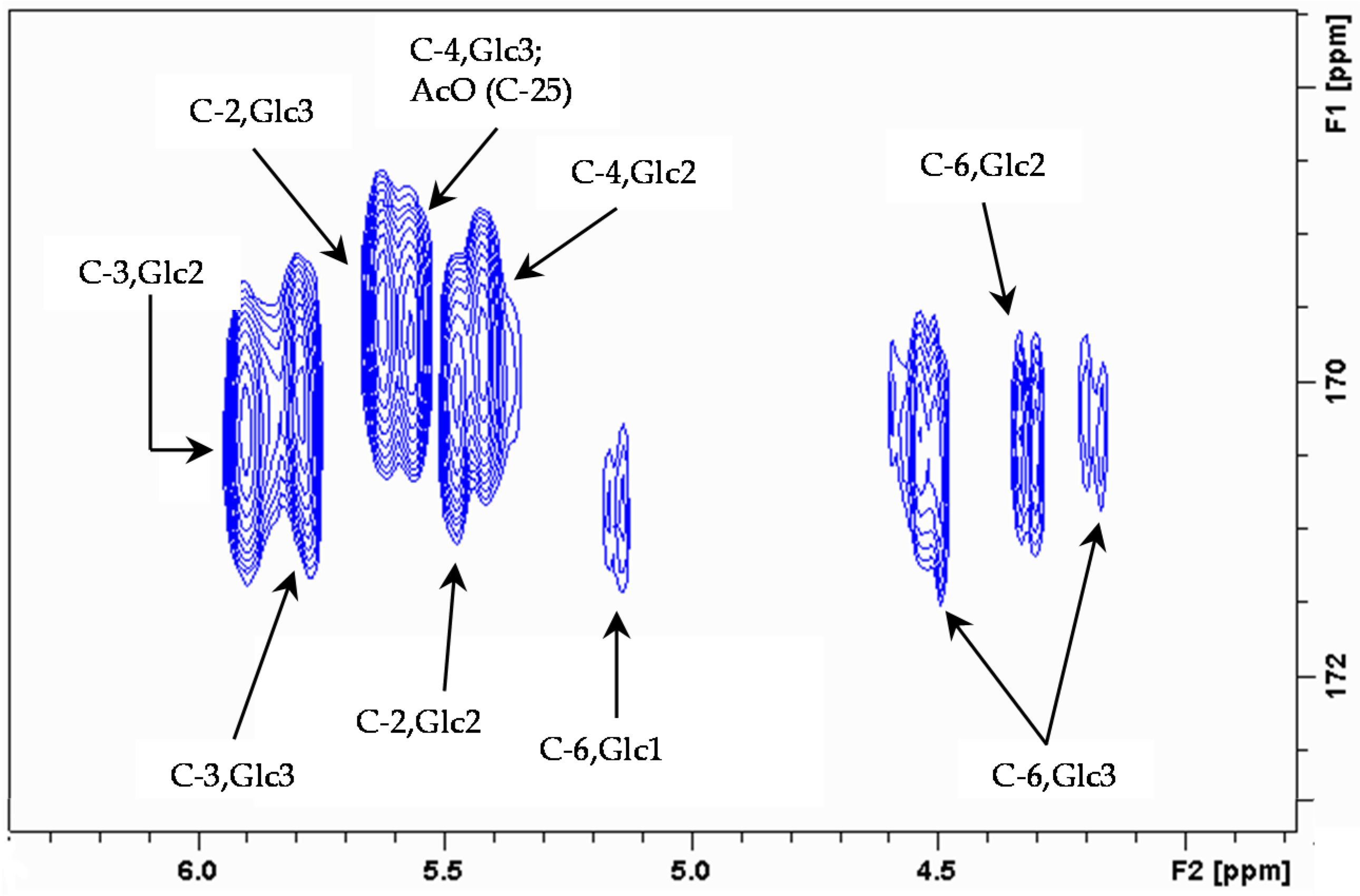


{kind=link}
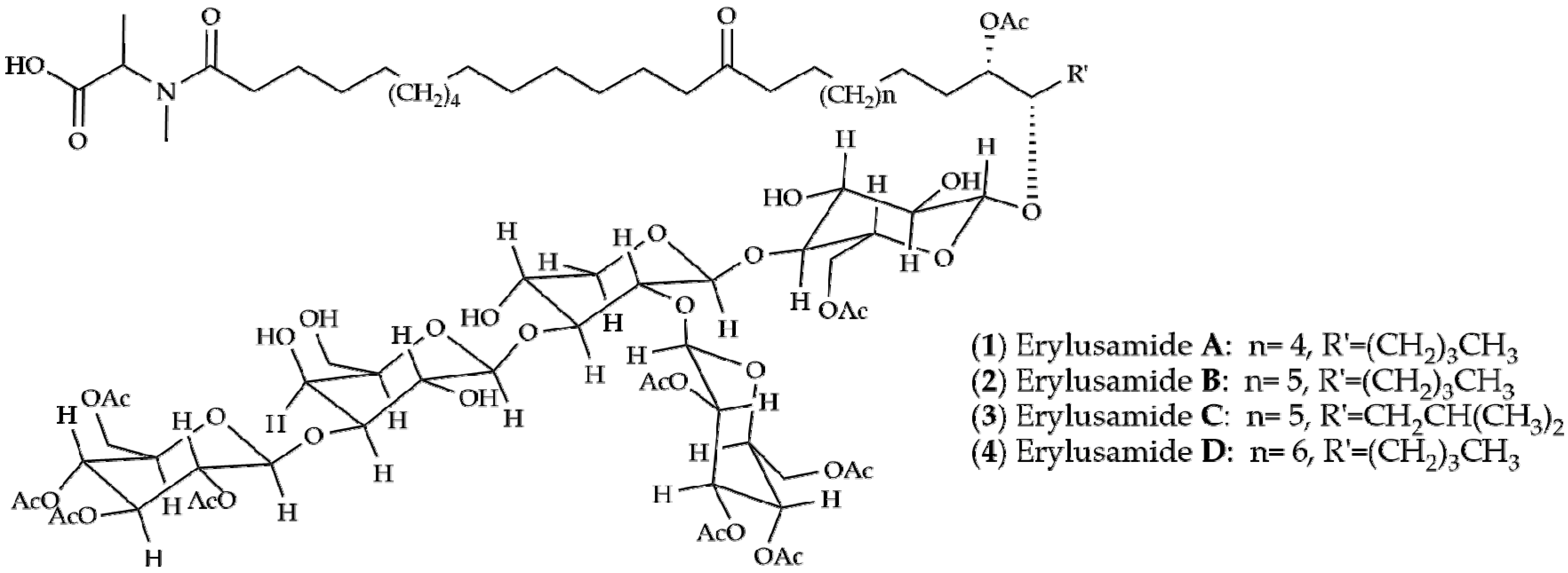
{kind=link}
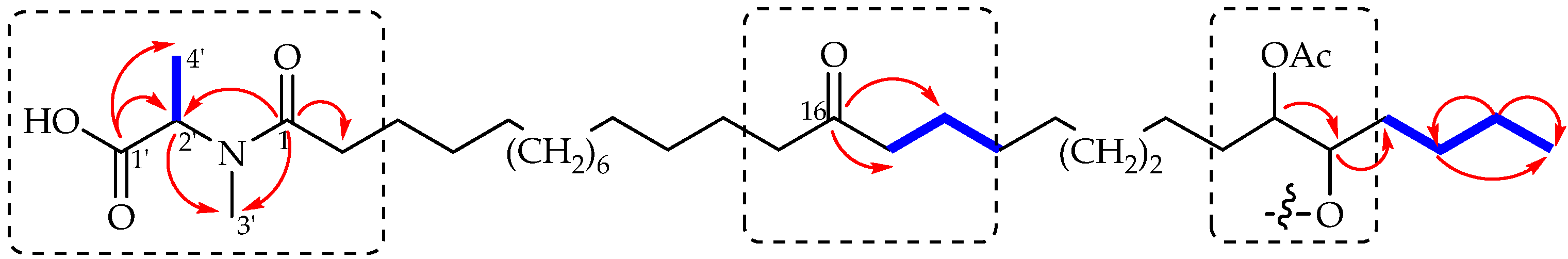
{kind=link}
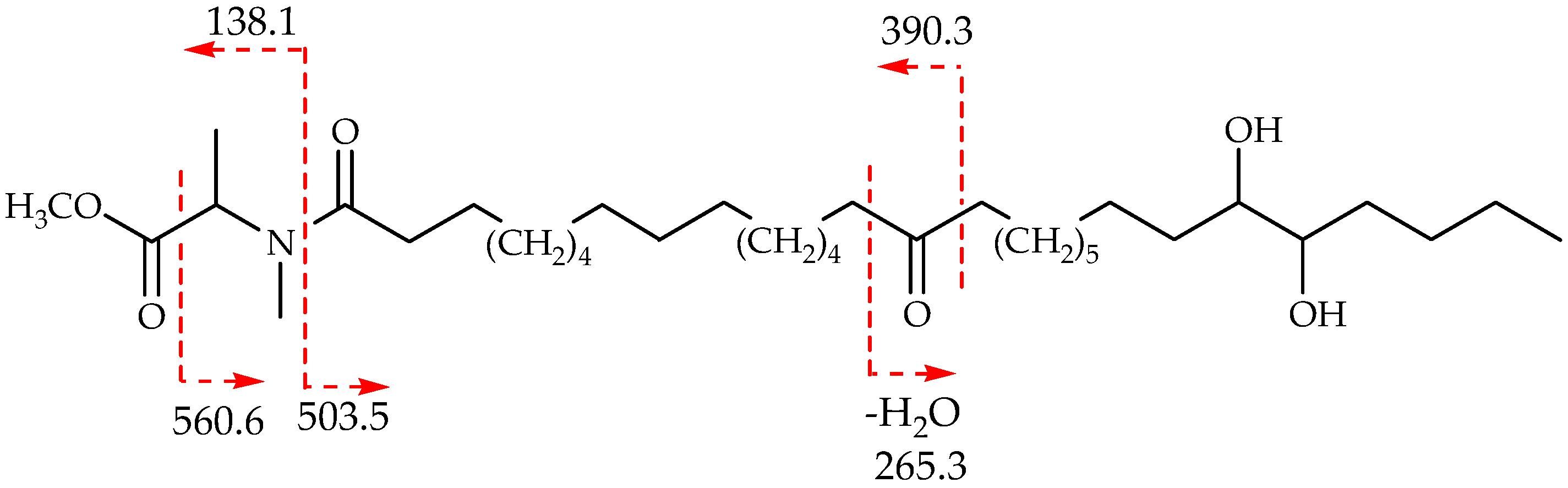
{kind=link}
{kind=link}
{kind=link}
| Sponge/Origin | Compounds | |||||
|---|---|---|---|---|---|---|
| Activity | ||||||
| Erylus formosus La Parguera, Puerto Rico [4] | Fatty acid: Tetradecanoic 13-Methyltetradecanoic 12-Methyltetradecanoic 3-Methylpentadecanoic Hexadecenoic Methylpentadecanoic Hexadecanoic 3-Methylhexadecanoic 15-Methylhexadecanoic 14-Methylhexadecanoic 5,9-Octadecadienoic Octadecenoic Octadecanoic | Methyloctadecanoic 5,9-Icosadienoic 19-Methyl-5,9-icosadienoic 18-Methyl-5,9-icosadienoic Heneicosanoic Tricosanoic Tetracosanoic Pentacosanoic 24-Methyl-5,9-pentacosadienoic 5,9-Hexacosadienoic 25-Methyl-5,9-hexacosadienoic 24-Methyl-5,9-hexacosadienoic 5,9-Octacosadienoic 5,9-Nonacosadienoic | ||||
| NR | ||||||
| Erylus goffrilleri Mona Island (Puerto Rico) [5] | Fatty acid: Tridecanoic 12-Methyltridecanoic Tetradecanoic 3-Methyltetradecanoic 13-Methyltetradecanoic 12-Methyltetradecanoic 9-Pentadecenoic Pentadecanoic 3-Methylpentadecanoic 14-Methylpentadecanoic 13-Methylpentadecanoic (Z)-9-Hexadecenoic (Z)-11-Hexadecenoic Hexadecanoic (Z)-15-Methyl-9-hexadecenoic 10-Methylhexadecanoic 15-Methylhexadecanoic 14-Methylhexadecanoic (5Z,9Z)-2-Methoxy-5,9-hexadecadienoic (Z)-9-Heptadecenoic (Z)-11-Heptadecenoic Heptadecanoic (5Z,9Z)-2-Methoxy-15-methyl-5,9-hexadeca-dienoic Methylheptadecanoic (5Z,9Z)-5,9-Octadecadienoic (9Z)-2-Methoxy-15-methyl-9-hexadecenoic (Z)-9-Octadecenoic (Z)-11-Octadecenoic 2-Methoxy-14-methylhexadecanoic Octadecanoic Methyl-6-octadecenoic (5Z,9Z)-17-Methyl-5,9-octadecadienoic 11-Methyloctadecanoic (5Z,9Z)-2-Methoxy-5,9-octadecadienoic | (5Z,9Z)-2-Methoxy-5,9-nonadecadienoic 11-Eicosenoic Eicosanoic (5Z,9Z)-19-Methyl-5,9-eicosadienoic (5Z,9Z)-18-Methyl-5,9-eicosadienoic Methyleicosanoic (5Z,9Z)-5,9-Heneicosadienoic 19-Methyleicosanoic 18-Methyleicosanoic (5Z,9Z)-2-Methoxy-5,9-eicosadienoic 11-Nonadecenoic Nonadecanoic 5,8,11,14-Eicosatetraenoic Docosanoic 16-Methyldocosanoic 21-Methyldocosanoic 20-Methyldocosanoic Tricosanoic Methyltricosanoic Tetracosanoic Methyltetracosanoic (5Z,9Z)-24-Methyl-5,9-pentacosadienoic (5Z,9Z)-23-Methyl-5,9-pentacosadienoic (5Z,9Z)-5,9-Hexacosadienoic (5Z,9Z)-25-Methyl-5,9-hexacosadienoic (5Z,9Z)-24-Methyl-5,9-hexacosadienoic (5Z,9Z)-5,9-Heptacosadienoic (5Z,9Z)-26-Methyl-5,9-heptacosadienoic (5Z,9Z)-25-Methyl-5,9-heptacosadienoic (5Z,9Z)-5,9-Octacosadienoic (5Z,9Z)-5,9-Nonacosadienoic Methylnonadecanoic 17-Methyloctadecanoic 16-Methyloctadecanoic | ||||
| NR | ||||||
| Erylus placenta Hachijojima Island (Japan) [6,7] | Erylusamine A: R1 = CH2CH2CH3, R2 = H Erylusamine B: R1 = CH2CH(CH3)2, R2 = H Erylusamine C: R1 = CH2CH(CH3)2 R2 = Ac Erylusamine D: R1 = CH2CH2CH2CH2CH3 R2 = Ac |  | ||||
| Interleukin-6 (IL-6) receptor antagonists | ||||||
| Erylus cf. Lendenfeidi Gulf of Eilat (Red sea) [8] | Erylusamine TA: R1 = Ac; R2 = (CH2)5N(CH3)2; R3 = H, n = 8, m = 2 Erylusine: R1 = Ac; R2 = (CH2)3NCH3(CH2)4N(CH3)2; R3 = H, n = 8, m = 2 Erylusidine R1 = H; R2 = (CH2)4NHC = NH(NH2); R3 = COCH2CH(CH3)2, n = 8, m = 3 |  | ||||
