Metabolomic Tools for Secondary Metabolite Discovery from Marine Microbial Symbionts

 ,
, 
Abstract
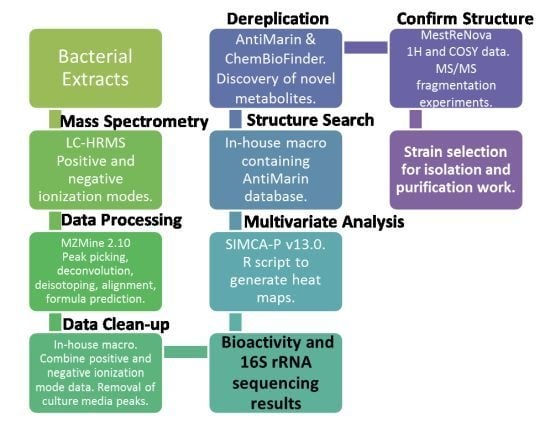
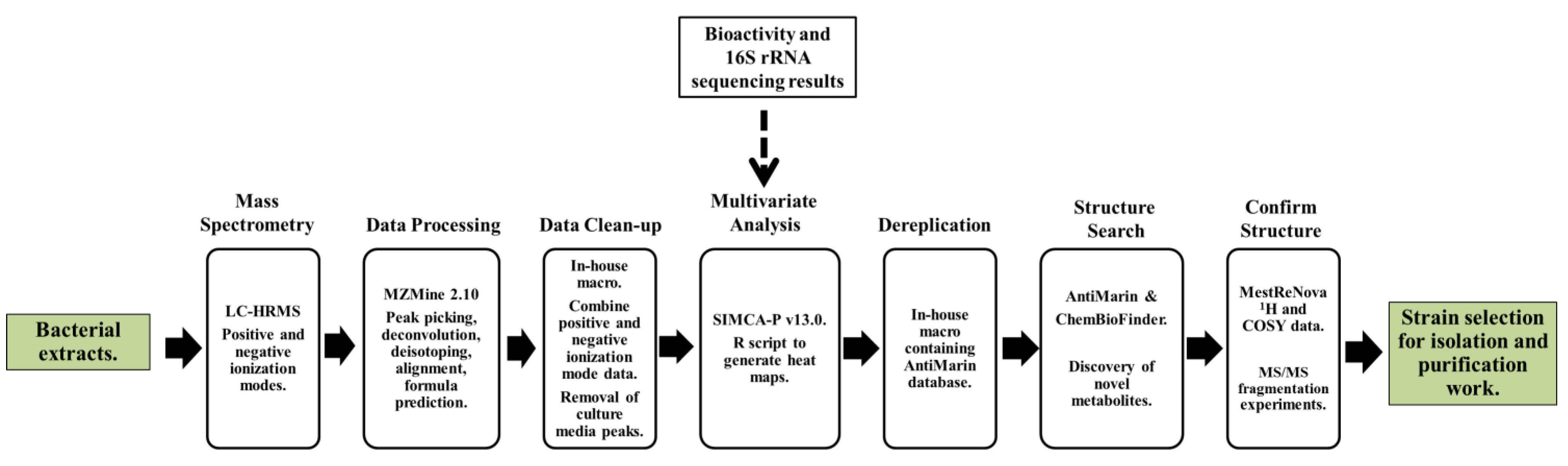
:
1. Introduction
2. Results and Discussion
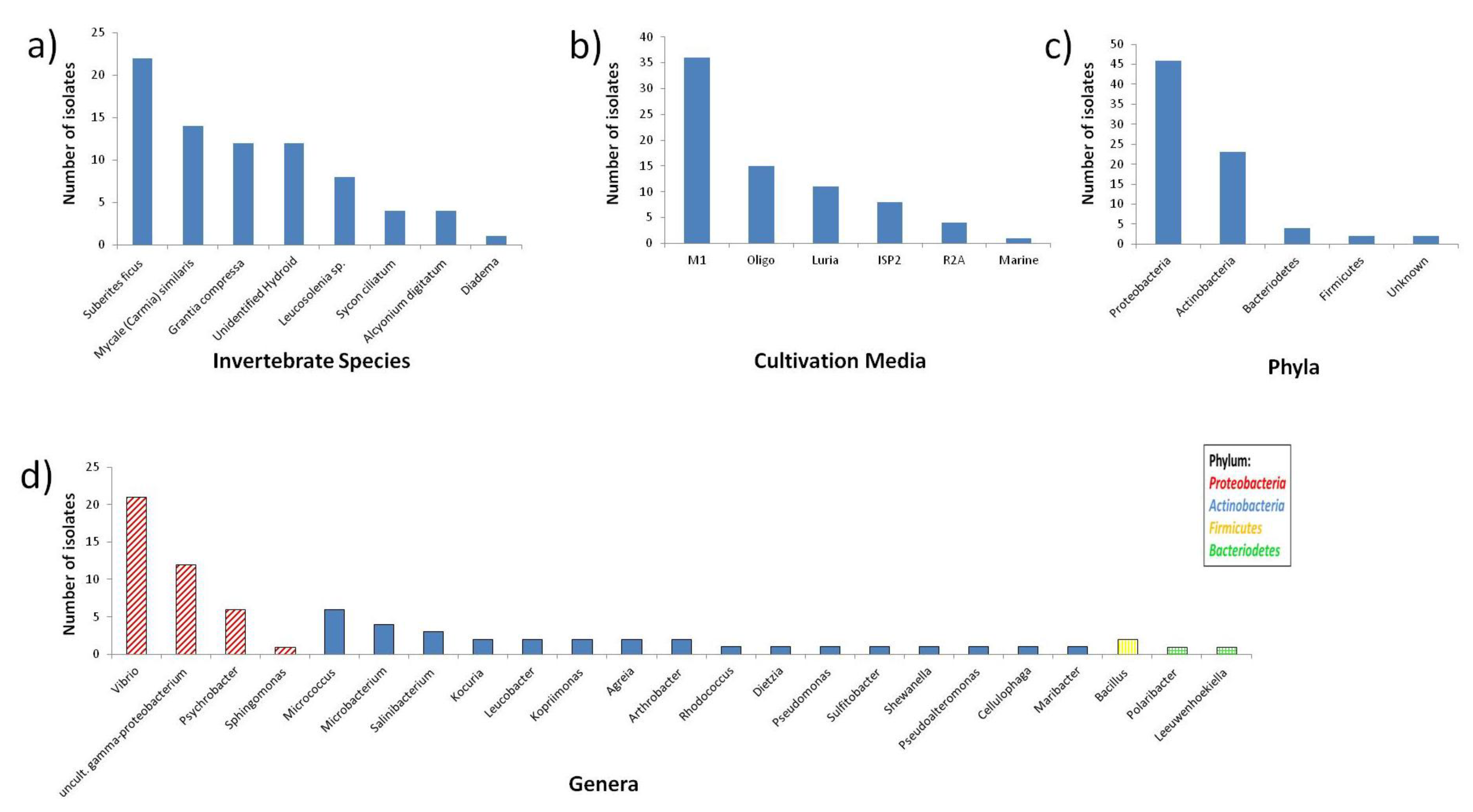
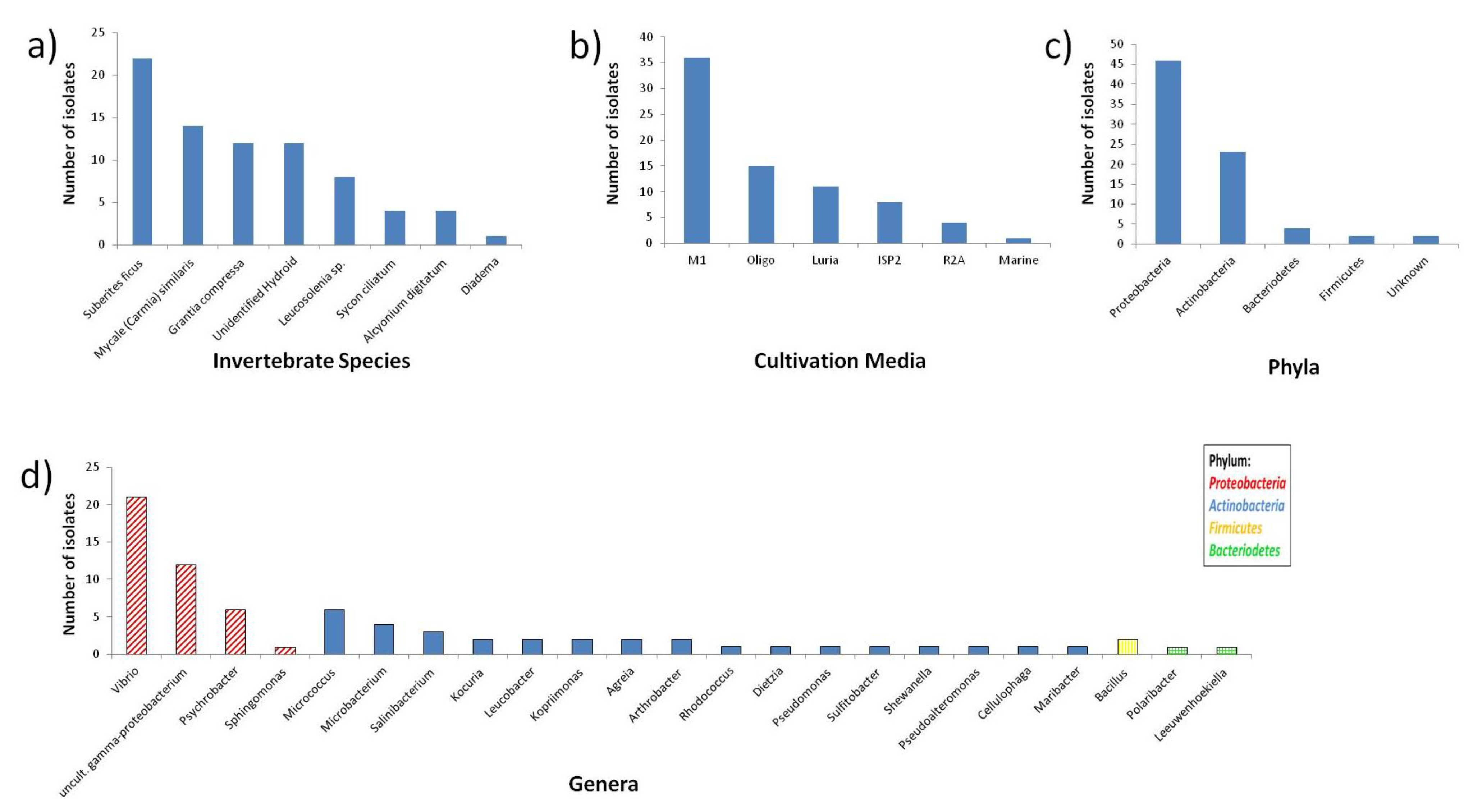
2.1. Diversity of Invertebrate-Associated Bacteria
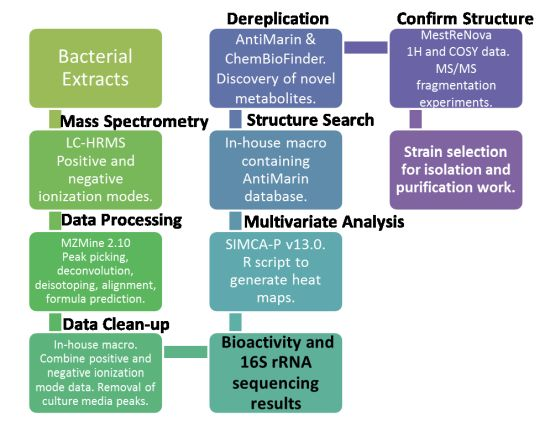
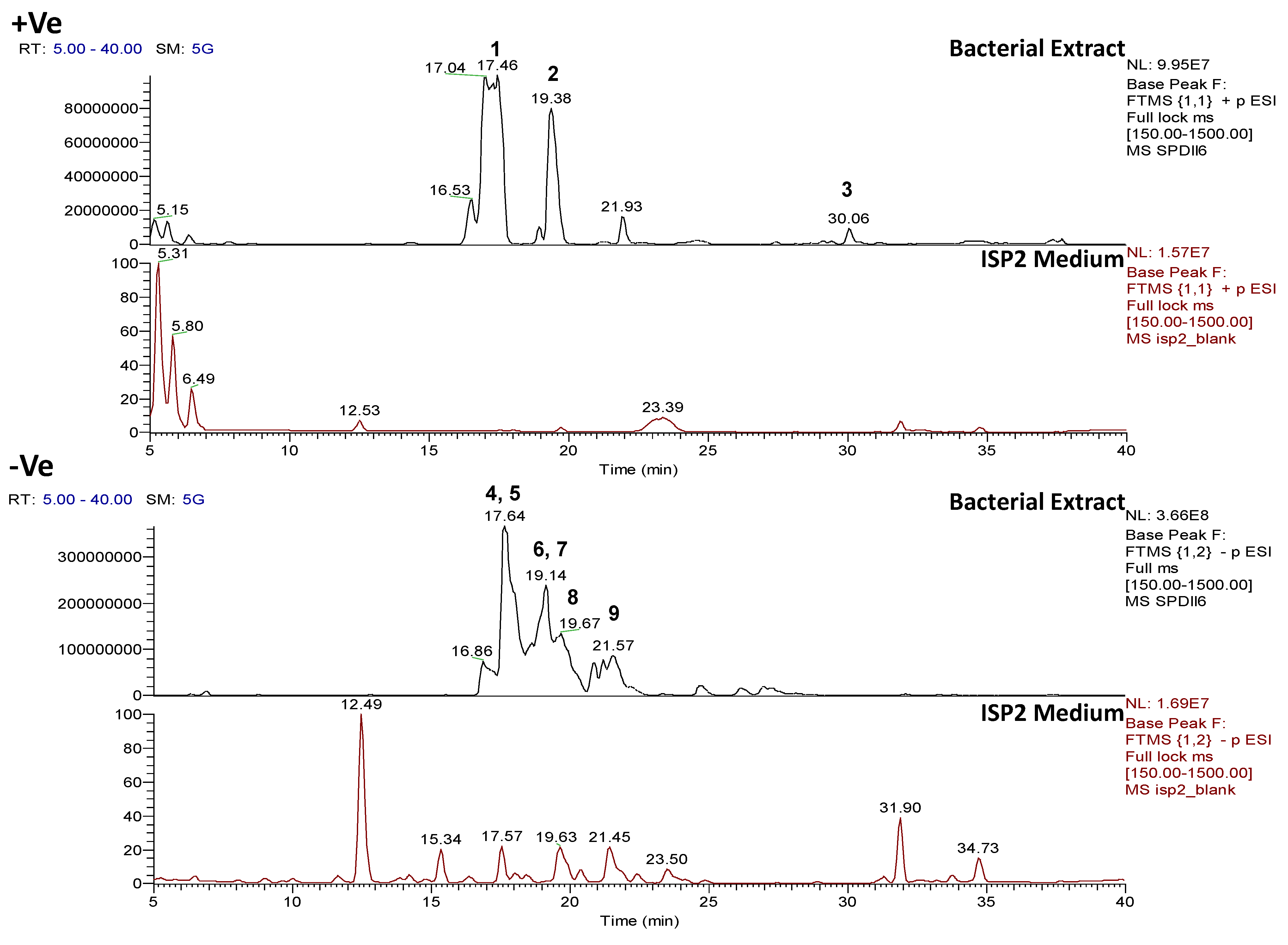
2.2. Data Processing and Data Clean-Up
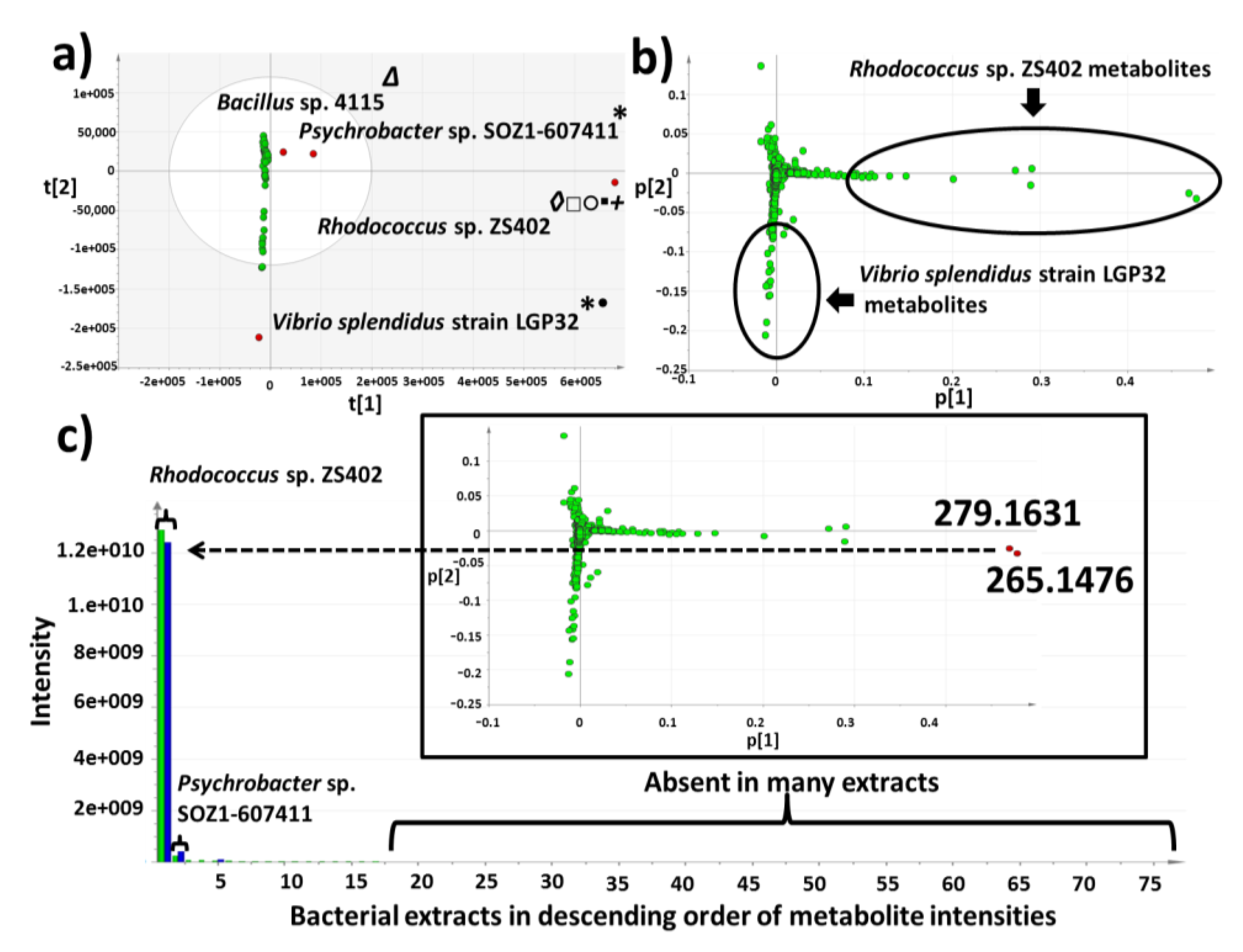
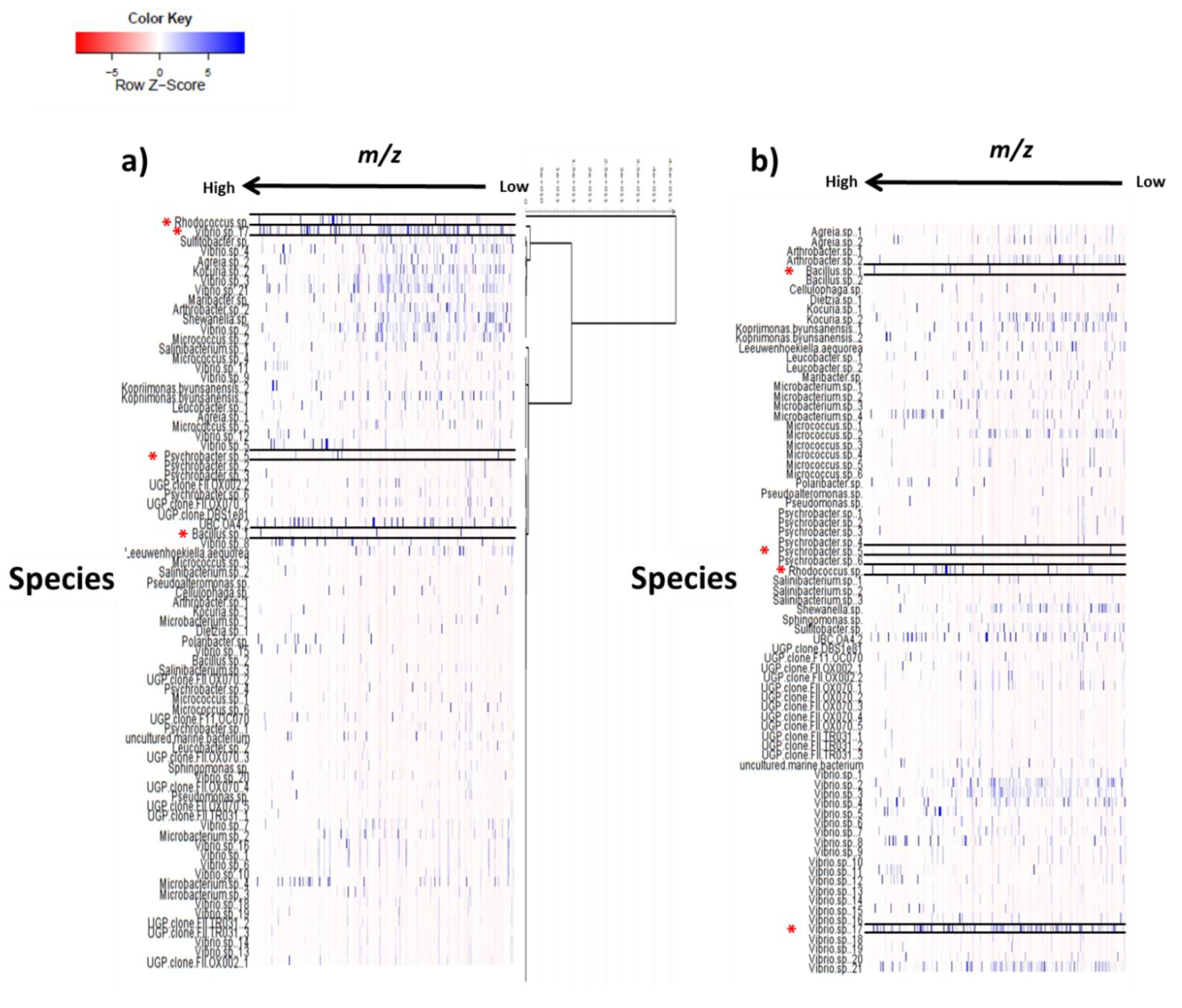
2.3. Multivariate Analysis for Strain Selection
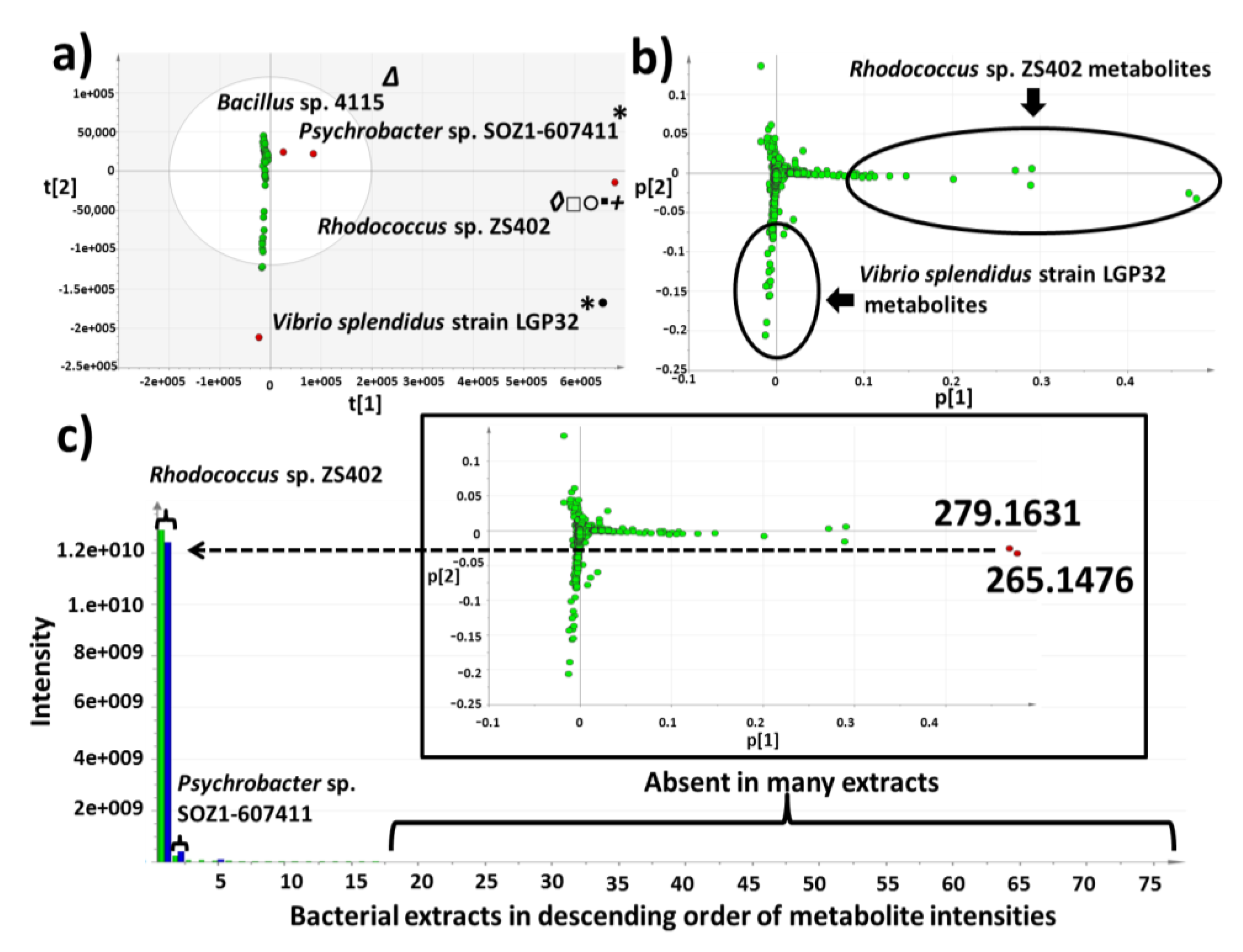
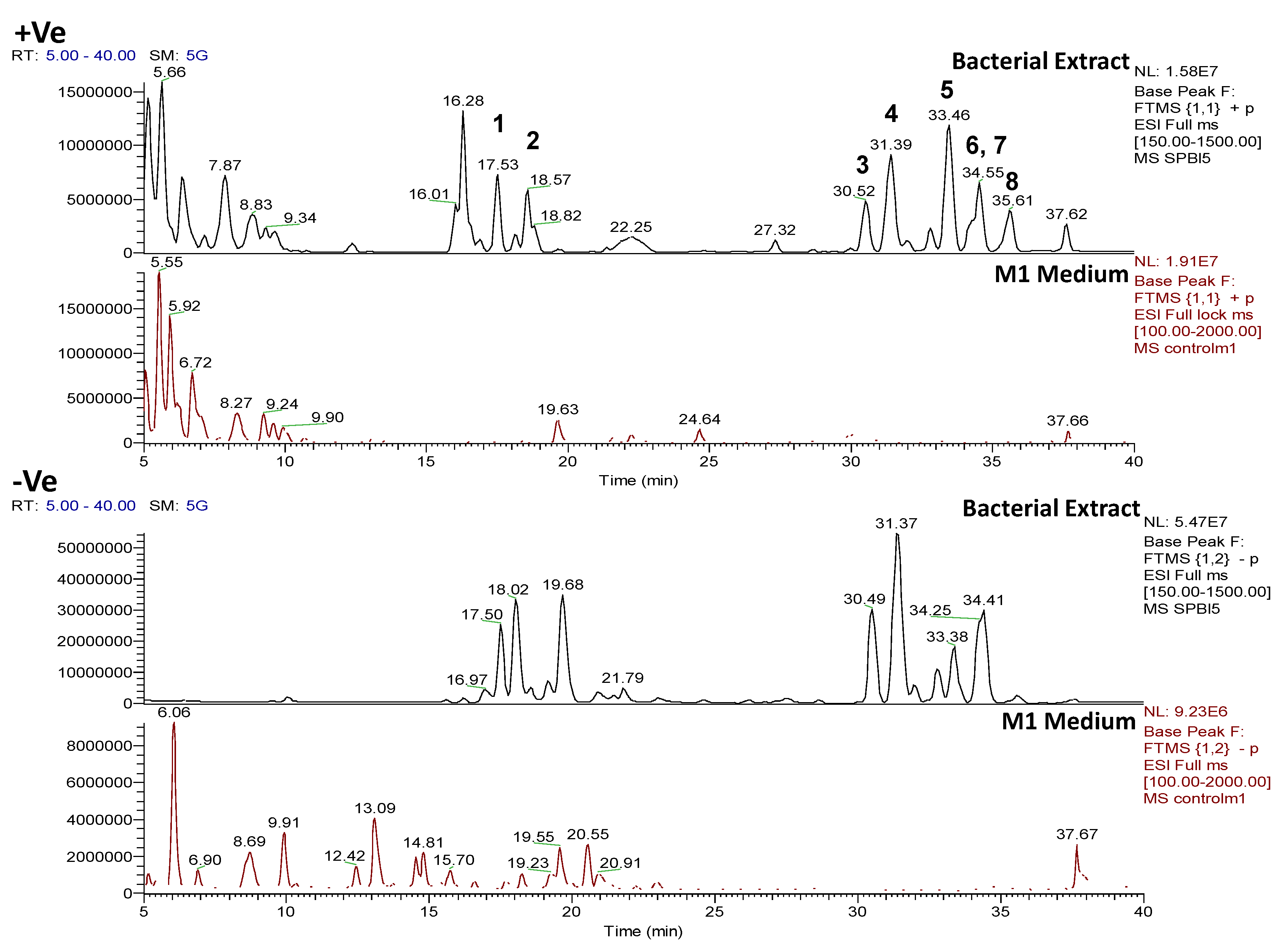
2.4. Chemical Diversity of Natural Products in Outlying Bacterial Extracts

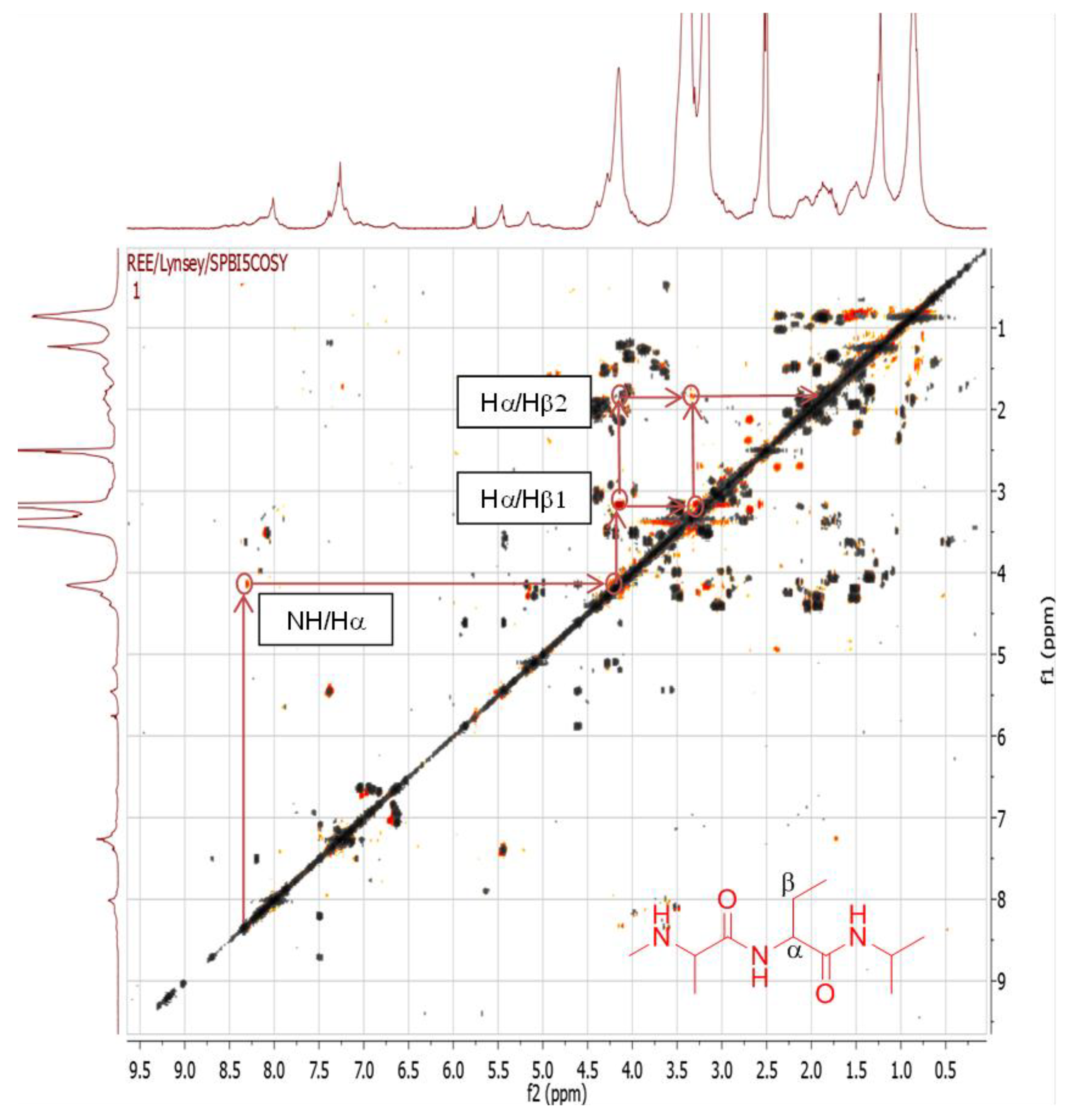
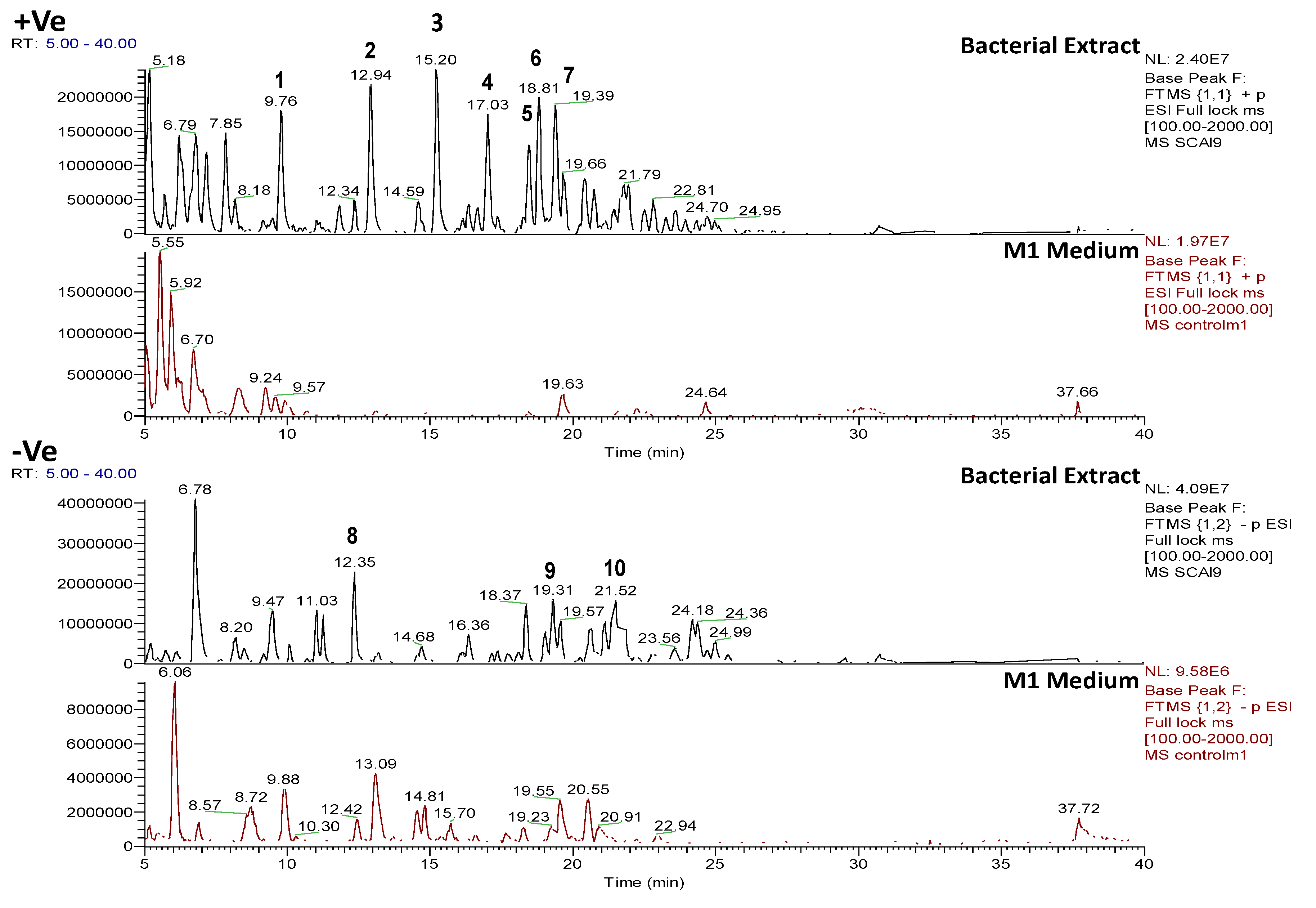
2.4.1. Dereplication of Bacillus sp. 4115

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
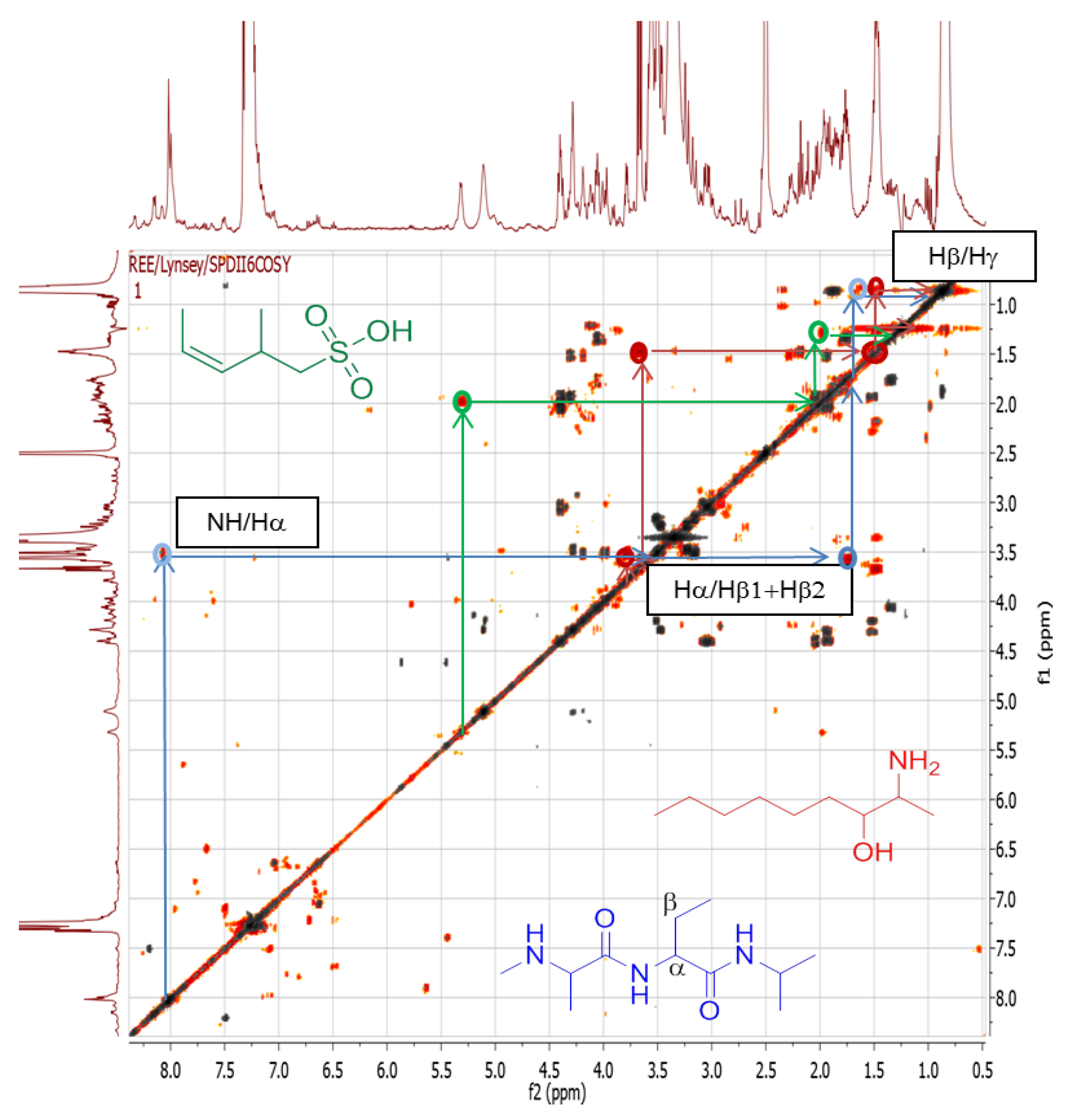{kind=link}
| Bacterial Strain | (a) Total number of features (m/z) | (b) Total number of features (m/z) after removal of features (m/z) from medium | (c) Total number of features identified by dereplication with AntiMarin | |||
|---|---|---|---|---|---|---|
| Positive ion mode | Negative ion mode | Positive ion mode | Negative ion mode | Putatively identified (positive and negative modes) | Unidentified (positive and negative modes) | |
| Bacillus sp. 4115 | 1220 | 1037 | 359 (29.4% remaining) | 438 (42.2% remaining) | 270 (51.3%) | 526 (48.7%) |
