An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent

Abstract
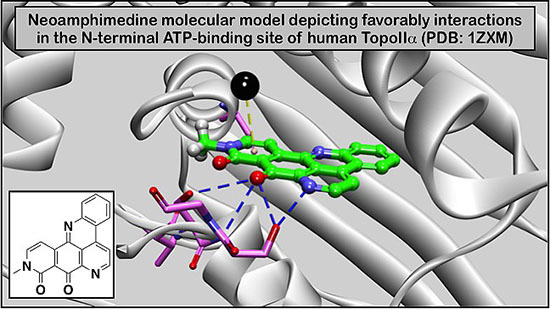
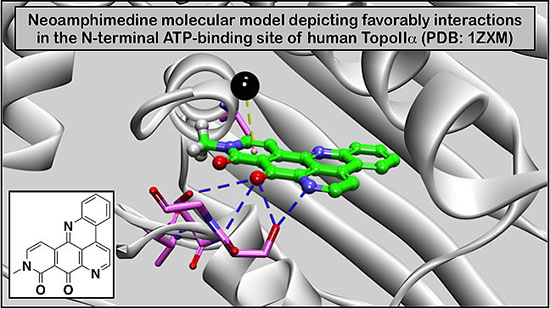
:
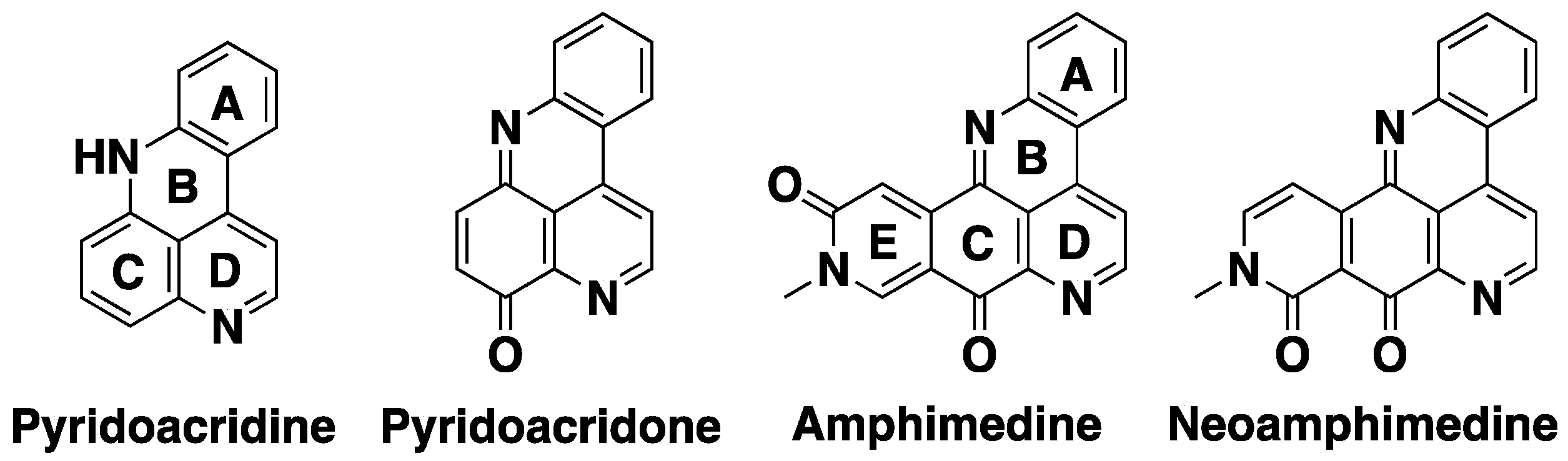
1. Introduction
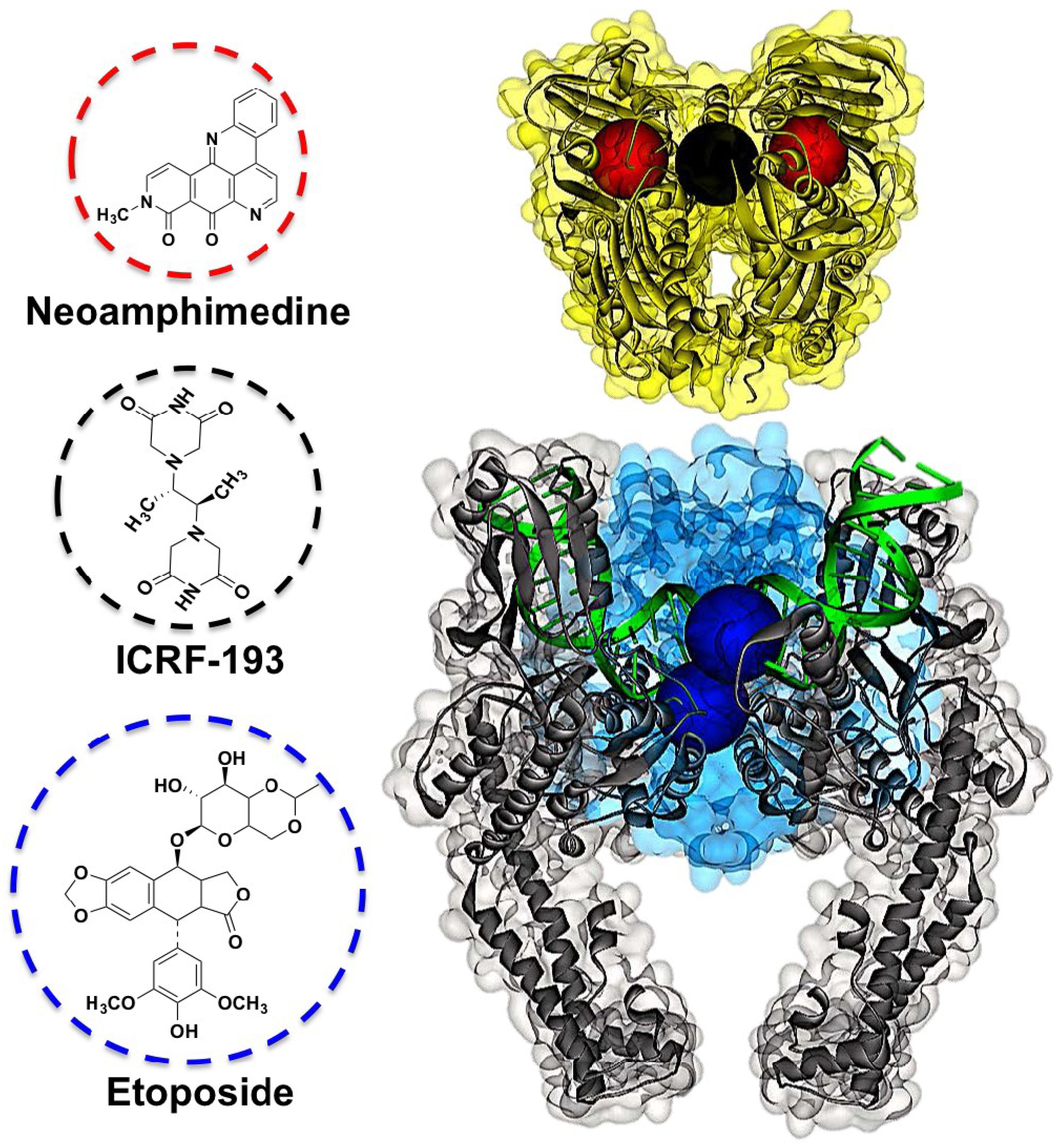
2. Results and Discussion
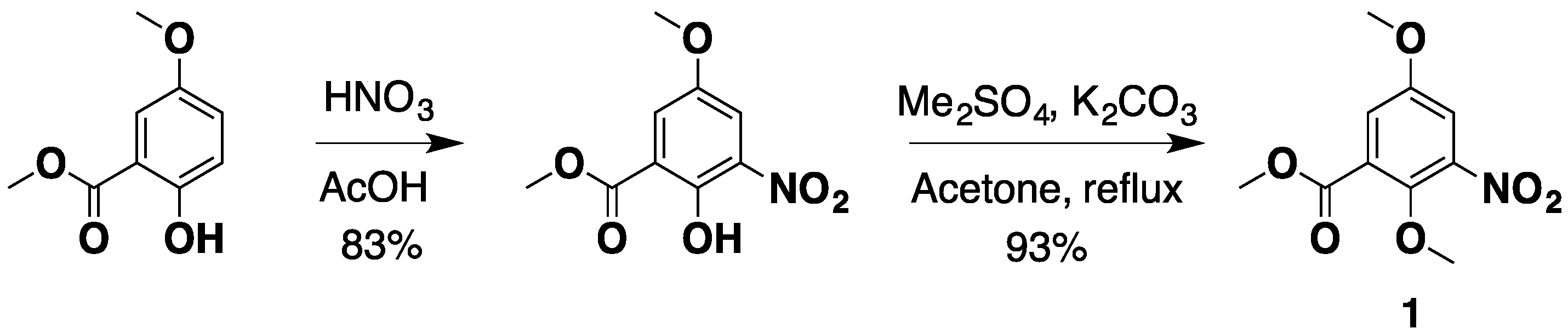
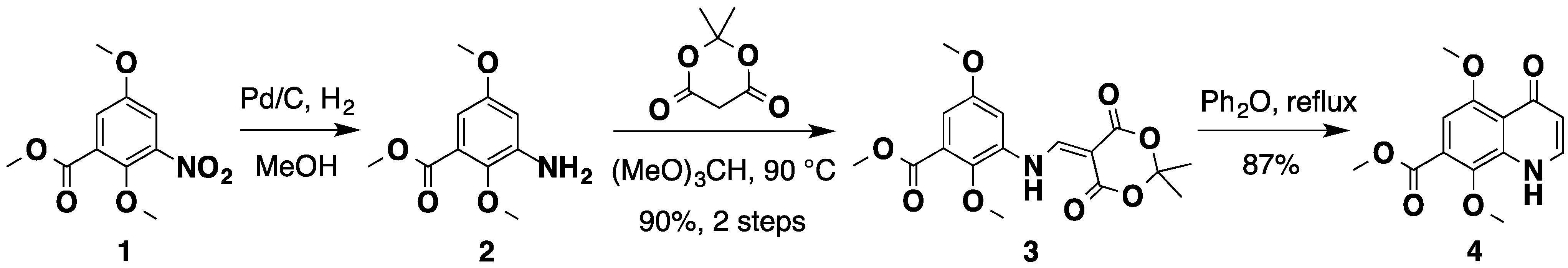
2.1. An Improved Total Synthesis of Neoamphimedine



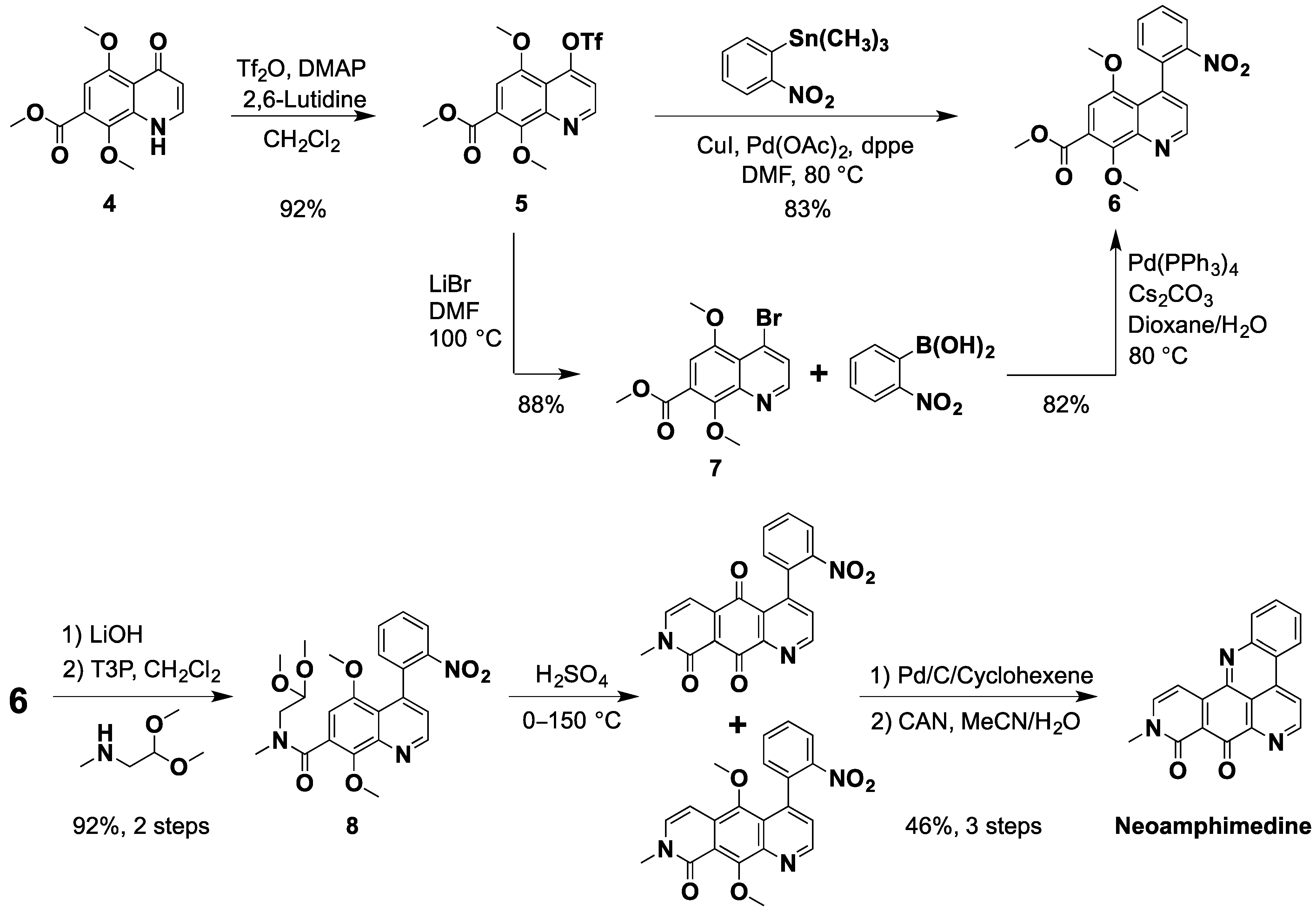

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
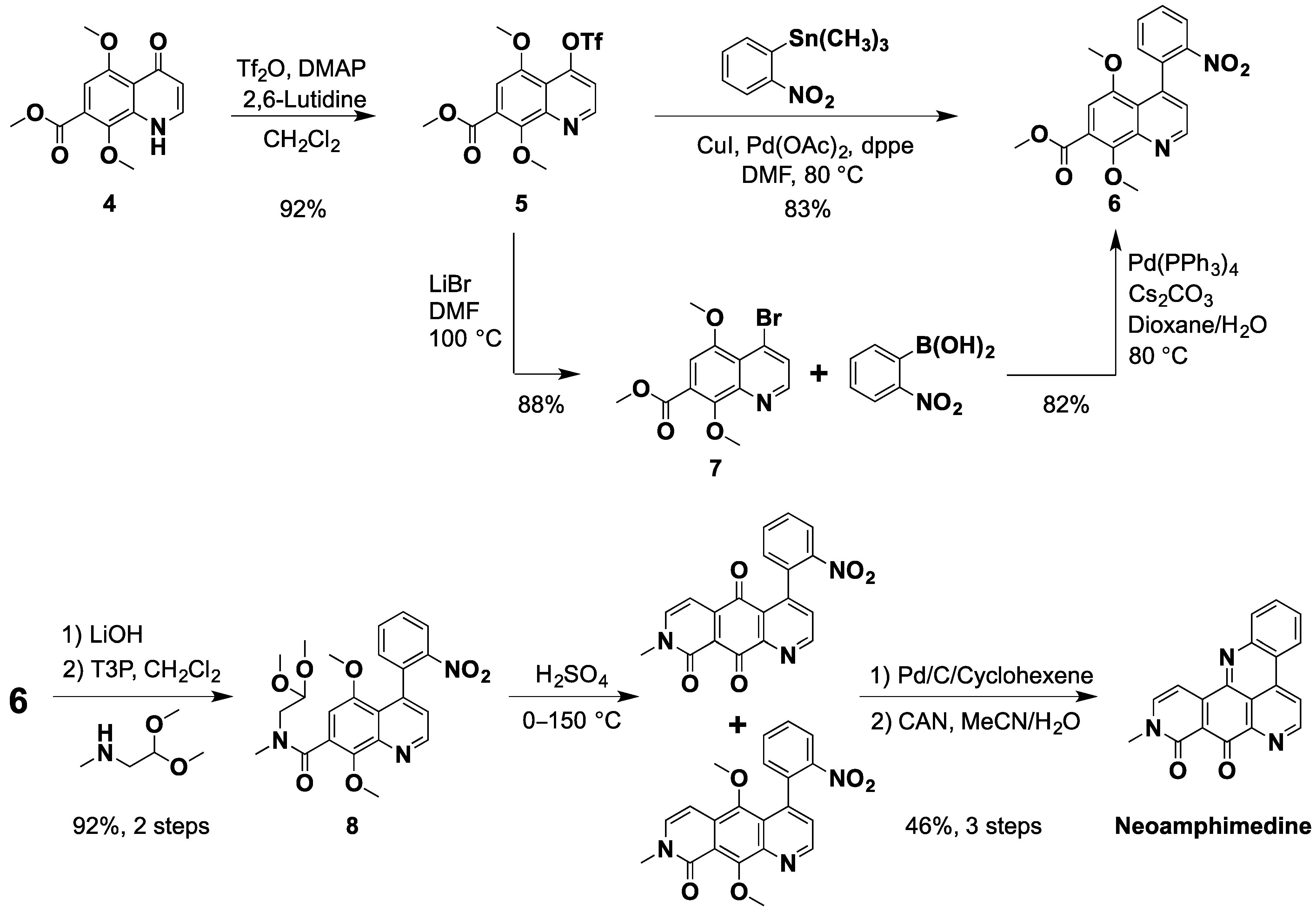{kind=link}
| Human Cancer Cells | Neo IC50 (μM) | Etoposide IC50 (μM) |
|---|---|---|
| Breast | ||
| MCF7 | 0.433 | 0.83 * |
| MDA-MB-231 | 0.76 | 9.03 * |
| PMC42LA | 0.723 | — |
| T47D | 0.740 | 11.58 * |
| Colorectal | ||
| HCT116 | 0.229 | 1.01 * |
| SW48 | 0.006 | 0.87 * |
| SW480 | 0.383 | 6.4 ** |
| SW620 | 0.060 | 0.39 * |
| Leukemia | ||
| HEL | 0.135 | 3.88 * |
| Kasumi I | 0.244 | 1.32 * |
| Molm13 | 0.018 | 0.38 * |
| OCI AML3 | 0.036 | — |
| Lung | ||
| A549 | 0.489 | 3.9 ** |
| H2009 | 1.095 | — |
| HCC827 | 2.957 | — |
| SW1573 | 0.157 | 2.25 * |
2.2. Cytotoxicity Studies with Neo Using a Panel of Human Colorectal Cancer Cell Lines
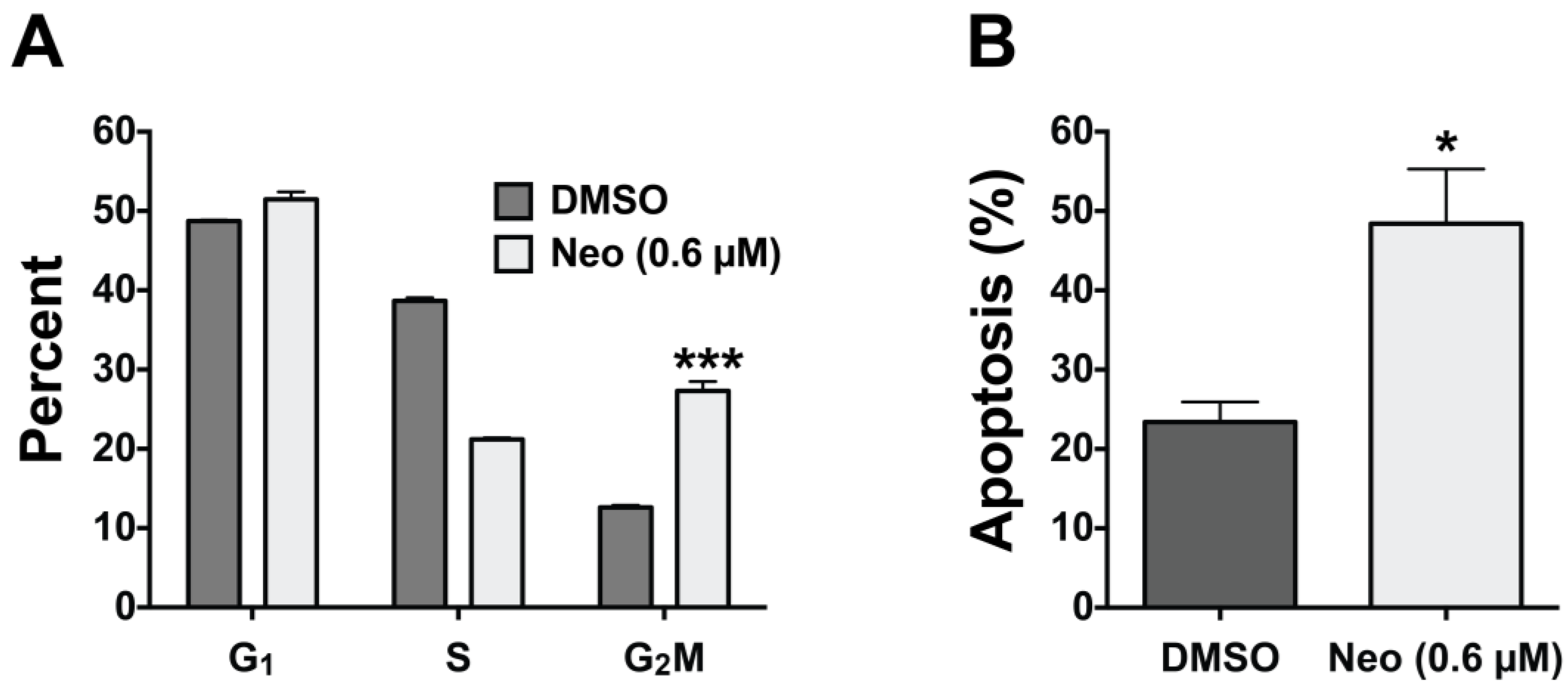
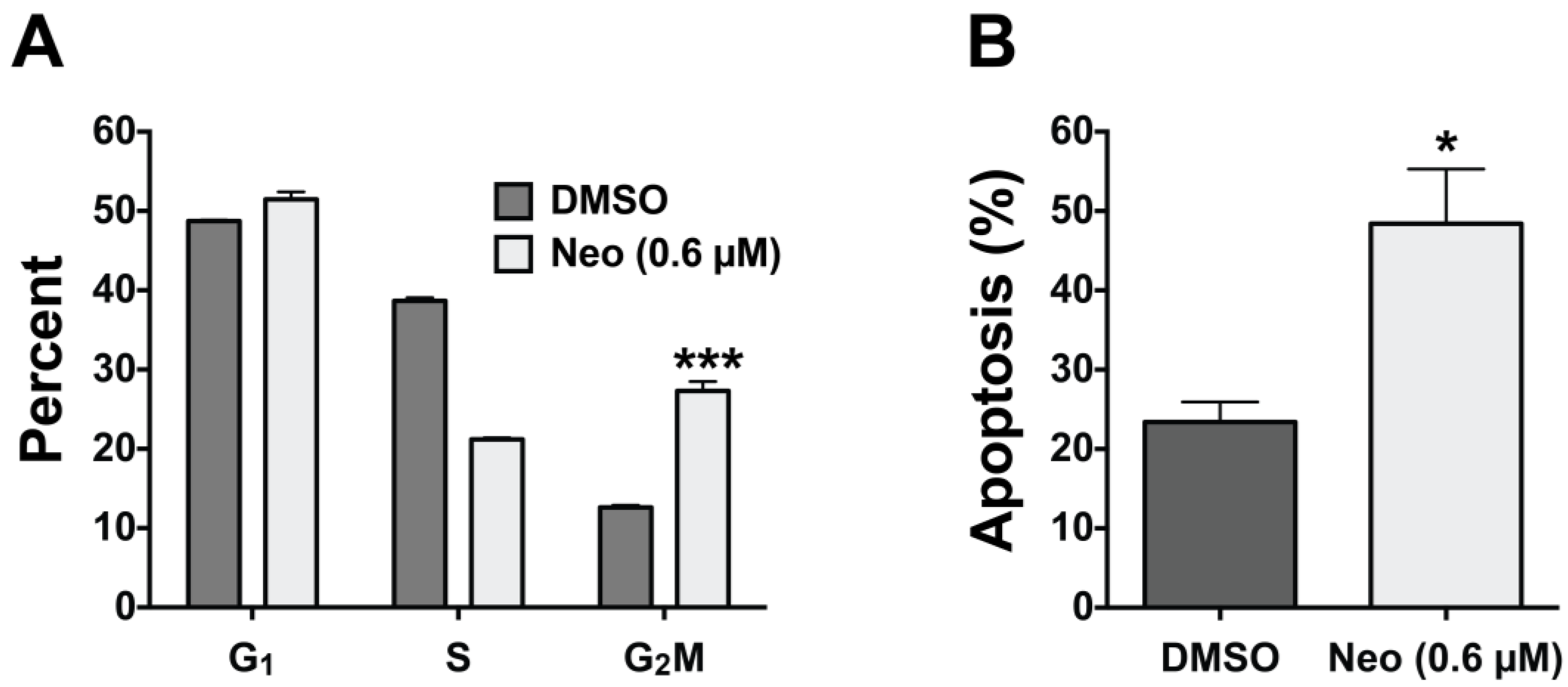
2.3. Neo Exerts Cytotoxicity by Inducing G2-M Cell Cycle Arrest and Apoptosis
3. Experimental Section
3.1. General Experimental Procedures
3.2. Cell Culture
3.3. Sulforhodamine B (SRB) Cytotoxicity Assay
3.4. SRB Recovery Assay
3.5. Acid Phosphatase (APH) Assay
3.6. FACS Analysis of Cell Cycle Distribution and Apoptosis
3.7. Synthetic Procedures
3.7.1. Methyl 2-hydroxy-5-methoxy-3-nitrobenzoate
3.7.2. Methyl 2,5-Dimethoxy-3-nitrobenzoate (1)
