
Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri


Abstract
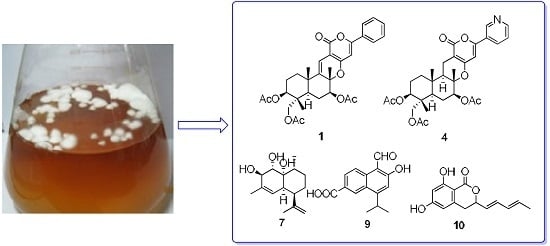
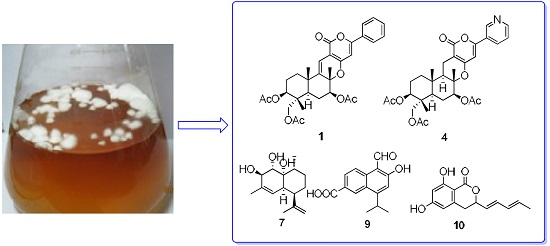
:
1. Introduction
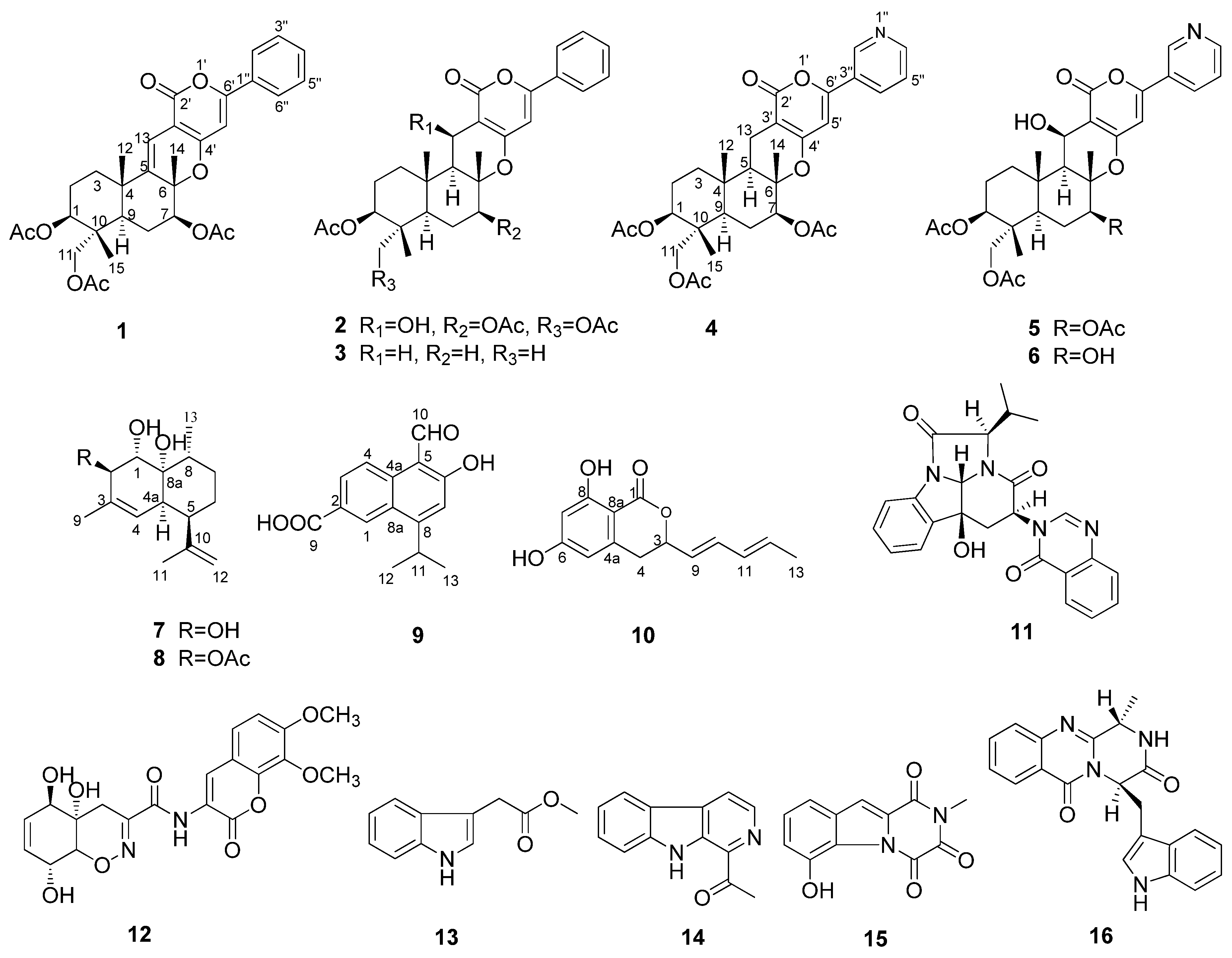
2. Results and Discussion
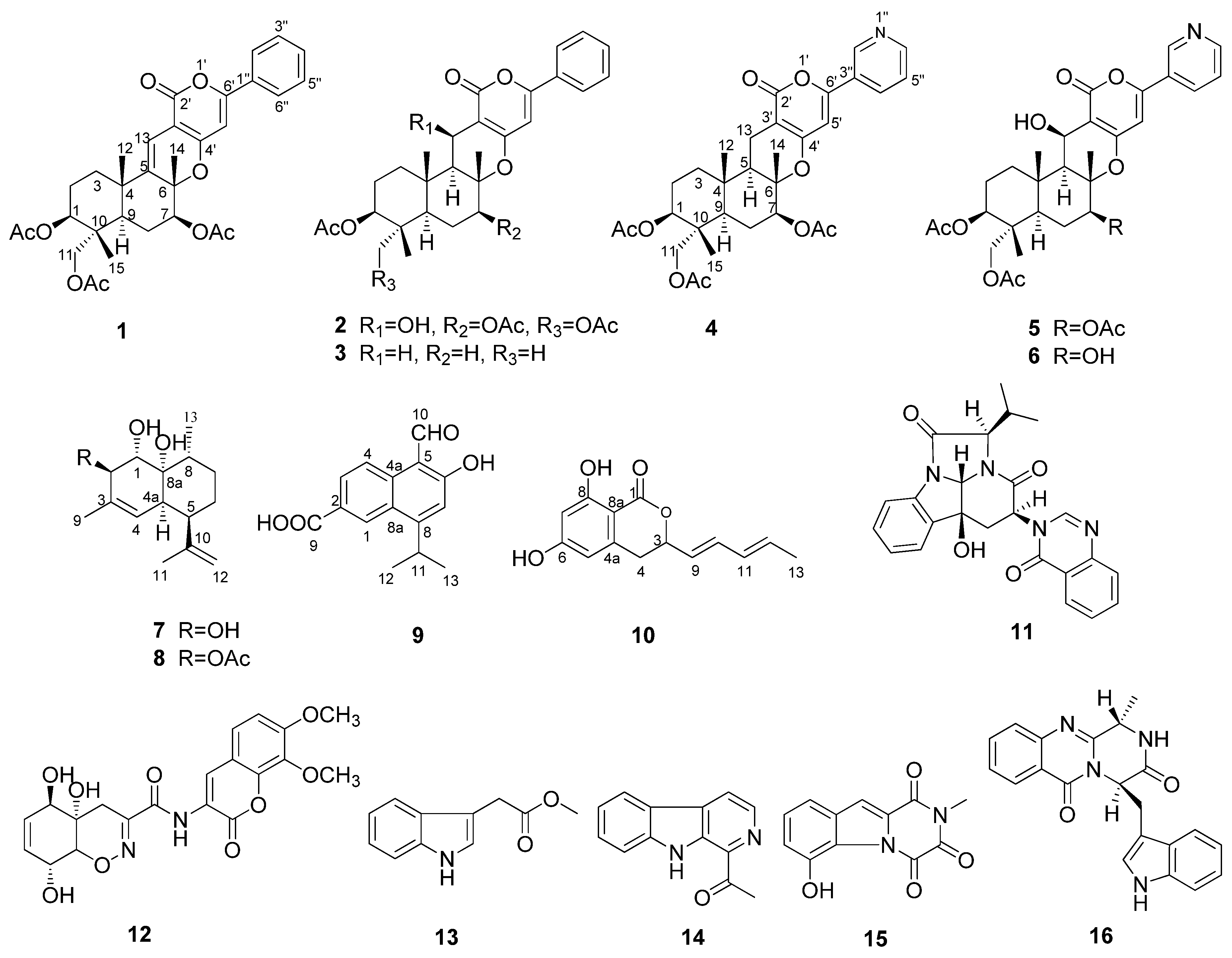
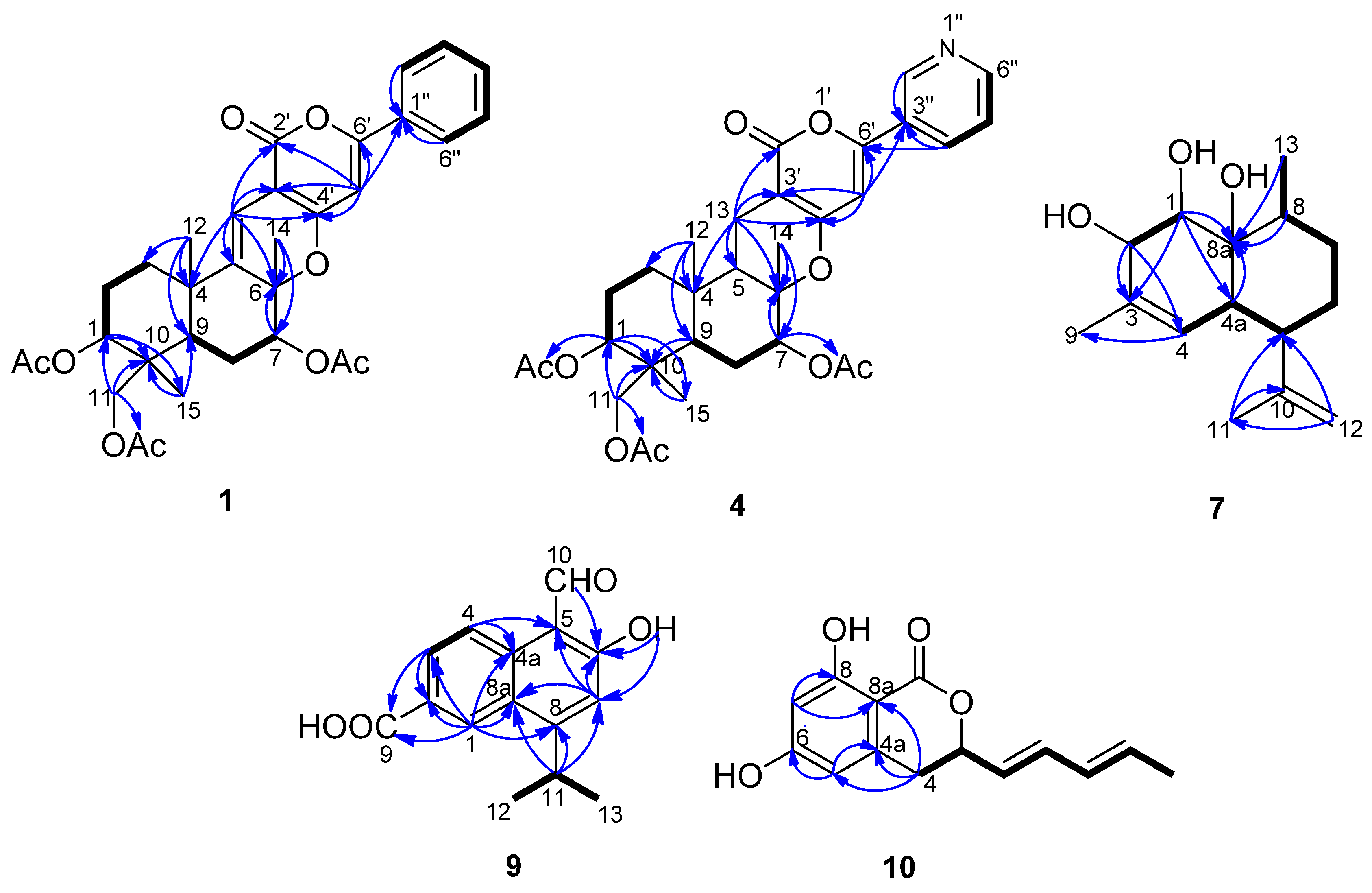
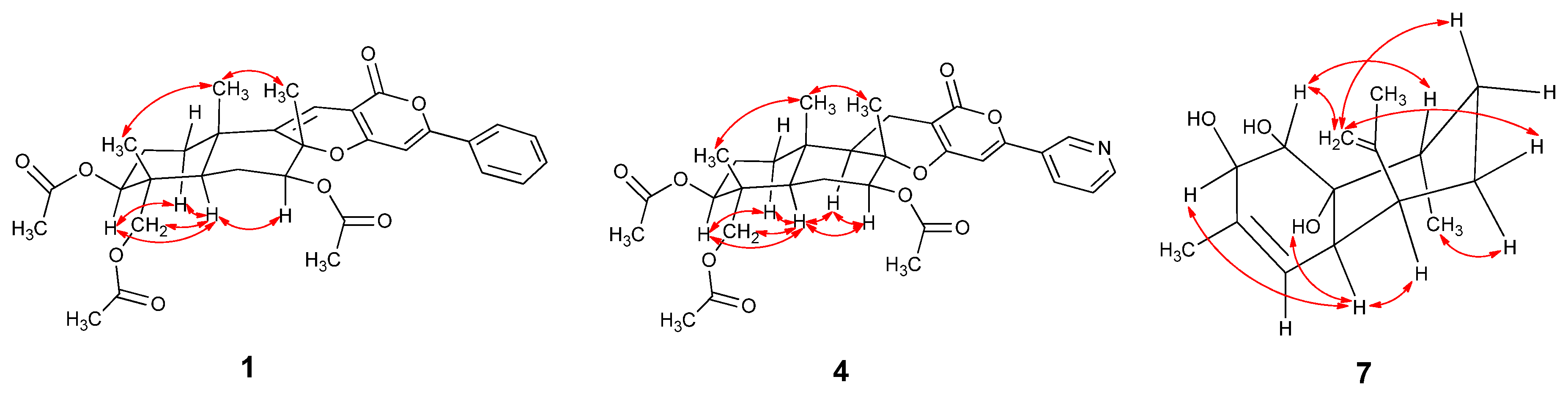
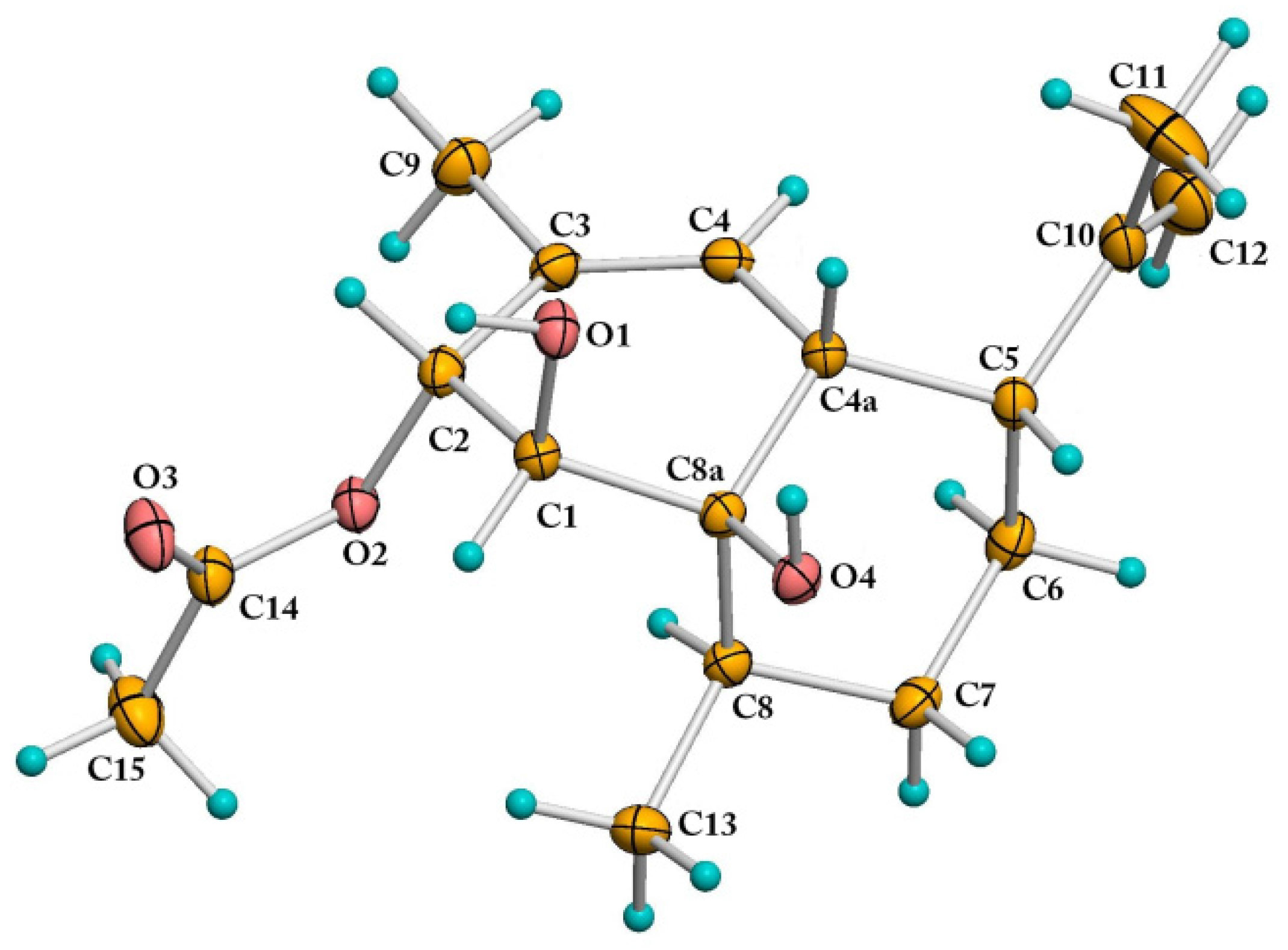
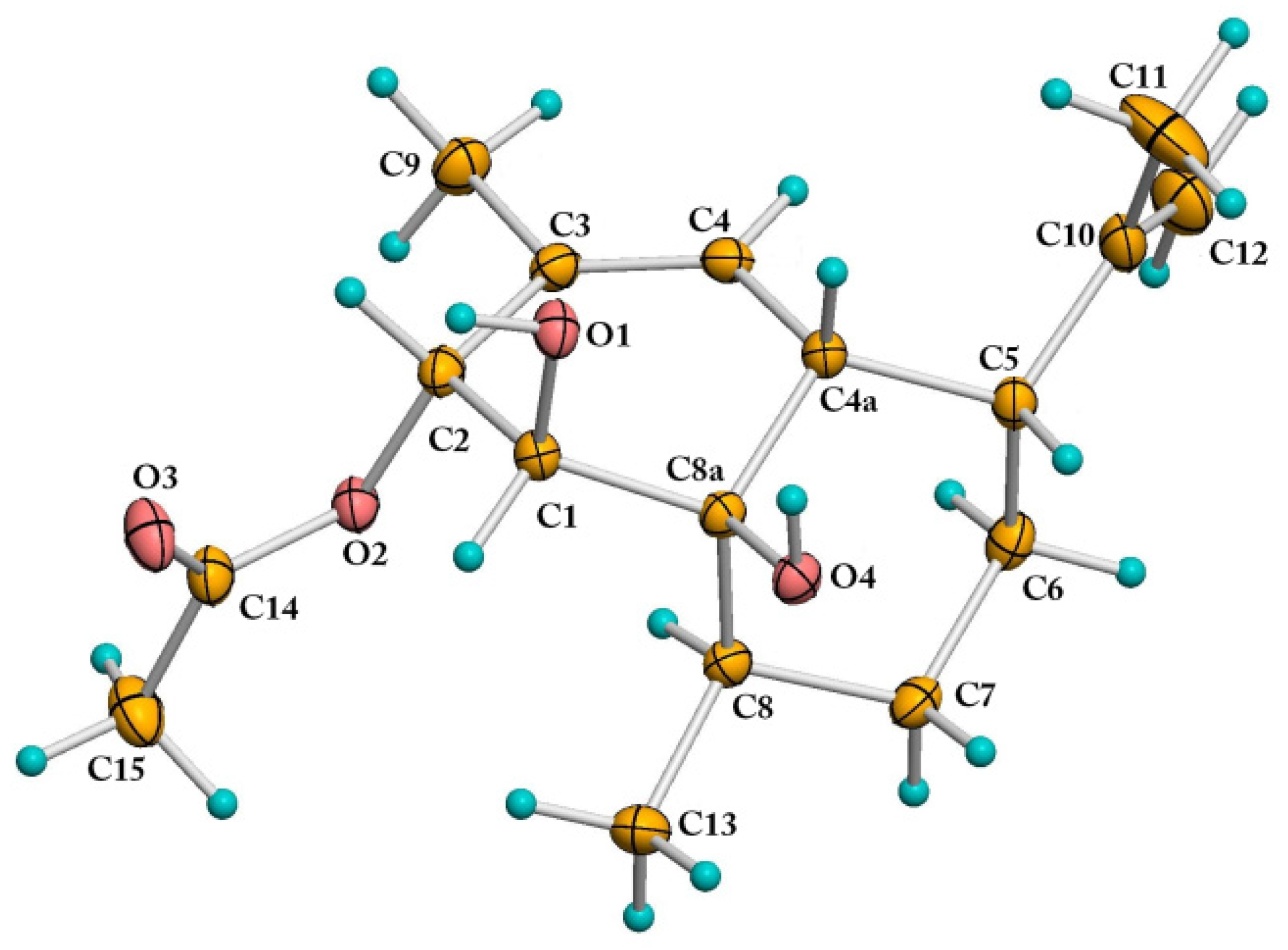
2.1. Structural Elucidation
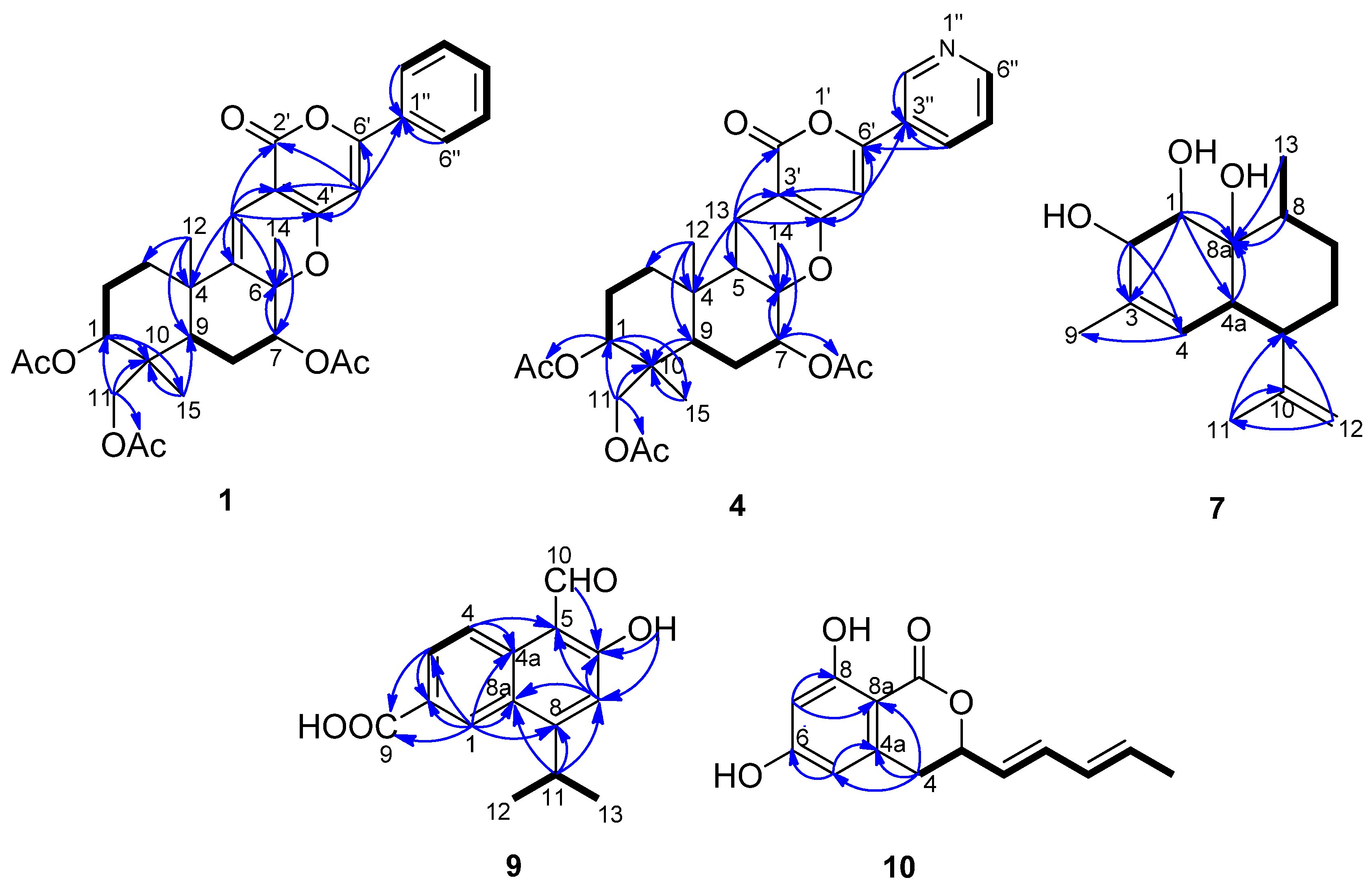

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 4 a | 6 b | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 73.3, CH | 4.79, dd (11.6, 4.4) | 73.5, CH | 4.79, dd (11.6, 4.8) | 74.4, CH | 4.79, dd (11.2, 5.6) |
| 2 | 23.2, CH2 | 1.98, m; 1.75, m | 22.8, CH2 | 1.88, m; 1.70, m | 23.6, CH2 | 1.84, m; 1.88, m |
| 3 | 35.4, CH2 | 2.08, m; 1.62, m | 36.6, CH2 | 1.83, m; 1.20, ddd (13.2, 13.2, 3.2) | 36.8, CH2 | 1.43, td (12.8, 4.8); 2.15, td (12.8, 4.8) |
| 4 | 38.7, C | 36.6, C | 38.8, C | |||
| 5 | 143.7, C | 50.3, CH | 1.60, dd (12.4, 4.8) | 55.0, CH | 1.66, d (3.6) | |
| 6 | 83.5, C | 81.9, C | 86.2, C | |||
| 7 | 77.7, CH | 5.22, d (12.0, 5.2) | 77.5, CH | 5.02, dd (11.6, 4.8) | 77.6, CH | 4.97, dd (12.4, 5.2) |
| 8 | 24.3, CH2 | 1.81, m; 1.63, m | 24.9, CH2 | 1.78, m; 1.50, m | 29.0, CH2 | 1.60, d (12.4); 1.82, m |
| 9 | 41.0, CH | 1.73, m | 45.1, CH | 1.66, d (12.0) | 46.2, CH | 1.53, d (3.6) |
| 10 | 40.5, C | 40.2, C | 41.3, C | |||
| 11 | 64.6, CH2 | 3.78, d (12.0); 3.74, d (12.0) | 64.7, CH2 | 3.78, d (12.0); 3.73, d (12.0) | 64.6, CH2 | 3.83, d (11.6); 3.77, d (11.6) |
| 12 | 24.2, CH3 | 1.25, s | 15.5, CH3 | 1.00, s | 17.9, CH3 | 1.48, s |