| Vibrio splendidus strain LGP32 | 2767 | 654 | 1102 (39.8% remaining) | 617 (94.3% remaining) | 699 (40.7%) | 1019 (59.3%) |
| Rhodococcus sp. ZS402 | 1198 | 2361 | 659 (55% remaining) | 1715 (72.6% remaining) | 519 (28%) | 1855 (72%) |
| Peak ID | ESI
Mode | m/z | Rt (min) | Molecular Formula
(Isotope Fit Score A0 to A3) | RDB | Hits | Fragmentation Data | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fragment ions MS2 +Ve | Chemical Formula | RDB | Fragment ions MS3 +Ve | Molecular Formula | RDB | ||||||||
| 1 | Pos | 445.29092 | 17.5 | C22H40O7N2 (99.49%) | 4 | No hits | 427.27921 399.28485 314.19589 232.15408 214.14343 186.14862 | C22H39O6N2 C21H39O5N2 C16H28O5N C11H22O4N C11H20O3N C10H20O2N | 5
4 4 2 3 2 | 168.13794
72.08067 | C10H18ON
C4H10N | 3
1 | |
| 2 | Pos | 459.30646 | 18.6 | C23H42O7N2 (71.70%) | 4 | No hits | 441.29553 413.30063 328.21176 228.15930 200.16431 | C23H41O6N2 C22H41O5N2 C17H30O5N C12H22O3N C11H22O2N | 5
4 4 3 2 | 146.11752
86.09630 | C7H16O2N
C5H12N | 1
1 | |
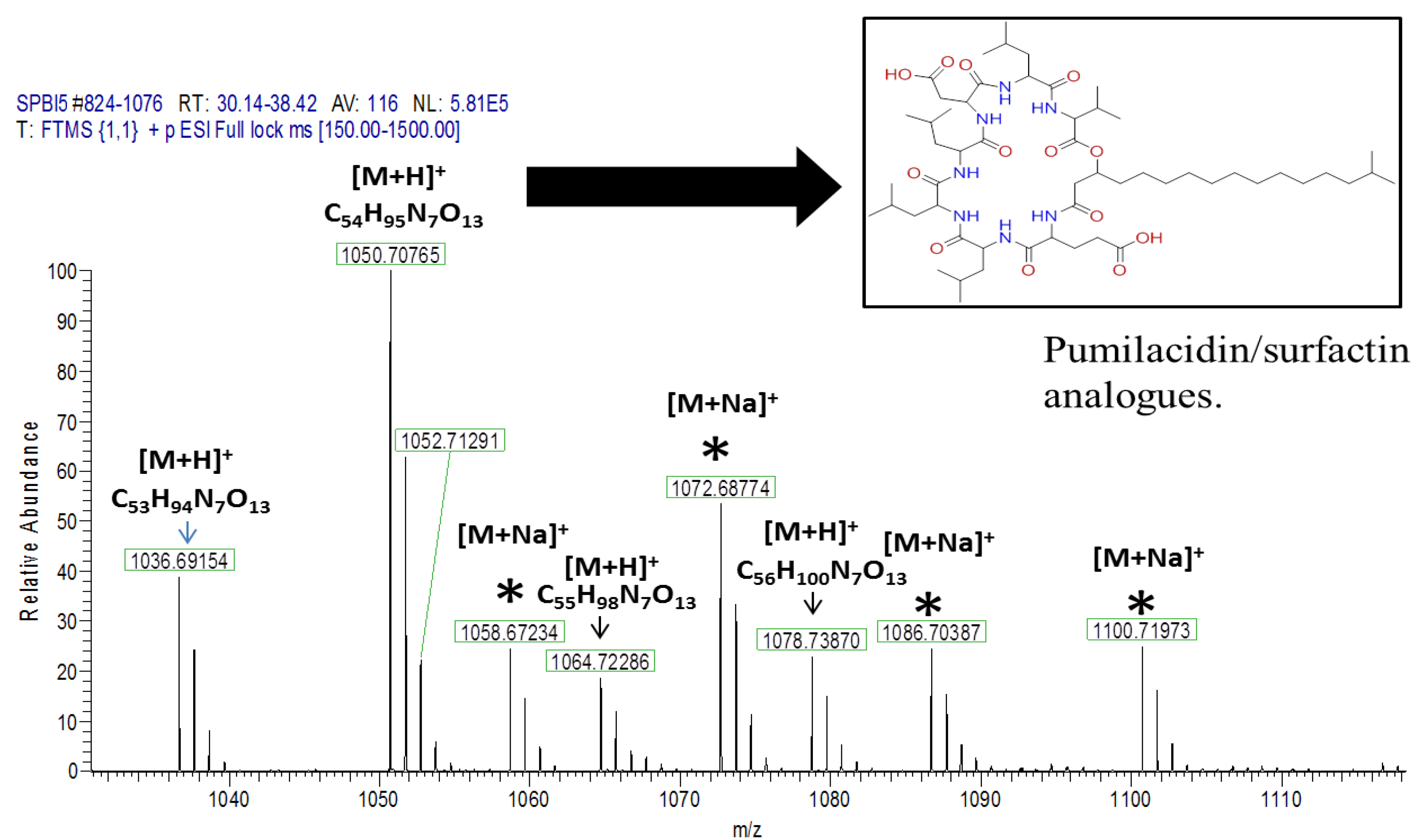
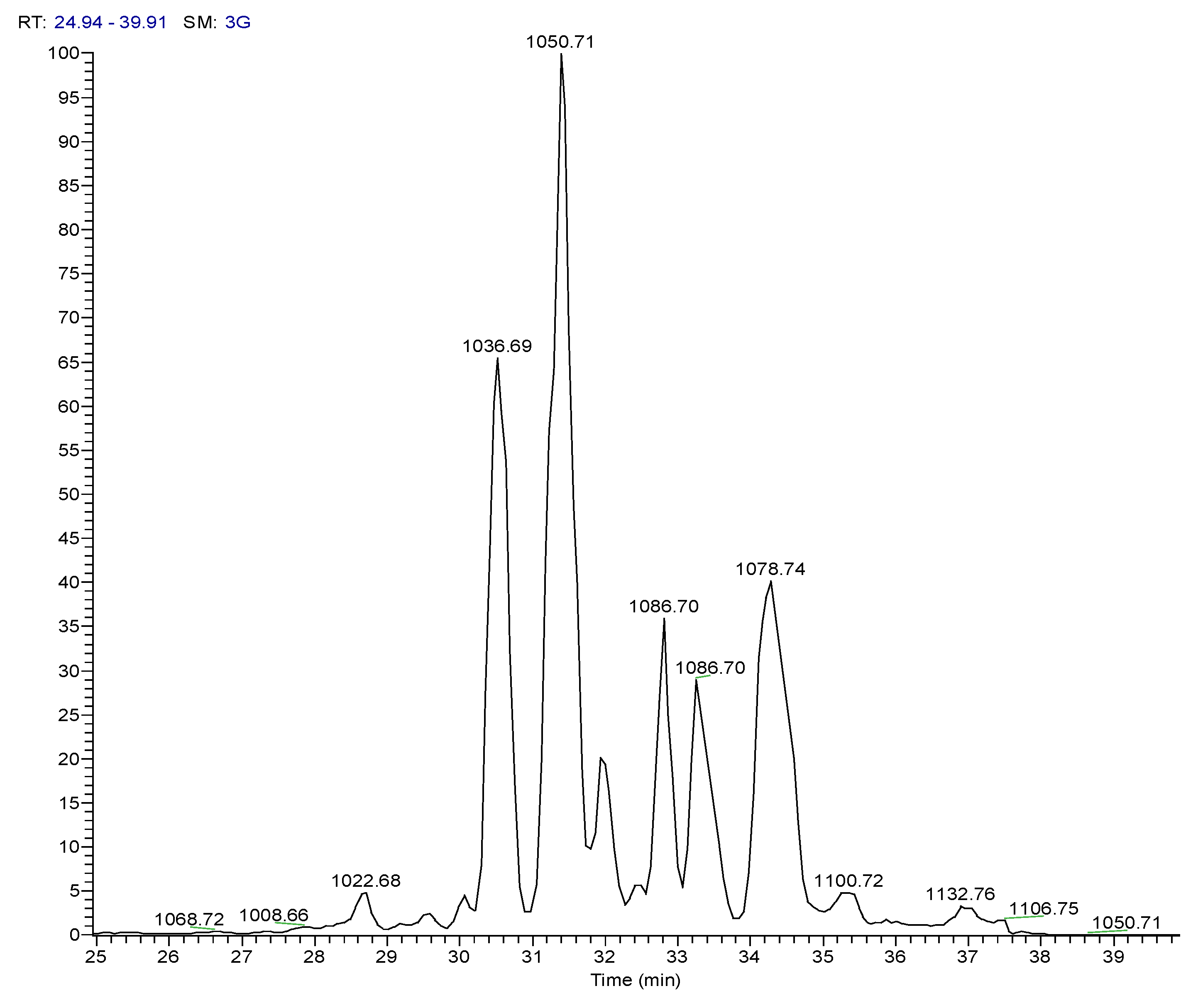
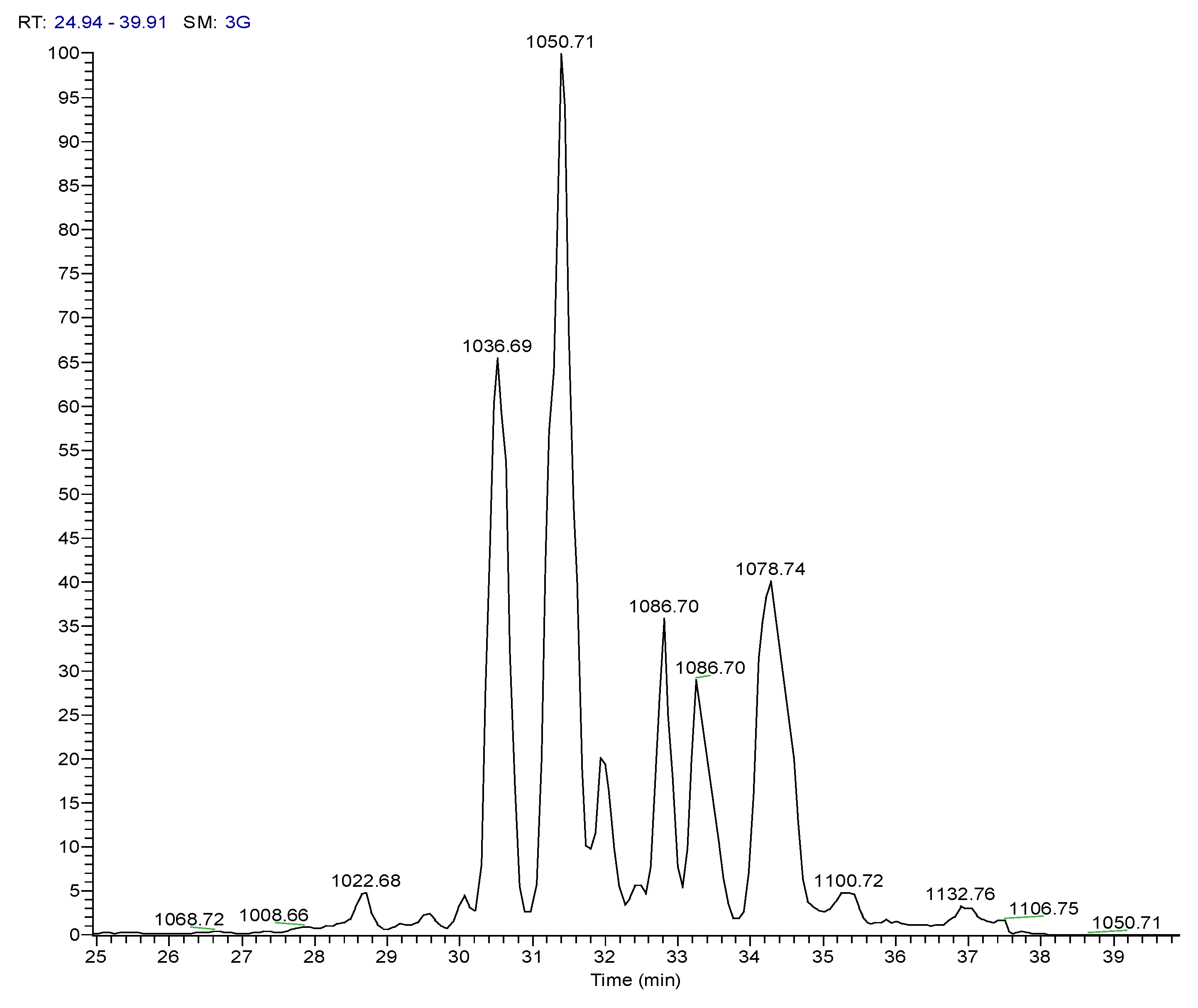
| 3 | Pos | 1036.69141 | 30.5 | C53H93O13N7 (99.82%) | 11 | Pumilacidin B// (surfactin-1) or other cyclic peptide | 1018.67596 937.61896 685.44714 667.43732 455.28571 | C53H92O12N7 C48H85O12N6 C33H61O9N6 C33H59O8N6 C22H39O6N4 | 12
10 7 8 6 | 568.36853
342.20117 | C28H50O7N5 C16H28O5N3 | 7
5 | |