| NR | ||||||
| Erylus trisphaerus Dominica [9] | Trisphaerolide |  | ||||
| Low in vitro cytotoxicity against MCF7 human breast cancer cells | ||||||
| Pachymatisma johnstonia Isle of Mann (UK) [3] | Pachymoside A |  | ||||
| Crude extract showed inhibitory activity of bacterial type III secretion | ||||||
| 1 | 2 | 3 | 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N° | δ 13C | δ 1H, m (J, Hz) | N° | δ 13C | δ 1H, m (J, Hz) | N° | δ 13C | δ 1H, m (J, Hz) | N° | δ 13C | δ 1H, m (J, Hz) |
| 1 | 173.2 | - | 1 | 173.2 | - | 1 | 173.2 | - | 1 | 173.2 | - |
| 173.1 | - | 173.1 | - | 173.1 | - | 173.1 | - | ||||
| 2 | 33.8 | 2.43, m | 2 | 33.8 | 2.43, m | 2 | 33.8 | 2.43, m | 2 | 33.8 | 2.43, m |
| 3 | 25.5 | 1.81, m | 3 | 25.5 | 1.81, m | 3 | 25.5 | 1.81, m | 3 | 25.5 | 1.82, m |
| 4 | 29.7 | 1.38, m | 4 | 29.8 | 1.38, m | 4 | 29.8 | 1.39, m | 4 | 29.7 | 1.38, m |
| 5–12 | 29.6–29.9 | 1.18–1.33 Overlap. | 5–12 | 29.6–29.9 | 1.18–1.33 Overlap. | 5–12 | 29.7–29.9 | 1.18–1.32 Overlap. | 5–12 | 29.6–29.9 | 1.19–1.33 Overlap. |
| 13 | 29.6 | 1.28, m | 13 | 29.6 | 1.28, m | 13 | 29.6 | 1.28, m | 13 | 29.6 | 1.28, m |
| 14 | 24.2 | 1.64, m | 14 | 24.2 | 1.64, m | 14 | 24.2 | 1.64, m | 14 | 24.2 | 1.65, m |
| 15 | 42.8 | 2.42, m | 15 | 42.7 | 2.42, m | 15 | 42.8 | 2.42, m | 15 | 42.8 | 2.42, m |
| 16 | 210.5 | - | 16 | 210.5 | - | 16 | 210.5 | - | 16 | 210.5 | - |
| 17 | 42.8 | 2.42, m | 17 | 42.7 | 2.42, m | 17 | 42.8 | 2.42, m | 17 | 42.8 | 2.42, m |
| 18 | 24.2 | 1.64, m | 18 | 24.2 | 1.64, m | 18 | 24.2 | 1.64, m | 18 | 24.2 | 1.65, m |