| 13 | 111.4, C | 6.36, s | 16.9, CH2 | 2.57, dd (17.2, 4.8); 2.33, dd (17.2, 12.4) | 60.6, CH | 4.95, d (3.6) |
| 14 | 21.2, CH3 | 1.57, s | 15.4, CH3 | 1.32, s | 15.9, CH3 | 1.76, s |
| 15 | 13.3, CH3 | 0.87, s | 13.3, CH3 | 0.86, s | 13.3, CH3 | 0.92, s |
| 2′ | 162.1, C | 163.7, C | 163.5, C | |||
| 3′ | 100.3, C | 99.8, C | 100.1, C | |||
| 4′ | 160.2, C | 162.2, C | 157.9, C | |||
| 5′ | 97.4, CH | 6.46, s | 99.2, CH | 6.43, s | 147.6, CH | |
| 6′ | 154.9, C | 155.9, C | 128.4, C | |||
| 1′′ | 131.0, C | |||||
| 2′′ | 125.6, CH | 7.81, m | 146.7, CH | 8.99, s | 147.6, CH | 9.07, dd (2.4, 0.8) |
| 3′′ | 128.9, CH | 7.44, m | 127.3, C | 127.5, C | ||
| 4′′ | 131.0, CH | 7.44, m | 132.8, CH | 8.09, brd (8.0) | 133.6, CH | 8.22, ddd (8.4, 2.4, 1.6) |
| 5′′ | 128.9, CH | 7.44, m | 123.6, CH | 7.39, dd (8.0, 4.8) | 124.6, CH | 7.53, ddd (8.4, 4.8, 0.8) |
| 6′′ | 125.6, CH | 7.81, m | 151.2, CH | 8.66, d (4.8) | 152.3, CH | 8.69, dd (4.8, 1.6) |
| 1-OCOCH3 | 21.1, CH3 | 2.04, s | 21.1, CH3 | 2.04, s | 21.0, CH3 | 2.00, s |
| 1-OCOCH3 | 170.0, C | 170.5, C | 170.6, C | |||
| 7-OCOCH3 | 21.1, CH3 | 2.16, s | 21.3, CH3 | 2.15, s | ||
| 7-OCOCH3 | 169.9, C | 170.1, C | ||||
| 11-OCOCH3 | 20.8, CH3 | 2.10, s | 20.8, CH3 | 2.11, s | 20.7, CH3 | 1.99, s |
| 11-OCOCH3 | 171.1, C | 171.0, C | 170.8, C | |||
| 13-OH | 4.28, brs | |||||
| 7-OH | 4.10, brs | |||||
| Position | 7 a | 9 b | 10 c | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult., (J in Hz) | δC, Type | δH, Mult., (J in Hz) | δC, Type | δH, Mult., (J in Hz) | |
| 1 | 74.6, CH | 3.98, d (1.2) | 127.9, CH | 8.92, d (1.5) | 169.1, C | |
| 2 | 74.2, CH | 4.05, s | 137.4, C | |||