3.7.3. Methyl 3-((2,2-Dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl-amino)-2,5-dimethoxy-benzoate (3)
3.7.4. Methyl 1,4-Dihydro-5,8-dimethoxy-4-oxo-quinoline-7-carboxylate (4)
3.7.5. Methyl 5,8-Dimethoxy-4-trifluoromethanesulfonyloxy-quinoline-7-carboxylate (5)
3.7.6. Methyl 4-Bromo-5,8-dimethoxy-quinoline-7-carboxylate (7)
3.7.7. Methyl 5,8-Dimethoxy-4-(2-nitrophenyl)-quinoline-7-carboxylate (6)
3.7.8. 7-[N-(2,2-Dimethoxyethyl)-N-methyl]-carboxamide-5,8-dimethoxy-4-(2-nitrophenyl) quinoline (8)
3.7.9. Neoamphimedine (neo)
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Osheroff, N.; Zechiedrich, E.L.; Gale, K.C. Catalytic function of DNA topoisomerase II. BioEssays: News Rev. Mol. Cell. Dev. Biol. 1991, 13, 269–275. [Google Scholar]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and mechanism of DNA topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Bates, A.D.; Berger, J.M.; Maxwell, A. The ancestral role of ATP hydrolysis in type II topoisomerases: Prevention of DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 6327–6339. [Google Scholar] [CrossRef] [PubMed]
- Linka, R.M.; Porter, A.C.G.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-Terminal regions of topoisomerase IIα and IIβ determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Topoisomerase II inhibitors. Update Cancer Ther. 2006, 1, 3–15. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.S.; Clarke, D.J.; Mullinger, A.M.; Gimenez-Abian, J.F.; Creighton, A.M.; Johnson, R.T. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature 1994, 372, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Abian, J.F.; Clarke, D.J.; Mullinger, A.M.; Downes, C.S.; Johnson, R.T. A postprophase topoisomerase II-dependent chromatid core separation step in the formation of metaphase chromosomes. J. Cell Biol. 1995, 131, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.J.; Karaca, G.F.; Zhou, Y.; Simpson, D.A.; Cordeiro-Stone, M.; Kaufmann, W.K. Topoisomerase IIα maintains genomic stability through decatenation G2 checkpoint signaling. Oncogene 2010, 29, 4787–4799. [Google Scholar] [CrossRef] [PubMed]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.N.; Wang, J.C.; Liu, L.F. From the Cover: Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Ishida, R. Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta 1998, 1400, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Ikegami, Y.; Ishida, R.; Andoh, T. Inhibition of topoisomerase II by antitumor agents bis(2,6-dioxopiperazine) derivatives. Cancer Res. 1991, 51, 4903–4908. [Google Scholar] [PubMed]
- Saltiel, E.; McGuire, W. Doxorubicin (adriamycin) cardiomyopathy. West. J. Med. 1983, 139, 332–341. [Google Scholar]
- Weiss, G.; Loyevsky, M.; Gordeuk, V.R. Dexrazoxane (ICRF-187). Gen. Pharmacol. 1999, 32, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L. Utility of dexrazoxane for the reduction of anthracycline-induced cardiotoxicity. Expert Rev. Cardiovasc. Ther. 2008, 6, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.H.; Nitiss, K.C.; Rose, A.; Dong, J.; Zhou, J.; Hu, T.; Osheroff, N.; Jensen, P.B.; Sehested, M.; Nitiss, J.L. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J. Biol. Chem. 2000, 275, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Vezin, H.; Lansiaux, A.; Bailly, C. Toposiomerase inhibitors of marine origin and their potential as anticancer agents. Top. Curr. Chem. 2005, 253, 89–108. [Google Scholar]