| 4 | Pos | 1050.70771 | 31.4 | C54H95O13N7 (99.79%) | 11 | Pumilacidin A// or other cyclic peptide | 1032.69104 937.61823 699.46234 681.45282 455.28555 | C54H94O12N7 C48H85O12N6 C34H63O9N6 C34H61O8N6 C22H39O6N4 | 12
10 7 8 6 | 568.36816
342.20087 | C28H50O7N5 C16H28O5N3 | 7
5 | |
| 5 | Pos | 875.53519 | 33.5 | C43H77O15N3 (98.53%) | 7 | No hits | 710.38348 685.41257 659.46952 654.51534 647.45954 615.44423 610.48905 | C32H58O15N2 C31H61O14N2 C31H67O12N2 C34H72O10N C34H65O10N C29H63O11N2 C32H68O9N | 5
3 1 1 3 1 1 | ||||
| 6 | Pos | 1078.73917 | 34.5 | C56H99O13N7 (99.80%) | 11 | Pumilacidin C// or other cyclic peptide | 1061.72498 966.65216 699.46283 681.45337 455.28549 | C55H99O13N6 C49H88O12N7 C34H63O9N6 C34H61O8N6 C22H39O6N4 | 10
10 7 8 6 | 568.36859
342.20135 | C28H50O7N5C
16H28O5N3 | 7
5 | |
| 7 | Pos | 889.55163 | 34.5 | C42H76O14N6 (84.86%) | 8 | No hits | 861.55371 817.49072 803.47546 790.47894 776.46429 757.47034 690.39093 676.41254 662.39655 590.33954 563.32874 | C41H77O13N6 C38H69O13N6 C37H67O13N6 C37H68O13N5 C36H66O13N5 C36H65O11N6 C31H56O12N5 C31H58O11N5 C30H56O11N5 C26H48O10N5 C25H47O10N4 | 7
8 8 7 7 8 7 6 6 6 5 | 449.26096
463.27667 577.34413 477.25619 | C19H37O8N4 C20H39O8N4 C26H49O10N4 C20H37O9N4 | 4
4 5 5 | |
| 8 | Pos | 903.56635 | 35.6 | C43H78O14N6 (99.76%) | 8 | No hits | 817.49097 804.49593 790.47988 718.42297 704.40740 690.42803 676.41285 604.35589 590.34010 577.34508 491.27243 463.27740 | C38H69O13N6 C38H70O13N5 C37H68O13N5 C33H60O13N5 C32H58O12N5 C32H60O11N5 C31H58O11N5 C27H50O10N5 C26H48O10N5 C26H49O10N4 C21H39O9N4 C20H39O8N4 | 8
7 7 7 7 6 6 6 6 5 5 4 | 364.20825 | C15H30O7N3 | 3 | |