| 3 | 132.3, C | 129.2, CH | 8.22, dd (9.0,1.5) | 78.4, CH | 5.13, ddd (10.0, 6.8, 4.0) | |
| 4 | 124.4, CH | 5.28, s | 121.6, CH | 8.77, d (9.0) | 32.5, CH2 | 3.00, dd (16.4, 4.0); 2.93, dd (16.4, 10.0) |
| 4a | 38.1, CH | 2.67, s | 127.3, C | 141.7, C | ||
| 5 | 41.4, CH | 2.38, brd (12.0) | 111.4, C | 107.1, CH | 6.24, d (2.0) | |
| 6 | 25.9, CH2 | 1.56, m; 1.33, m | 167.1, C | 163.4, C | ||
| 7 | 30.7, CH2 | 1.48, m; 1.40, m | 117.0, CH | 7.21, s | 101.0, CH | 6.18, d (2.0) |
| 8 | 31.2, CH | 1.99, m | 159.5, C | 164.8, C | ||
| 8a | 73.8, C | 126.3, C | 100.1, C | |||
| 9 | 20.1, CH3 | 1.78, s | 167.6, C | 127.0, CH | 5.71, dd (15.2, 6.8) | |
| 10 | 147.2, C | 195.3, CH | 11.42, brs | 133.3, CH | 6.35, dd (15.2, 10.0) | |
| 11 | 22.4, CH3 | 1.74, brs | 30.1, CH | 3.90, heptet (7.0) | 130.4, CH | 6.10, ddd (15.2, 10.0, 1.2) |
| 12 | 111.0, CH2 | 4.94, q (1.2 ); 4.72, s | 23.4, CH3 | 1.45, d (7.0) | 131.7, CH | 5.81, dq (15.2, 6.8) |
| 13 | 15.0, CH3 | 0.93, d (6.8) | 23.4, CH3 | 1.45, d (7.0) | 18.0, CH3 | 1.74, dd (6.8, 1.2) |
| 1-OH | 2.00, s | |||||
| 2-OH | 2.00, s | |||||
| 2-OCOCH3 | ||||||
| 2-OCOCH3 | ||||||
| 6-OH | 13.42, s | 11.06, brs | ||||
| 8-OH | 11.06, brs | |||||
| 8a-OH | 2.00, s | |||||
| COOH | 10.94, brs | |||||
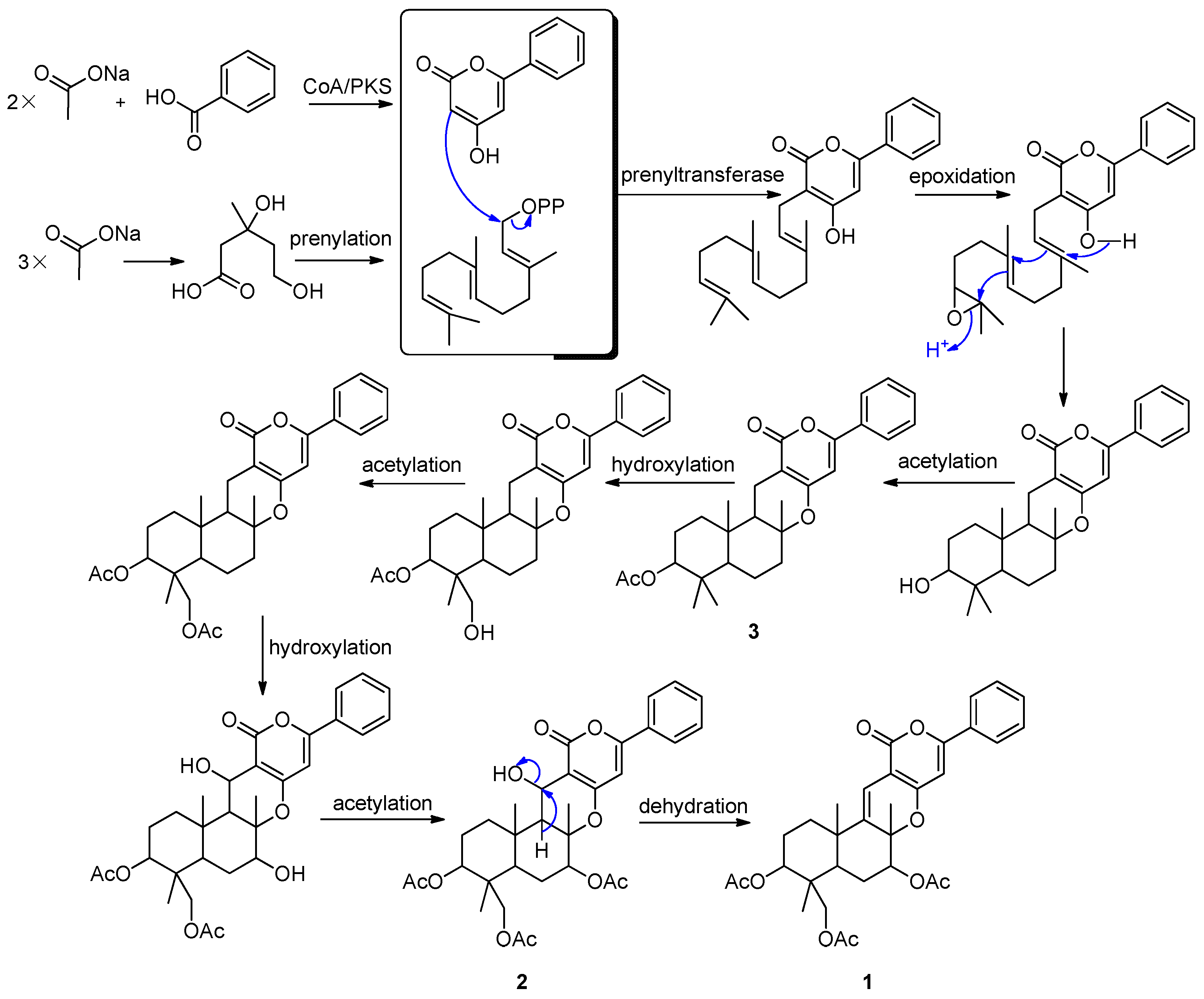
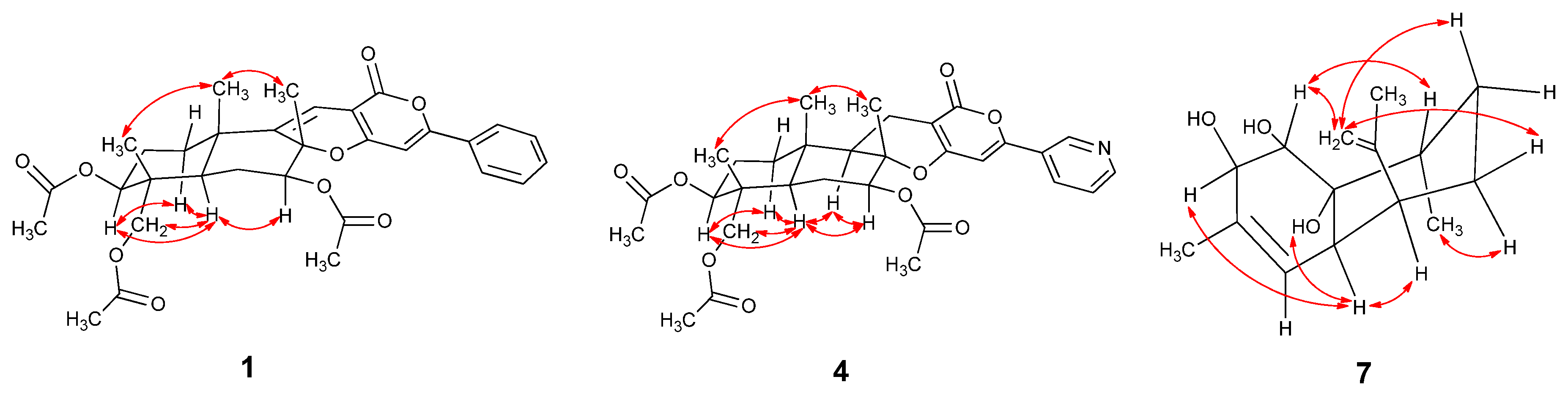
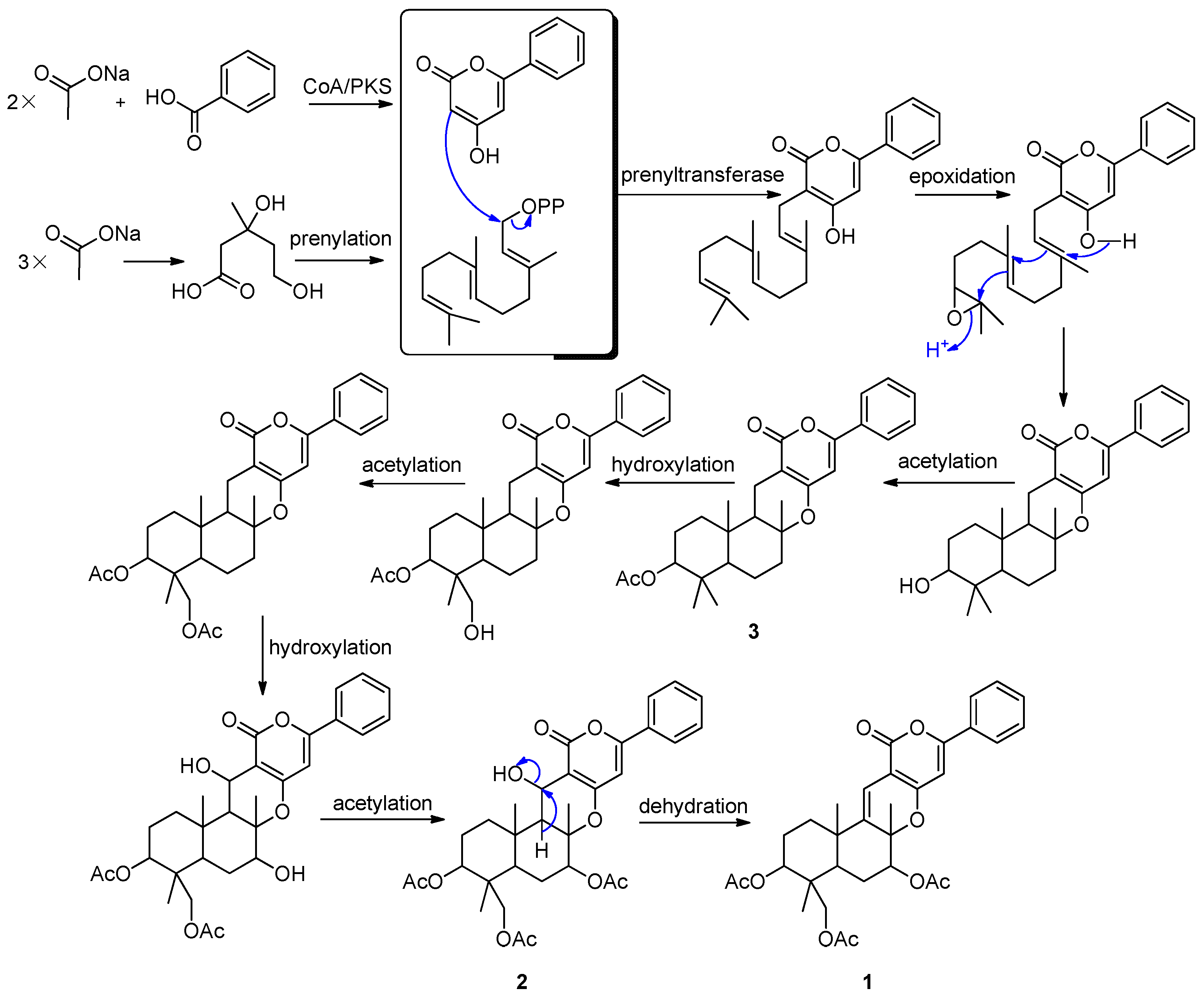
2.2. Proposed Biosynthetic Pathways
2.3. Cytotoxicity
| Compounds | 6 h | 12 h | 24 h | 48 h |
|---|---|---|---|---|
| 1 | 65.79 | 68.26 | 76.16 | 85.24 |
| 2 | 60.75 | 68.86 | 81.21 | 95.01 |
| 3 | 72.55 | 72.15 | 73.08 | 93.73 |
| 4 | 47.53 | 57.41 | 73.21 | 85.37 |
| 5 | 41.05 | 54.60 | 58.04 | 71.77 |