| 19–23 | 29.6–29.9 | 1.19–132 Overlap | 19–24 | 29.6–29.9 | 1.18–1.33 Overlap | 19–24 | 29.7–29.9 | 1.18–1.32 Overlap | 19–25 | 29.7–29.9 | 1.19–1.33 Overlap |
| 24 | 29.7 | 1.83, m | 25 | 29.7 | 1.82, m | 25 | 29.7 | 1.81, m | 26 | 29.7 | 1.83, m |
| 25 | 74.9 | 5.53, m | 26 | 74.9 | 5.53, m | 26 | 74.9 | 5.54, m | 27 | 74.9 | 5.54, m |
| AcO | 169.7 * | AcO | 169.7 * | AcO | 169.7* | AcO | 169.7 * | ||||
| 26 | 80.7 | 4.03, m | 27 | 80.8 | 4.03, m | 27 | 80.7 | 4.03, m | 28 | 80.8 | 4.05, m |
| 27 | 30.9 | 1.79, m | 28 | 30.9 | 1.79, m | 28 | 39.1 | 1.16, m | 29 | 30.9 | 1.80, m |
| 28 | 32.0 | 1.23, m | 29 | 32.0 | 1.23, m | 29 | 28.2 | 1.51, m | 30 | 32.0 | 1.25, m |
| 29 | 22.8 | 1.28, m | 30 | 22.8 | 1.28, m | 30 | 22.8 | 0.85, d (6.0) | 31 | 22.9 | 1.27, m |
| 30 | 14.7 | 0.84, t (7.0) | 31 | 14.2 | 0.86, t (6.5) | 31 | 22.8 | 0.85, d (6.0) | 32 | 14.2 | 0.86, t (6.6) |
| 32 | 22.8 | 0.85, d (6.0) | |||||||||
| 1’ | 174.9 | - | 1’ | 174.9 | - | 1’ | 174.9 | - | 1’ | 175.0 | - |
| 174.5 | - | 174.5 | - | 174.5 | - | 174.6 | - | ||||
| 2’ | 52.7 | 5.75, q (7.2) | 2’ | 52.6 | 5.74, q (7.3) | 2’ | 52.7 | 5.74, q (7.3) | 2’ | 52.7 | 5.75, q (7.3) |
| 55.8 | 4.97, q (7.3) | 4.97, q (7.4) | 55.7 | 4.97, q (7.3) | 55.8 | 4.97, q (7.2) | |||||
| 3’ | 31.5 | 3.06, s | 3’ | 31.5 | 3.06, s | 3’ | 31.5 | 3.06, s | 3’ | 31.5 | 3.07, s |
| 28.9 | 3.14, s | 28.9 | 3.13, s | 28.9 | 3.14,s | 28.9 | 3.15,s | ||||
| 4’ | 15.0 | 1.54, d (7.3) | 4’ | 15.0 | 1.55, d (7.3) | 4’ | 15.0 | 1.55, d (7.3) | 4’ | 15.0 | 1.54, d (7.4) |
| 16.0 | 1.60, d (7.2) | 16.0 | 1.69, d (7.1) | 16.0 | 1.60, d (7.3) | 16.0 | 1.61, d (7.2) | ||||