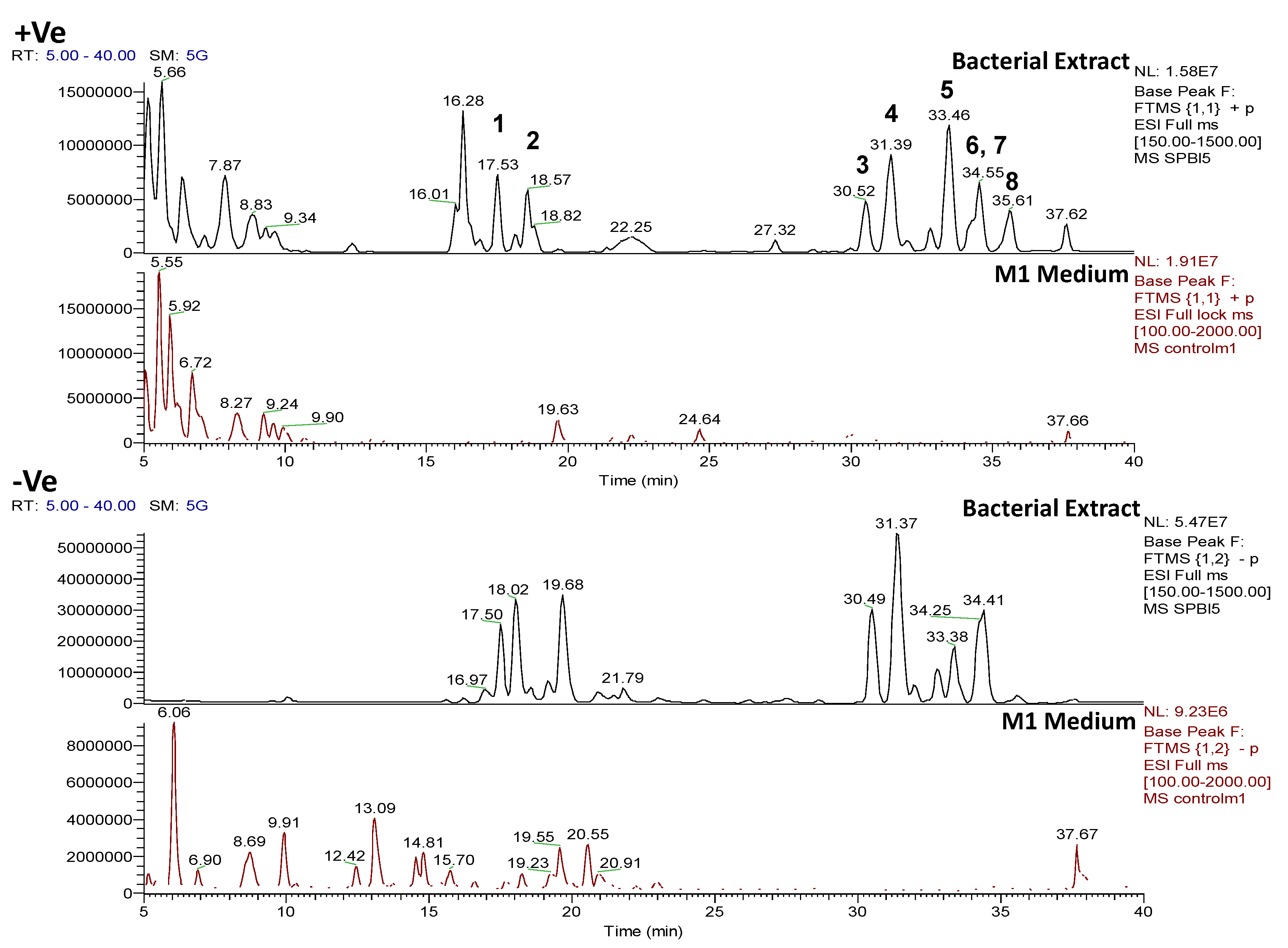


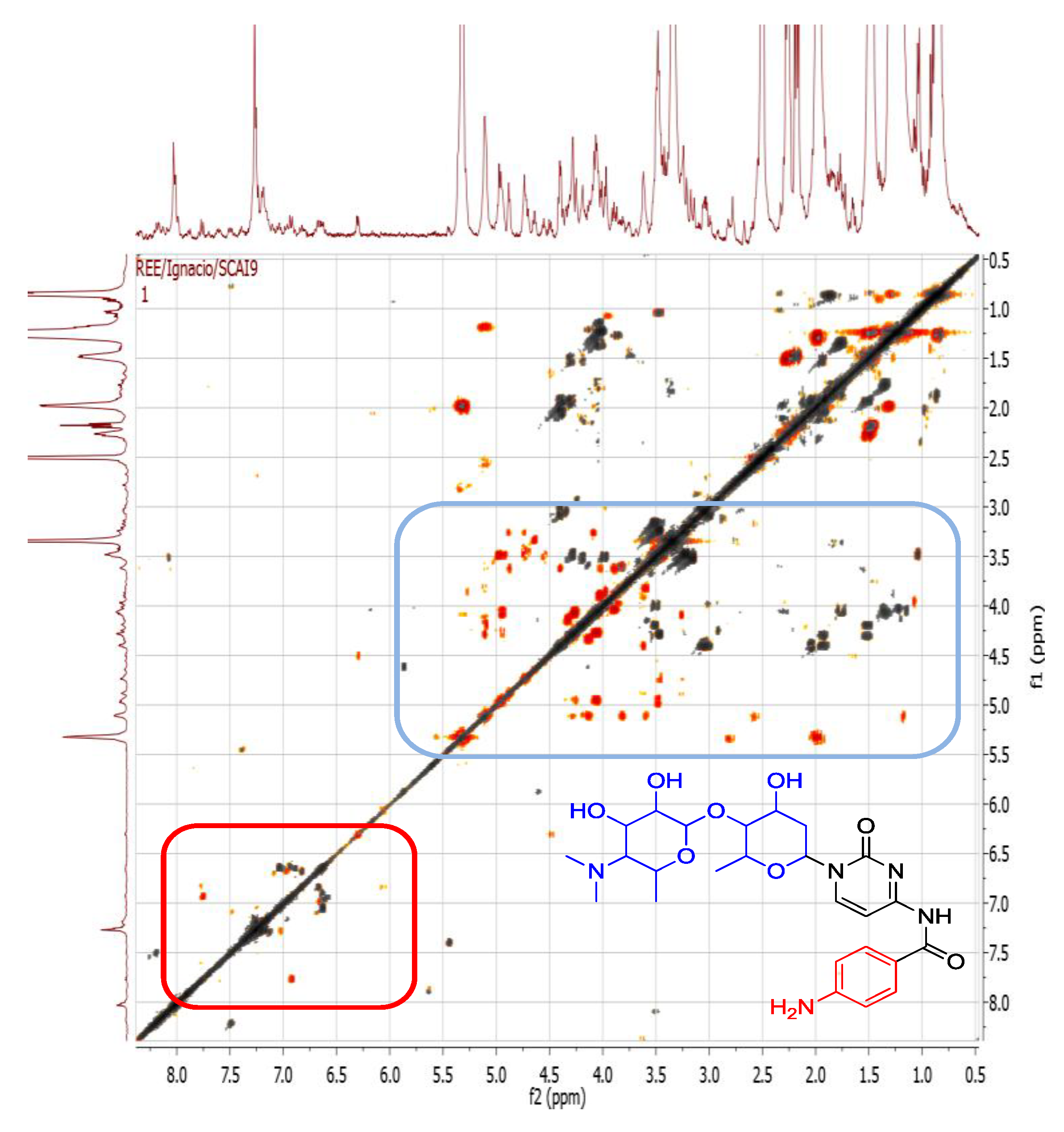
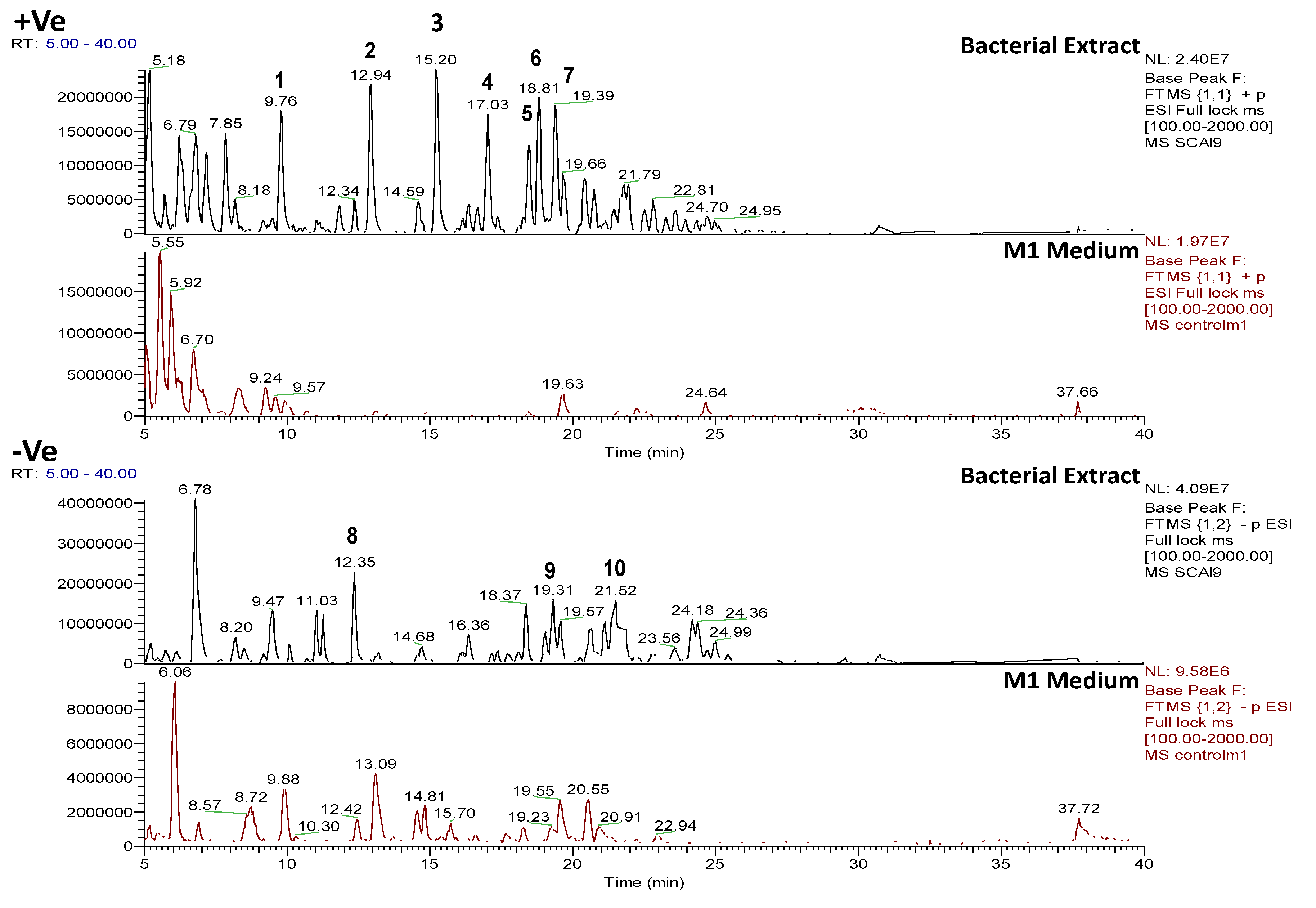
2.4.2. Dereplication of Vibrio splendidus Strain LGP32
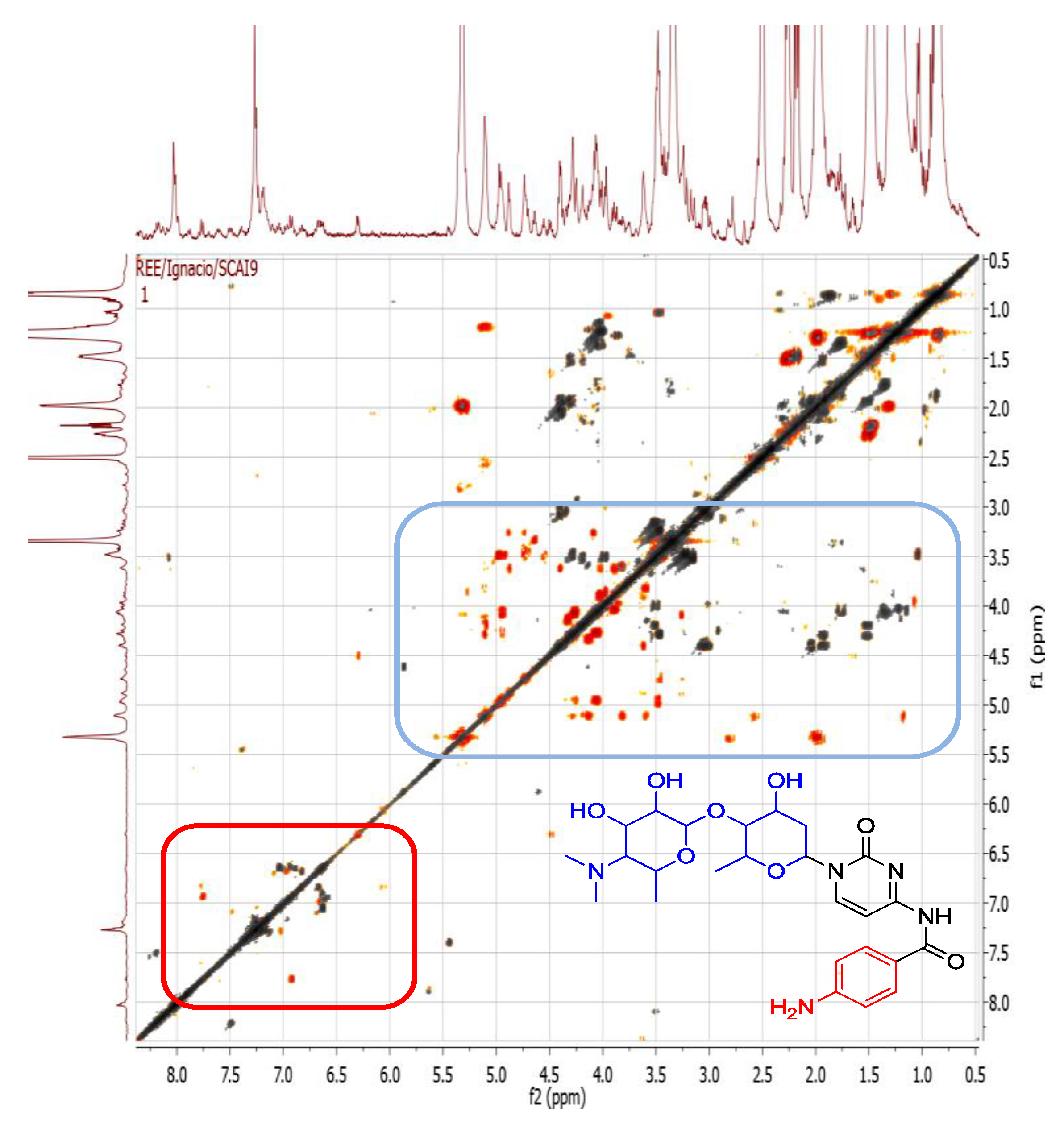
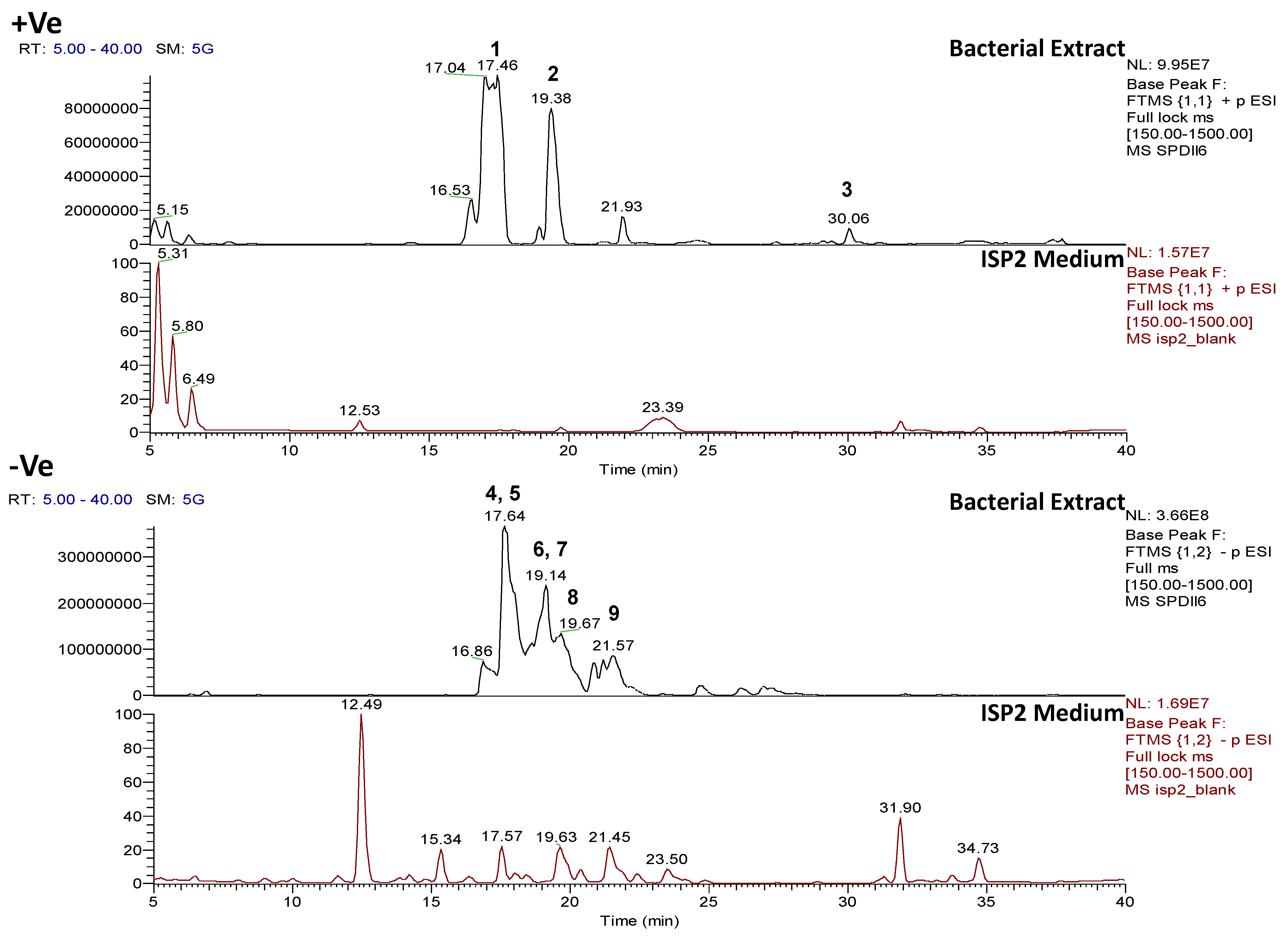
2.4.3. Dereplication of Rhodococcus sp. ZS402
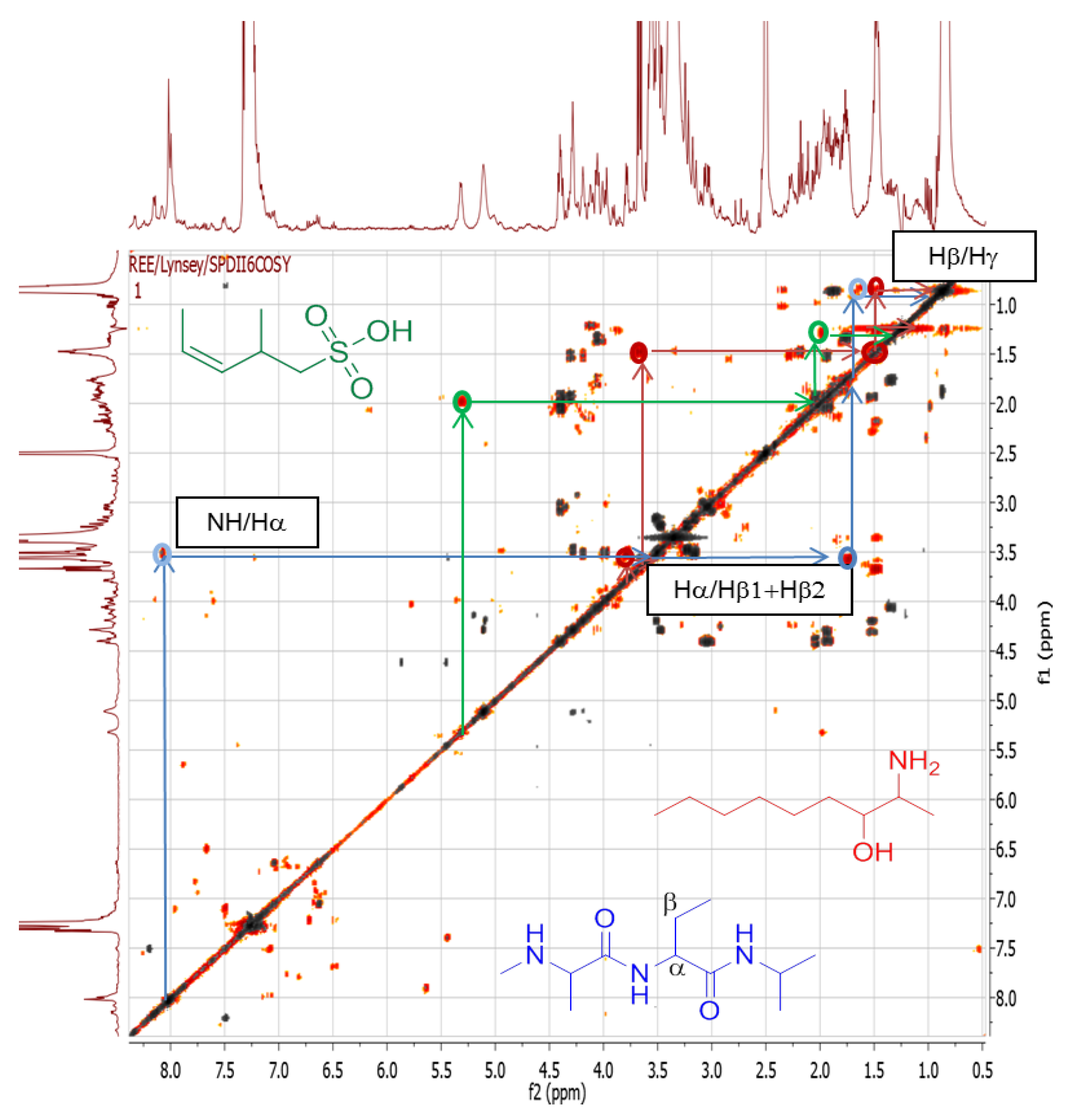
3. Experimental Section
3.1. Sample Collection and Bacterial Isolation
| Peak ID | ESI Mode | m/z | Rt (min) | Molecular Formula (Isotope Fit Score A0 to A3) | RDB | Hits | Fragment Ions MS2 | Molecular Formula | RDB | Fragment ions MS3 | Molecular Formula | RDB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | P | 219.12266 | 9.8 | C10H18O5 (99.97%) | 2 | (4E)-6,7,9-Trihydroxydec-4-enoic acid | 173.08047 133.08556 115.07513 87.04388 73.06467 | C8H13O4 C6H13O3 C6H11O2 C4H7O2 C4H9O | 3 1 2 2 1 | |||
| 2 | P | 305.1590 | 12.9 | C14H24O7 (99.99%) | 3 | No hits | 259.11685 219.12209 173.08040 155.06985 133.08549 115.07516 87.04391 | C12H19O6 C10H19O5 C8H13O4 C8H11O3 C6H13O3 C6H11O2 C4H7O2 | 4 2 3 4 1 3 2 | |||
| 3 | P | 408.22407 | 15.2 | C18H33O9N (Ammonium adduct of C18H31O9) (60.57%) | 3 | No hits | 392.19974 305.15979 259.11786 219.12292 | undetermined C14H25O7 C12H19O6 C10H19O5 | 3 4 2 | |||
| 3 | P | 408.22407 | 15.2 | C19H29O5N5 (99.50%) | 8 | No hits | 392.19969 305.15978 259.11783 219.12292 | undetermined C15H21O3N4 C13H15O2N4 C10H1905 | 8 9 2 | 173.08086 155.07023 133.08593 115.07541 | C8H13O4 C8H11O3 C6H1303 C6H11O2 | 3 4 1 2 |