| 6 | 25.58 | 29.46 | 35.30 | 56.03 |
| 7 | 79.58 | 81.89 | 90.46 | 98.68 |
| 8 | 64.52 | 69.33 | 77.98 | 91.87 |
| 9 | 57.96 | 65.32 | 69.92 | 90.97 |
| 10 | 36.47 | 38.25 | 52.36 | 61.67 |
| 11 | 63.70 | 69.16 | 71.65 | 94.53 |
| 15 | 56.97 | 61.43 | 80.33 | 92.52 |
| Rotenone | 69.21 | 79.43 | 90.93 | 98.54 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction and Isolation
3.4. Bioassay
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ansante, T.F.; Ribeiro, L.D.; Bicalho, K.U.; Fernandes, J.B.; da Silva, M.F.D.F.; Vieira, P.C.; Vendramim, J.D. Secondary metabolites from Neotropical Annonaceae: Screening, bioguided fractionation, and toxicity to Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae). Ind. Crops Prod. 2015, 74, 969–976. [Google Scholar] [CrossRef]
- Munoz, E.; Escalona, D.; Salazar, J.R.; Alarcon, J.; Cespedes, C.L. Insect growth regulatory effects by diterpenes from Calceolaria talcana Grau & Ehrhart (Calceolariaceae: Scrophulariaceae) against Spodoptera frugiperda and Drosophila melanogaster. Ind. Crop. Prod. 2013, 45, 283–292. [Google Scholar]
- Devappa, R.K.; Angulo-Escalante, M.A.; Makkar, H.P.S.; Becker, K. Potential of using phorbol esters as an insecticide against Spodoptera frugiperda. Ind. Crop. Prod. 2012, 38, 50–53. [Google Scholar] [CrossRef]
- Sarria, A.L.F.; Soares, M.S.; Matos, A.P.; Fernandes, J.B.; Vieira, P.C.; da Silva, M.F.G.F. Effect of triterpenoids and limonoids isolated from Cabralea canjerana and Carapa guianensis (Meliaceae) against Spodoptera frugiperda (J.E. Smith). Z. Naturforschung C 2011, 66, 245–250. [Google Scholar] [CrossRef]
