
Bioactive Isopimarane Diterpenes from the Fungus, Epicoccum sp. HS-1, Associated with Apostichopus japonicus

Abstract
:1. Introduction
2. Results and Discussion
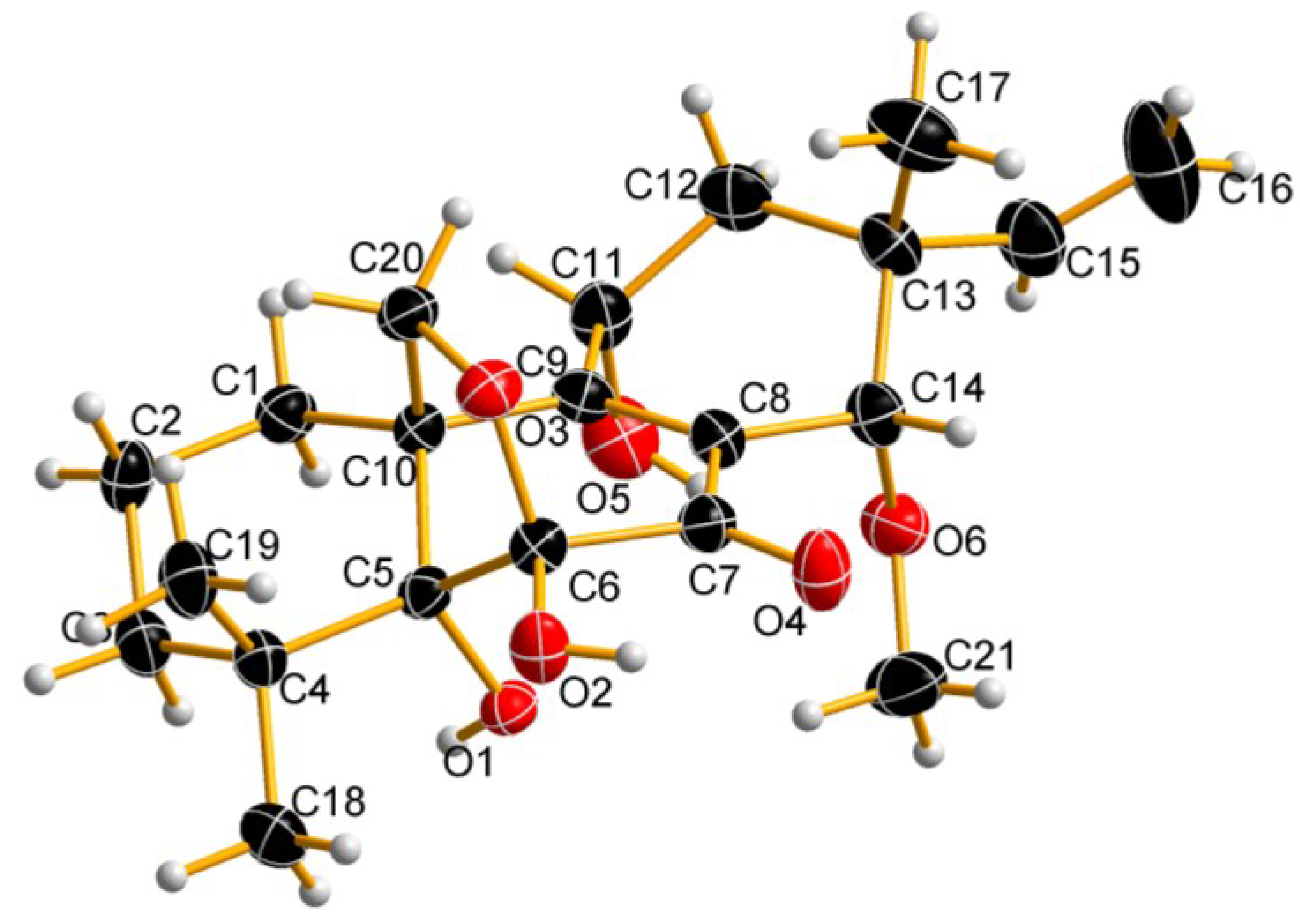
2.1. Chemical Structure Elucidation
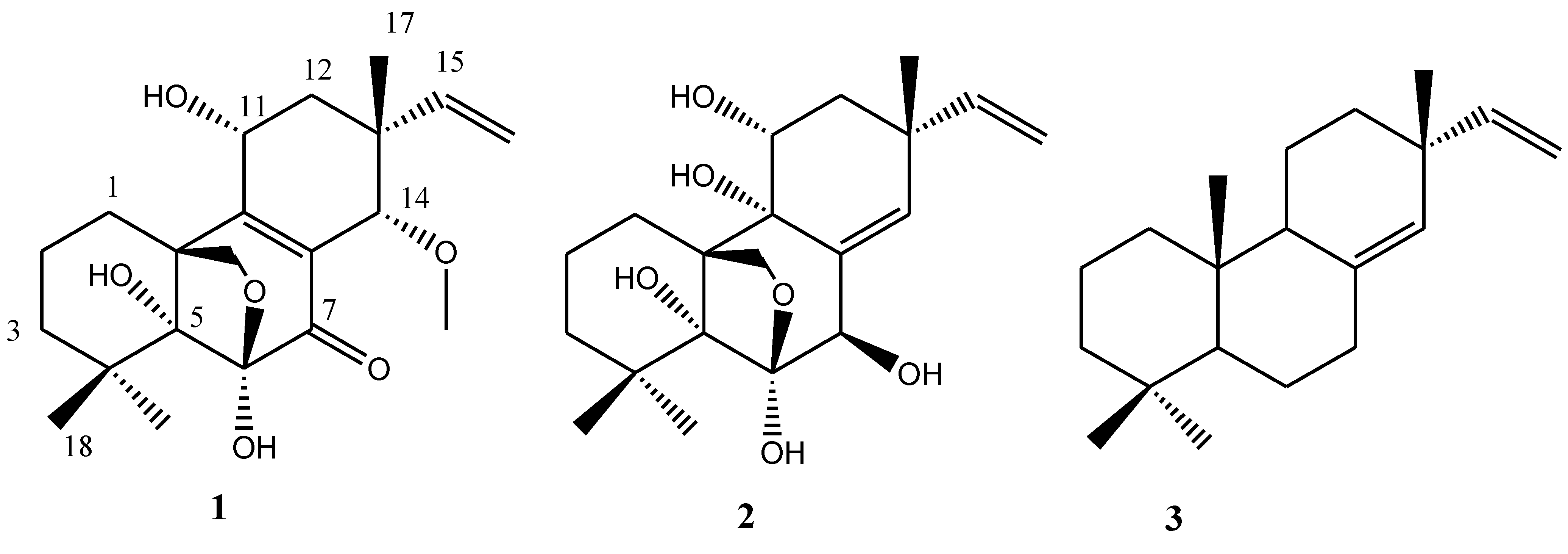

{kind=link}
{kind=link}
| Position | δC, mult. | δH | HMBC (H→C) |
|---|---|---|---|
| 1 | 23.3, CH2 | a, 2.15 (dt, 4.8, 13.8); b, 1.67 (m) | C-2, 10 |
| 2 | 17.8, CH2 | 1.82 (2H, dt, 6.0,13.0) | C-1, 3 |
| 3 | 37.1, CH2 | a, 1.60 (dt, 3.0, 13.8); b, 1.28 (m) | C-2, 5 |
| 4 | 36.8, C | ||
| 5 | 81.2, C | ||
| 6 | 105.0, C | ||
| 7 | 193.4, C | ||
| 8 | 133.2, C | ||
| 9 | 165.7, C | ||
| 10 | 52.9, C | ||
| 11 | 62.8, CH | 4.42 (dd, 2.0, 8.4) | C-8, 9, 12 |
| 12 | 40.8, CH2 | a, 2.30 (dd, 2.0, 14.4); b, 1.78 (dd, 9.0, 14.4) | C-9, 11, 13, 15 |
| 13 | 41.2, C | ||
| 14 | 80.0, CH | 4.15 (s) | C-7, 9, 21-OCH3 |
| 15 | 145.3, CH | 6.20 (dd, 11.4, 18.6) | C-16 |
| 16 | 112.3, CH2 | a, 5.07 (d, 12.0); b, 5.11 (d, 18.0) | C-14, 15 |
| 17 | 25.5, CH3 | 0.81 (s) | C-13, 14, 15 |
| 18 | 23.8, CH3 | 1.51 (s) | C-4, 5, 19 |
| 19 | 28.1, CH3 | 1.18 (s) | C-4, 5, 18 |
| 20 | 69.8, CH2 | a, 4.43 (d, 9.0); b, 3.22 (d, 9.0) | C-5, 6, 9 |
| 21 | 57.6, CH3 | 3.23 (s) | C-14 |
2.2. Biological Activity
| Compounds | IC50 (μM) |
|---|---|
| 1 | 4.6 ± 0.1 |
| 2 | 11.9 ± 0.4 |
| 3 | >200.0 |
| Resveratrol a | 31.2 ± 4.4 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.4. Assays for Inhibitory Activity
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lyssenko, V.; Laakso, M. Genetic screening for the risk of Type 2 diabetes. Diabetes Care 2013, 36, 5120–5126. [Google Scholar] [CrossRef]