| 4 | P | 494.25967 | 17.0 | C22H39O11N (Ammonium adduct of C22H37O11) (99.97%) | 4 | No hits | 477.23270 | C22H37O11 | 5 | 459.22238 431.19101 373.18582 345.15396 305.15924 259.11740 219.12263 155.07021 | C22H35O10 C20H31O10 C18H29O8 C16H25O8 C14H25O7 C12H19O6 C10H19O5 C8H11O3 | 6 6 5 5 3 4 2 3 |
| 5 | P | 580.2965 | 18.5 | C26H45O13N (Ammonium adduct of C26H43O13) (99.96%) | 5 | No hits | 563.26880 477.23288 431.19122 345.15424 305.15945 | C26H43O13 C22H37O11 C20H31O10 C16H25O8 C14H25O7 | 6 5 6 5 3 | 259.11752 219.12265 155.07025 | C12H19O6 C10H19O5 C8H11O3 | 4
2 4 |
| 6 | P | 448.2180 | 18.8 | C20H33O10N (Ammonium adduct of C20H30O10) (91.50%) | 5 | No hits | 431.18991 345.15372 259.11725 241.10663 155.07002 | C20H31O10 C16H25O8 C12H19O6 C12H17O5 C8H11O3 | 6 5 4 5 4 | |||
| 7 | P | 534.2550 | 19.4 | C24H39O12N (Ammonium adduct of C24H37O12) (99.13%) | 6 | No hits | 517.22723 431.19070 345.15402 259.11737 241.10681 155.07013 | C24H37O12 C20H31O10 C16H25O8 C12H19O6 C12H17O5 C8H11O | 7 6 5 4 5 4 | |||
| 7 | P | 534.2550 | 19.4 | C25H35O8N5 (71.66%) | 11 | Oxyplicacetin; 3′-Hydroxy-plicacetin | 517.22723 431.19070 345.15402 259.11737 241.10681 | C25H33O8N4 C21H27O6N4 C17H21O4N4 C10H17O5N3 C10H15O4N3 | 12 11 10 4 5 | |||
| 8 | N | 269.13940 | 12.3 | C14H22O5 (99.99%) | 5 | No hits | 251.12892 225.14969 | C14H19O4 C13H21O3 | 6 4 | |||
| 9 | N | 405.24944 | 19.3 | C20H38O8 (99.92%) | 3 | No hits | 359.24274 267.19690 | C19H35O6 C16H27O3 | 3 4 | |||