| Position | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δ 13C | δ 1H, m (J, Hz) | δ 13C | δ 1H, m (J, Hz) | δ 13C | δ 1H, m (J, Hz) | δ 13C | δ 1H, m (J, Hz) | |
| Gal | ||||||||
| 1 | 104.4 | 5.16, d (7.5) | 104.4 | 5.16, d (7.7) | 104.4 | 5.15, d (7.6) | 104.4 | 5.16, d (7.7) |
| 2 | 70.7 | 4.54, m | 70.7 | 4.53, m | 70.7 | 4.53, m | 70.72 | 4.53, m |
| 3 | 84.4 | 4.24, m | 84.3 | 4.25, m | 84.3 | 4.25, m | 84.32 | 4.24, m |
| 4 | 69.0 | 4.70, brs | 68.9 | 4.71, brs | 68.7 | 4.71, brs | 68.91 | 4.72, brs |
| 5 | 77.3 | 4.21, m | 77.4 | 4.20, m | 77.3 | 4.21, m | 77.27 | 4.20, m |
| 6 | 61.6 | 4.38, dd | 61.6 | 4.38, dd | 61.6 | 4.38, dd | 61.62 | 4.38, dd |
| (5.1; 10.6) | (5.2; 10.6) | (4.9; 10.9) | (5.2; 10.6) | |||||
| 4.44, dd | 4.43, dd | 4.44, dd | 4.44, dd | |||||
| (6.8; 10.6) | (6.9; 10.6) | (6.7; 10.9) | (6.9; 10.6) | |||||
| Xyl | ||||||||
| 1 | 103.2 | 4.90,d (7.3) | 103.2 | 4.89, d (7.3) | 103.2 | 4.90, d (7.6) | 103.2 | 4.90, d (7.4) |
| 2 | 78.3 | 4.22, m | 78.2 | 4.22, m | 78.3 | 4.21, m | 78.3 | 4.21, m |
| 3 | 88.2 | 4.25, m | 88.1 | 4.24, m | 88.0 | 4.24, m | 88.0 | 4.26, m |
| 4 | 69.0 | 4.00, m | 68.9 | 3.99, m | 69.0 | 3.99, m | 69.0 | 4.00, m |
| 5 | 65.9 | 3.51, t (10.7) | 65.9 | 3.50, t (9.8) | 65.9 | 3.51, t (10.3) | 65.9 | 3.51, t (10.7) |
| 4.14, m | 4.14, m | 4.14, m | 4.13, m | |||||
| Glc1 | ||||||||
| 1 | 104.6 | 4.86, d (7.9) | 104.6 | 4.87, d (7.8) | 104.6 | 4.88, d (8.0) | 104.6 | 4.87, d (8.0) |
| 2 | 74.9 | 3.97, m | 74.6 | 3.96, m | 74.6 | 3.97, m | 74.6 | 3.98, m |
| 3 | 76.6 | 4.18, m | 76.6 | 4.18, m | 76.6 | 4.18, m | 76.6 | 4.18, m |