- Van de Laar, F.A.; Lucassen, P.L.; Akkermans, R.P.; van de Lisdonk, E.H.; de Grauw, W.J. Alpha-glucosidase inhibitors for people with impaired glucose tolerance or impaired fasting blood glucose. Cochrane Database Syst. Rev. 2006, 18, CD005061. [Google Scholar]
- Ahamad, J.; Naquvi, K.J.; Mir, S.R.; Ali, M.; Shuaib, M. Review on role of natural α-glucosidase inhibitors for management of diabetes mellitus. Int. J. Biomed. Res. 2011, 2, 374–380. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Mitra, A.; Dey, B.; Pal, A. Exploration of Anti-diabetic Potentials Amongst Marine Species. Indo Global J. Pharmaceut. Sci. 2014, 4, 65–73. [Google Scholar]
- Xiao, Z.E.; Huang, H.R.; Shao, C.L.; Xia, X.K.; Ma, L.; Huang, X.S.; Lu, Y.J.; Lin, Y.C.; Long, Y.H.; She, Z.G. Asperterpenols A and B, new sesterterpenoids isolated from a mangrove endophytic fungus Aspergillus sp. 085242. Org. Lett. 2013, 15, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.K.; Zhang, J.Y.; Zhang, Y.G.; Wei, F.; Liu, X.; Jia, A.R.; Liu, C.H.; Li, W.; She, Z.G.; Lin, Y.C. Pimarane diterpenes from the fungus Epicoccum sp. HS-1 associated with Apostichopus japonicus. Bioorg. Med. Chem. Lett. 2012, 22, 3017–3019. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Xia, G.X.; Li, H.X.; Ma, L.; Ding, B.; Lu, Y.J.; He, L.; Xia, X.K.; She, Z.G. Vermistatin derivatives with α-Glucosidase inhibitory activity from the mangrove endophytic fungus Penicillium sp. Planta Med. 2014, 80, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Bashyal, B.P.; Wijeratne, E.M.K.; U’Ren, J.M.; Liu, M.X.; Gunatilaka, M.K.; Arnold, A.E.; Gunatilaka, A.A.L. Smardaesidins A–G, isopimarane and 20-nor-isopimarane diterpenoids from Smardaea sp., a fungal endophyte of the moss Ceratodon purpureus. J. Nat. Prod. 2011, 74, 2052–2061. [Google Scholar]
- Maslovskaya, L.A.; Savchenko, A.I.; Gordon, V.A.; Reddell, P.W.; Pierce, C.J.; Parsons, P.G.; Williams, C.M. EBC-316, 325–327, and 345: New pimarane diterpenes from Croton insularis found in the Australian rainforest. Aust. J. Chem. 2015. doi: org/10.1071/CH14550. [Google Scholar]
- Yilmaz, A.; Caglar, P.; Dirmenci, T.; Goren, N.; Topcu, G. A novel isopimarane diterpenoid with acetylcholinesterase inhibitory activity from Nepeta sorgerae, an endemic species to the Nemrut Mountain. Nat. Prod. Commun. 2012, 7, 693–696. [Google Scholar] [PubMed]
- Huang, S.Z.; Ma, Q.Y.; Fang, W.W.; Xu, F.Q.; Peng, H.; Dai, H.F.; Zhou, J.; Zhao, Y.X. Three new isopimarane diterpenoids from Excoecaria acerifolia. J. Asian Nat. Prod. Res. 2013, 15, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kito, K.; Ooi, T.; Kanoh, K.; Shizuri, Y.; Kusumi, T. Four pimarane diterpenes frommarine fungus: Chloroform incorporated in crystal lattice for absolute configuration analysis by X-ray. Chem. Lett. 2007, 36, 1386–1387. [Google Scholar] [CrossRef]
- Dettrakul, S.; Kittakoop, P.; Isaka, M.; Nophichai, S.; Suyarnsestakorn, C.; Tanticharoen, M.; Thebtaranonth, Y. Antimycobacterial pimarane diterpenes from the fungus Diaporthe sp. Bioorg. Med. Chem. Lett. 2003, 13, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Bromanm, K.; Viljanen, K.; Moreira, V.M.; Yli-Kauhaluoma, J.; Ruohonen, L.; Nakari-Setala, T. Isolation and purification of ent-pimara-8(14), 15-diene from engineered Aspergillus nidulans by accelerated solvent extraction combined with HPLC. Anal. Methods 2014, 6, 1227–1234. [Google Scholar] [CrossRef]
- Du, Z.Y.; Liu, R.R.; Shao, W.Y.; Mao, X.P.; Ma, L.; Gu, L.Q.; Huang, Z.S.; Chan, A.S.C. α-Glucosidase inhibition of natural curcuminoids and curcumin analogs. Eur. J. Med. Chem. 2006, 41, 213–218. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X.; Qi, J.; Liu, Y.; Jia, A.; Zhang, Y.; Liu, C.; Gao, C.; She, Z. Bioactive Isopimarane Diterpenes from the Fungus, Epicoccum sp. HS-1, Associated with Apostichopus japonicus. Mar. Drugs 2015, 13, 1124-1132. https://doi.org/10.3390/md13031124
Xia X, Qi J, Liu Y, Jia A, Zhang Y, Liu C, Gao C, She Z. Bioactive Isopimarane Diterpenes from the Fungus, Epicoccum sp. HS-1, Associated with Apostichopus japonicus. Marine Drugs. 2015; 13(3):1124-1132. https://doi.org/10.3390/md13031124
Chicago/Turabian StyleXia, Xuekui, Jun Qi, Yayue Liu, Airong Jia, Yonggang Zhang, Changheng Liu, Cuiling Gao, and Zhigang She. 2015. "Bioactive Isopimarane Diterpenes from the Fungus, Epicoccum sp. HS-1, Associated with Apostichopus japonicus" Marine Drugs 13, no. 3: 1124-1132. https://doi.org/10.3390/md13031124
APA StyleXia, X., Qi, J., Liu, Y., Jia, A., Zhang, Y., Liu, C., Gao, C., & She, Z. (2015). Bioactive Isopimarane Diterpenes from the Fungus, Epicoccum sp. HS-1, Associated with Apostichopus japonicus. Marine Drugs, 13(3), 1124-1132. https://doi.org/10.3390/md13031124