| 10 | N | 285.20719 | 21.5 | C16H30O4 (88.36%) | 3 | Hexadecanedioic acid/ethyl plakortide Z/ethyl didehydro-seco-plakortide Z | 267.19641 | C16H27O3 | 4 | 125.09721 141.12836 185.11803 223.20638 | C8H13O C9H17O C10H17O3 C15H27O | 3 2 3 3 |
| Peak ID | ESI Mode | m/z | Rt (min) | Molecular Formula (Isotope Fit Score A0 to A3) | Hits | RDB | Fragment Ions MS2 | Molecular Formula | RDB | Fragment Ions MS3 | Molecular Formula | RDB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | P | 230.2481 | 17.3 | C14H31ON (99.77%) | Xestoaminol C | 1 | 212.23662 | C14H30N | 1 | |||
| 2 | P | 258.2793 | 19.4 | C16H35ON (99.95%) | No hits | 1 | 240.26793 | C16H34N | 1 | |||
| 3 | P | 597.5208 | 30.1 | C35H68O5N2 (99.01%) | No hits | 3 | 337.28409 355.29462 351.29974 369.31042 | C20H37O2N2 C20H39O3N2 C21H39O2N2 C21H41O3N2 | 4 3 4 3 | 319.27350
295.27368 | C20H35ON2 C18H35ON2 | 5
3 |
| 4 | N | 265.1476 | 17.6 | C12H26O4S (99.25%) | No hits | 1 | 96.9590 | [HSO4]− | 1 | |||
| 5 | N | 760.54162 | 17.6 | C42H75O5N5S (90.86%) | No hits | 9 | 531.30280 | C30H45O5NS | 9 | 96.9590 | [HSO4]− | |
| 6 | N | 279.1631 | 19.1 | C13H28O4S (98.04%) | No hits | 1 | 96.9590 | [HSO4]− | 1 | 96.9590 | ||
| 7 | N | 816.60400 | 19.1 | C46H83O5N5S (87.43%) | No hits | 9 | 279.16318 | C32H49O5NS C13H27O4S | 9 1 | 96.9590 | [HSO4]− | |
| 8 | N | 309.17358 | 19.7 | C14H30O5S (98.04%) | No hits | 1 | 96.9590 | [HSO4]− | 1 | |||
| 9 | N | 293.1790 | 21.6 | C14H30O4S (99.86%) | No hits | 1 | 96.9590 | [HSO4]− | 1 |
| Rt (min) | m/z [M + H]+ | Molecular Formula | Isotope Fit Score A0 to A3 (%) | RDB | Predictions to Calculated RDB |
|---|---|---|---|---|---|