| 4 | 81.9 | 3.97, m | 81.8 | 3.97, m | 81.9 | 3.97, m | 81.9 | 3.97, m |
| 5 | 73.0 | 4.09, m | 72.9 | 4.08, m | 73.0 | 4.10, m | 73.0 | 4.11, m |
| 6 | 64.3 | 4.92, m | 64.3 | 4.93, m | 64.1 | 4.93, m | 64.3 | 4.93, m |
| 5.16, m | 5.14, m | 5.16, m | 5.15, m | |||||
| Ac (C-6) | 171.0 | - | - | 171.0 | 171.0 | - | ||
| Glc2 | ||||||||
| 1 | 102.8 | 5.55, d (8.0) | 102.8 | 5.55, d (8.2) | 102.8 | 5.55, d (8.5) | 102.8 | 5.55, d (8.1) |
| 2 | 72.2 | 5.48, dd | 72.2 | 5.47,dd | 72.2 | 5.48, t | 72.1 | 5.48, dd |
| (8.2; 9.5) | (8.4; 9.3) | (9.3) | (8.4;9.3) | |||||
| 3 | 73.5 | 5.78, t (9.6) | 73.5 | 5.78, t (9.6) | 73.5 | 5.78, t (9.9) | 73.4 | 5.78, t (9.5) |
| 4 | 69.4 | 5.42, t (9.8) | 69.4 | 5.42, t (9.7) | 69.4 | 5.42, t (9.7) | 69.4 | 5.42, t (9.7) |
| 5 | 72.1 | 4.22, m | 72.1 | 4.21, m | 72.1 | 4.22, m | 72.1 | 4.22, m |
| 6 | 62.7 | 4.32, dd | 62.7 | 4.32, dd | 62.6 | 4.31, dd | 62.7 | 4.31, dd |
| (2.3; 12.1) | (2.1; 11.8) | (~2; 11.8) | (2.3;12.1) | |||||
| 4.51, dd | 4.50,dd, | 4.52, dd | 4.51, dd | |||||
| (5.4; 12.1) | (5.3; 12.0) | (4.8; 11.8) | (5.4;12.1) | |||||
| Ac (C-2) | 170.2 | - | 170.2 | - | 170.2 | - | 170.2 | - |
| Ac (C-3) | 170.1 | - | 170.1 | - | 172.2 | - | 170.2 | - |
| Ac (C4) | 169.8 | - | 169.9 | - | 169.9 | - | 169.9 | - |
| Ac (C-6) | 170.4 | - | 170.5 | - | 170.5 | - | 170.5 | - |
| Glc3 | ||||||||
| 1 | 99.6 | 6.37, d (7.9) | 99.6 | 6.35, d (8.0) | 99.6 | 6.36, d (7.6) | 99.6 | 6.36, d (7.9) |
| 2 | 72.3 | 5.62, m | 72.2 | 5.61, t (10) | 72.2 | 5.62, m | 72.2 | 5.62, m |
| 3 | 73.7 | 5.90, t (9.4) | 73.7 | 5.89, t (9.5) | 73.7 | 5.9, t (9.3) | 73.7 | 5.90, t (9.5) |