- Ponder, J.; Yoo, B.H.; Abraham, A.D.; Li, Q.; Ashley, A.K.; Amerin, C.L.; Zhou, Q.; Reid, B.G.; Reigan, P.; Hromas, R.; et al. Neoamphimedine circumvents metnase-enhanced DNA topoisomerase IIα activity through ATP-competitive inhibition. Mar. Drugs 2011, 9, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Bastide, J. Marine pyridoacridine alkaloids and synthetic analogues as antitumor agents. Med. Res. Rev. 2003, 23, 234–252. [Google Scholar] [CrossRef] [PubMed]
- De Guzman, F.S.; Carte, B.; Troupe, N.; Faulkner, D.J.; Harper, M.K.; Concepcion, G.P.; Mangalindan, G.C.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Neoamphimideine: A new pyridoacridine topoismerase II inhibitor which cantenates DNA. J. Org. Chem. 1999, 64, 1400–1402. [Google Scholar] [CrossRef]
- Marshall, K.M.; Barrows, L.R. Biological activities of pyridoacridines. Nat. Prod. Rep. 2004, 21, 731. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.M.; Matumoto, S.S.; Holden, J.A.; Concepcion, G.P.; Tasdemir, D.; Ireland, C.M.; Barrows, L.R. The anti-neoplastic and novel topoisomerase II-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem. Pharmacol. 2003, 66, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Thale, Z.; Johnson, T.; Tenney, K.; Wenzel, P.J.; Lobkovsky, E.; Clardy, J.; Media, J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Structures and cytotoxic properties of sponge-derived bisannulated acridines. J. Org. Chem. 2002, 67, 9384–9391. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.M.; Andjelic, C.D.; Tasdemir, D.; Concepcion, G.P.; Ireland, C.M.; Barrows, L.R. Deoxyamphimedine, a pyridoacridine alkaloid, damages DNA via the production of reactive oxygen species. Mar. Drugs 2009, 7, 196–209. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, D.V.; Bugni, T.S.; Ireland, C.M. The total synthesis of neoamphimedine. J. Org. Chem. 2007, 72, 8501–8505. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Mukai, Y.; Kubo, A. Synthesis of neoamphimedine. Heterocycles 2012, 85, 993–940. [Google Scholar] [CrossRef]
- Molinski, T.F. Marine pyridoacridine alkaloids: Structure, synthesis, and biological chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Tasdemir, D.; Marshall, K.M.; Mangalindan, G.C.; Concepcion, G.P.; Barrows, L.R.; Harper, M.K.; Ireland, C.M. Deoxyamphimedine, a new pyridoacridine alkaloid from two tropical Xestospongia sponges. J. Org. Chem. 2001, 66, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Skyler, D.; Heathcock, C.H. The pyridoacridine family tree: A useful scheme for designing synthesis and predicting undiscovered natural products. J. Nat. Prod. 2002, 65, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Melzer, B.; Plodek, A.; Bracher, F. Total synthesis of the marine pyridoacridine alkaloid demethyldeoxyamphimedine. J. Org. Chem. 2014, 79, 7239–7242. [Google Scholar] [CrossRef] [PubMed]
- Echavarren, A.M.; Stille, J.K. Total synthesis of amphimedine. J. Am. Chem. Soc. 1988, 110, 4051–4053. [Google Scholar] [CrossRef]
- Kubo, A.; Nakahara, S. Synthesis of amphimedine, a new fused aromatic alkaloid from a pacific sponge, Amphimedon sp. Heterocycles 1988, 27, 2095–2098. [Google Scholar] [CrossRef]
- Nakahara, S.; Tanaka, Y.; Kubo, A. Total synthesis of amphimedine. Heterocycles 1996, 43, 2113–2123. [Google Scholar] [CrossRef]