- Nihei, K.I.; Asaka, Y.; Mine, Y.; Ito, C.; Furukawa, H.; Motoharu, J.I.; Kubo, I. Insect antifeedants from tropical plants: Structures of dumnin and dumsenin. J. Agric. Food Chem. 2004, 52, 3325–3328. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.L.; Goodwin, R.H.; Tompkins, G.J.; McCawley, P. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 1977, 13, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhang, Y.M.; Weng, Q.F.; Hu, M.Y.; Zhong, G.H. Cytotoxic and insecticidal activities of derivatives of harmine, a natural insecticidal component isolated from Peganum harmala. Molecules 2010, 15, 7775–7791. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.H.; Seifert, R. Sf9 cells: A versatile model system to investigate the pharmacological properties of G protein-coupled receptors. Pharmacol. Ther. 2010, 128, 387–418. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Jiang, W.H.; Liang, W.L.; Huang, J.X.; Mo, Y.F.; Ding, Y.Q.; Lam, C.K.; Qian, X.J.; Zhu, X.F.; Lan, W.J. Induced marine fungus Chondrostereum sp. as a means of producing new sesquiterpenoids chondrosterins I and J by using glycerol as the carbon source. Mar. Drugs 2014, 12, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.J.; Liu, W.; Liang, W.L.; Xu, Z.; Le, X.; Xu, J.; Lam, C.K.; Yang, D.P.; Li, H.J.; Wang, L.Y. Pseudaboydins A and B: Novel isobenzofuranone derivatives from marine fungus Pseudallescheria boydii associated with starfish Acanthaster planci. Mar. Drugs 2014, 12, 4188–4199. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Chen, T.; Xie, Y.L.; Chen, W.D.; Zhu, X.F.; Lan, W.J. Isolation and structural elucidation of chondrosterins F–H from the marine fungus Chondrostereum sp. Mar. Drugs 2013, 11, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A–E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.L.; Le, X.; Li, H.J.; Yang, X.L.; Chen, J.X.; Xu, J.; Liu, H.L.; Wang, L.Y.; Wang, K.T.; Hu, K.C.; et al. Exploring the chemodiversity and biological activities of the secondary metabolites from the marine fungus Neosartorya pseudofischeri. Mar. Drugs 2014, 12, 5657–5676. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.E.; Rho, M.C.; Song, H.Y.; Lee, S.W.; Chung, M.Y.; Lee, J.H.; Kim, Y.H.; Lee, H.S.; Kim, Y.K. Phenylpyropene A and B, new inhibitors of acyl-CoA: Cholesterol acyltransferase produced by Penicillium griseofulvum F1959. J. Antibiot. 2002, 55, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.C.; Lee, H.S.; Chang, K.T.; Song, H.Y.; Kwon, O.E.; Lee, S.W.; Ko, J.S.; Hong, S.G.; Kim, Y.K. Phenylpyropene C, a new inhibitor of acyl-CoA: Cholesterol acyltransferase produced by Penicillium griseofulvum F1959. J. Antibiot. 2002, 55, 211–214. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, J.H.; Rho, M.C.; Lee, S.W.; Choi, J.N.; Lee, H.J.; Bae, K.S.; Kim, K.; Kim, Y.K. Penicillium griseofulvum F1959, high-production strain of pyripyropene A, specific inhibitor of Acyl-CoA: Cholesterol acyltransferase 2. J. Microbiol. Biotechnol. 2008, 18, 1663–1665. [Google Scholar] [PubMed]
- Obata, R.; Sunazuka, T.; Li, Z.; Tian, Z.; Harigaya, Y.; Tabata, N.; Tomoda, H.; Omura, S. Chemical modification and structure-activity relationships of pyripyropenes. 1. Modification at the four hydroxyl groups. J. Antibiot. 1996, 49, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Didier, C.; Critcher, D.J.; Walshe, N.D.; Kojima, Y.; Yamauchi, Y.; Barrett, A.G.M. Full stereochemical assignment and synthesis of the potent anthelmintic pyrrolobenzoxazine natural product CJ-12662. J. Org. Chem. 2004, 69, 7875–7879. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Andolfi, A.; Mathieu, V.; Boari, A.; Cimmino, A.; Moreno Y Banuls, L.; Vurro, M.; Kornienko, A.; Kiss, R.; Evidente, A. Fischerindoline, a pyrroloindole sesquiterpenoid isolated from Neosartorya pseudofischeri, with in vitro growth inhibitory activity in human cancer cell lines. Tetrahedron 2013, 69, 7466–7470. [Google Scholar] [CrossRef]
- Kanokmedhakul, K.; Kanokmedhakul, S.; Suwannatrai, R.; Soytong, K.; Prabpai, S.; Kongsaeree, P. Bioactive meroterpenoids and alkaloids from the fungus Eurotium chevalieri. Tetrahedron 2011, 67, 5461–5468. [Google Scholar] [CrossRef]
- Liao, L.J.; You, M.J.; Chung, B.K.; Oh, D.C.; Oh, K.B.; Shin, J. Alkaloidal metabolites from a marine-derived Aspergillus sp. fungus. J. Nat. Prod. 2015, 78, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Garo, E.C.; Starks, M.; Jensen, P.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Jiang, Y.; Han, L.; Lou, K.; Chen, Y.; Xu, L.; Huang, X. Chemical constituents from the mycelium of a new streptomycete. Chin. J. Med. Chem. 2010, 20, 201–205. [Google Scholar]
- Lee, D.S.; Eom, S.H.; Jeong, S.Y.; Shin, H.J.; Je, J.Y.; Lee, E.W.; Chung, Y.H.; Kim, Y.M.; Kang, C.K.; Lee, M.S. Anti-methicillin-resistant Staphylococcus aureus (MRSA) substance from the marine bacterium Pseudomonas sp. UJ-6. Environ. Toxicol. Phar. 2013, 35, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.; Taylor, A.; Vining, L.C. Sporidesmins. VI. Isolation and structure of dehydrogliotoxin a metabolite of Penicillium terlikowskii. J. Chem. Soc. Perkin Trans.1 1966, 20, 1799–1803. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines A–G, novel metabolites of a fungus separated from a Pseudolabrus marine fish. J. Chem. Soc. Perkin Trans. 1995, 18, 2345–2353. [Google Scholar] [CrossRef]
- Tomoda, H.; Tabata, N.; Nakata, Y.; Nishida, H.; Kaneko, T.; Obata, R.; Sunazuka, T.; Omura, S. Biosynthesis of pyripyropene A. J. Org. Chem. 1996, 61, 882–886. [Google Scholar] [CrossRef]
- Huang, J.F.; Tian, M.; Lv, C.J.; Li, H.Y.; Muhammad, R.H.; Zhong, G.H. Preliminary studies on induction of apoptosis by abamectin in Spodoptera frugiperda (Sf9) cell line. Pestic. Biochem. Physiol. 2011, 100, 256–263. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, W.-J.; Fu, S.-J.; Xu, M.-Y.; Liang, W.-L.; Lam, C.-K.; Zhong, G.-H.; Xu, J.; Yang, D.-P.; Li, H.-J. Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri. Mar. Drugs 2016, 14, 18. https://doi.org/10.3390/md14010018
Lan W-J, Fu S-J, Xu M-Y, Liang W-L, Lam C-K, Zhong G-H, Xu J, Yang D-P, Li H-J. Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri. Marine Drugs. 2016; 14(1):18. https://doi.org/10.3390/md14010018
Chicago/Turabian StyleLan, Wen-Jian, Sheng-Jiao Fu, Meng-Yang Xu, Wan-Ling Liang, Chi-Keung Lam, Guo-Hua Zhong, Jun Xu, De-Po Yang, and Hou-Jin Li. 2016. "Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri" Marine Drugs 14, no. 1: 18. https://doi.org/10.3390/md14010018
APA StyleLan, W.-J., Fu, S.-J., Xu, M.-Y., Liang, W.-L., Lam, C.-K., Zhong, G.-H., Xu, J., Yang, D.-P., & Li, H.-J. (2016). Five New Cytotoxic Metabolites from the Marine Fungus Neosartorya pseudofischeri. Marine Drugs, 14(1), 18. https://doi.org/10.3390/md14010018