| 4 | 69.7 | 5.58, m | 69.7 | 5.56, m | 69.7 | 5.56, m | 69.7 | 5.56, m |
| 5 | 72.3 | 3.93, m | 72.2 | 3.92, m | 72.2 | 3.92, m | ||
| 6 | 62.5 | 4.19, m | 62.5 | 4.56, m | 62.4 | 4.19, m | 62.4 | 4.18, m |
| 4.57, m | 4.18, m | 4.56, m | 4.57, m | |||||
| Ac(C-2) | 169.6 | - | 169.6 | - | 169.7 | - | 169.7 | - |
| Ac(C-3) | 170.5 | - | 170.5 | - | 170.5 | - | 170.5 | - |
| Ac(C-4) | 169.7 | - | 169.7 | - | 169.8 | - | 169.7 | - |
| Ac(C-6) | 170.2 | - | 170.4 | - | 170.4 | - | 170.4 | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspar, H.; Cutignano, A.; Grauso, L.; Neng, N.; Cachatra, V.; Fontana, A.; Xavier, J.; Cerejo, M.; Vieira, H.; Santos, S. Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens. Mar. Drugs 2016, 14, 179. https://doi.org/10.3390/md14100179
Gaspar H, Cutignano A, Grauso L, Neng N, Cachatra V, Fontana A, Xavier J, Cerejo M, Vieira H, Santos S. Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens. Marine Drugs. 2016; 14(10):179. https://doi.org/10.3390/md14100179
Chicago/Turabian StyleGaspar, Helena, Adele Cutignano, Laura Grauso, Nuno Neng, Vasco Cachatra, Angelo Fontana, Joana Xavier, Marta Cerejo, Helena Vieira, and Susana Santos. 2016. "Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens" Marine Drugs 14, no. 10: 179. https://doi.org/10.3390/md14100179
APA StyleGaspar, H., Cutignano, A., Grauso, L., Neng, N., Cachatra, V., Fontana, A., Xavier, J., Cerejo, M., Vieira, H., & Santos, S. (2016). Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens. Marine Drugs, 14(10), 179. https://doi.org/10.3390/md14100179