- Godard, A.; Rocca, P.; Guillier, F.; Duvey, G.; Nivoliers, F.; Marsais, F.; Quéguiner, G. Ortho-directed metallation of π-deficient heterocycles in connection with palladium-catalyzed biaryl cross-coupling Synthesis of marine alkaloids of the pyridoacridine series. Can. J. Chem. 2001, 79, 1754–1761. [Google Scholar]
- Prager, R.H.; Tsopelas, C.; Heisler, T. A simple synthesis of amphimedine. Aust. J. Chem. 1991, 44, 277–285. [Google Scholar] [CrossRef]
- Bracher, F.; Papke, T. Polycyclic aromatic alkaloids, XI a convenient formal total synthesis of the cytotoxic marine alkaloid amphimedine. Liebigs Ann. 1996, 1996, 115–116. [Google Scholar] [CrossRef]
- Kang, B.-R.; Shan, A.-L.; Li, Y.-P.; Xu, J.; Lu, S.-M.; Zhang, S.-Q. Discovery of 2-aryl-8-hydroxy (or methoxy)-isoquinolin-1(2H)-ones as novel EGFR inhibitor by scaffold hopping. Biorg. Med. Chem. 2013, 21, 6956–6964. [Google Scholar] [CrossRef]
- Chuang, T.-H.; Wu, P.-L. Synthesis and mechanistic study of isoquinolinones from cinnamoyl azides. J. Chin. Chem. Soc. 2006, 53, 413–420. [Google Scholar]
- Yan, Y.; Qin, B.; Ren, C.; Chen, X.; Yip, Y.K.; Ye, R.; Zhang, D.; Su, H.; Zeng, H. Synthesis, structural investigations, hydrogen–deuterium exchange studies, and molecular modeling of conformationally stablilized aromatic oligoamides. J. Am. Chem. Soc. 2010, 132, 5869–5879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wilke, B.I.; Zhan, J.; Watanabe, K.; Boddy, C.N.; Tang, Y. A new mechanism for benzopyrone formation in aromatic polyketide biosynthesis. J. Am. Chem. Soc. 2007, 129, 9304–9305. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.J.; Gaudino, J. A general synthesis of 5-arylnicotinates. J. Org. Chem. 1984, 49, 5237–5243. [Google Scholar] [CrossRef]