| 19.23 | 462.1727 | C19H23O7N7 | 87.66 | 12 | Cyclic with Phe/Tyr |
| 21.43 | 499.1871 | C18H26O9N8 | 87.64 | 10 | Cyclic |
| 22.15 | 587.2399 | C22H34O11N8 | 82.51 | 10 | Linear |
| 28.14 | 569.4893 | C33H64O5N2 | 99.99 | 3 | Linear |
| 29.46 | 583.5048 | C34H66O5N2 | 99.95 | 3 | Linear |
| 33.77 | 1078.7151 | C59H95O11N7 | 99.86 | 16 | Cyclic with Trp |
| 35.30 | 1118.7461 | C62H99O11N7 | 98.83 | 17 | Cyclic with Trp/Arg |
| 35.64 | 1092.7308 | C60H97O11N7 C55H97O13N9 | 99.91 85.21 | 16 12 | Cyclic with Trp Cyclic |
| 36.80 | 1106.7460 | C61H99O11N7 C56H99O13N9 | 99.50 99.05 | 16 12 | Cyclic with Trp Cyclic |
| 37.55 | 849.6953 | C51H88O4N6 C56H88O2N4 | 99.92 58.43 | 11 15 | Cyclic with Trp/Arg Cyclic with Trp/Arg |
3.2. Bacterial Culture and Extraction
3.3. Mass Spectrometry
3.4. NMR Spectroscopy
3.5. Data Analysis Tools for Mass Spectrometry Data
3.6. Molecular Identification
3.7. Bioassay Screening
3.7.1. Anti-Infectives
3.7.2. Metabolic Disease and Inflammation
3.7.3. Cell-based Functional Assays
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, J.H.; Singh, A.J.; Northcote, P.T. Microtubule-stabilizing drugs from marine sponges: Focus on peloruside A and zampanolide. Mar. Drugs 2010, 8, 1059–1079. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef]
- Vinothkumar, S.; Parameswaran, P.S. Recent advances in marine drug research. Biotechnol. Adv. 2013, 31, 1826–1845. [Google Scholar] [CrossRef]
- Wang, G. Diversity and biotechnological potential of the sponge-associated microbial consortia. J. Ind. Microbiol. Biotechnol. 2006, 33, 545–551. [Google Scholar] [CrossRef]