- Pagliarani, A.; Nesci, S.; Ventrella, V. Toxicity of organotin compounds: Shared and unshared biochemical targets and mechanisms in animal cells. Toxicol. Vitro 2013, 27, 978–990. [Google Scholar] [CrossRef]
- Hui, J.; Zhao, Y.; Zhu, L. Synthesis and in vitro anticancer activities of novel aryl-naphthalene lignans. Med. Chem. Res. 2011, 21, 3994–4001. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Gupta, S.; Singh, H.; Kumar, S.; Gautam, A.; Kapoor, P.; Raghava, G.P. CancerDR: Cancer drug resistance database. Sci. Rep. 2013, 3, 1445. [Google Scholar] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.T.; Sinai, P.; Kain, S.R. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Anal. Biochem. 1996, 241, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, R.E.; McMarlin, A.; Kleiner, D.; Wersto, R.; Martin, P.; Tsokos, M.; Stamp, G.W.; Stetler-Stevenson, W.G. Validation of a model of colon cancer progression. J. Pathol. 2000, 192, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Global-Data. Colorectal Cancer-Global Drug Forecasts and Treatment Analysis to 2020-Q4 2010; MarketPublishers, Ltd.: Limassol, Cyprus, 2010. [Google Scholar]
- Lee, C.C.; Houghton, P. Cytotoxicity of plants from Malaysia and Thailand used traditionally to treat cancer. J. Ethnopharmacol. 2005, 100, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.; Fang, R.; Techatanawat, I.; Steventon, G.; Hylands, P.J.; Lee, C.C. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 2007, 42, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Bruno, S.; Del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, D.V.; Modzelewska, K.; Glazar, A.I.; Gray, P.D.; Kaur, M.; Liu, T.; Grossman, D.; Harper, M.K.; Kuwada, S.K.; Moghal, N.; et al. The marine alkaloid naamidine A promotes caspase-dependent apoptosis in tumor cells. Anticancer Drugs 2009, 20, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; Lacroix, F.B.; Andreassen, P.R.; Wilson, L.; Margolis, R.L. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol. Cell 2004, 15, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar]
- Stover, J.S.; Shi, J.; Jin, W.; Vogt, P.K.; Boger, D.L. Discovery of inhibitors of aberrant gene transcription from libraries of DNA binding molecules: Inhibition of LEF-1-mediated gene transcription and oncogenic transformation. J. Am. Chem. Soc. 2009, 131, 3342–3348. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, L.; Abraham, A.D.; Zhou, Q.; Ali, H.; O'Brien, J.V.; Hamill, B.D.; Arcaroli, J.J.; Messersmith, W.A.; LaBarbera, D.V. An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent. Mar. Drugs 2014, 12, 4833-4850. https://doi.org/10.3390/md12094833
Li L, Abraham AD, Zhou Q, Ali H, O'Brien JV, Hamill BD, Arcaroli JJ, Messersmith WA, LaBarbera DV. An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent. Marine Drugs. 2014; 12(9):4833-4850. https://doi.org/10.3390/md12094833
Chicago/Turabian StyleLi, Linfeng, Adedoyin D. Abraham, Qiong Zhou, Hadi Ali, Jeremy V. O'Brien, Brayden D. Hamill, John J. Arcaroli, Wells A. Messersmith, and Daniel V. LaBarbera. 2014. "An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent" Marine Drugs 12, no. 9: 4833-4850. https://doi.org/10.3390/md12094833
APA StyleLi, L., Abraham, A. D., Zhou, Q., Ali, H., O'Brien, J. V., Hamill, B. D., Arcaroli, J. J., Messersmith, W. A., & LaBarbera, D. V. (2014). An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent. Marine Drugs, 12(9), 4833-4850. https://doi.org/10.3390/md12094833