- Hentschel, U.; Hopke, J.; Horn, M.; Friedrich, A.B.; Wagner, M.; Hacker, J.; Moore, B.S. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 2002, 68, 4431–4440. [Google Scholar] [CrossRef]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Kennedy, J.; Baker, P.; Piper, C.; Cotter, P.D.; Walsh, M.; Mooij, M.J.; Bourke, M.B.; Rea, M.C.; O’Connor, P.M.; Ross, R.P.; et al. Isolation and analysis of bacteria with antimicrobial activities from the marine sponge Haliclona simulans collected from Irish waters. Mar. Biotechnol. 2009, 11, 384–396. [Google Scholar] [CrossRef]
- Hildebrand, M.; Waggoner, L.E.; Liu, H.; Sudek, S.; Allen, S.; Anderson, C.; Sherman, D.H.; Haygood, M. bryA: An unusual modular polyketide synthase gene from the uncultivated bacterial symbiont of the marine bryozoan Bugula neritina. Chem. Biol. 2004, 11, 1543–1552. [Google Scholar] [CrossRef]
- Zhang, L.; An, R.; Wang, J.; Sun, N.; Zhang, S.; Hu, J.; Kuai, J. Exploring novel bioactive compounds from marine microbes. Curr. Opin. Microbiol. 2005, 8, 276–281. [Google Scholar] [CrossRef]
- Thomas, T.R.; Kavlekar, D.P.; LokaBharathi, P.A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar] [CrossRef]
- Proksch, P.; Edrada, R.A.; Ebel, R. Drugs from the seas—Current status and microbiological implications. Appl. Microbiol. Biotechnol. 2002, 59, 125–134. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.G. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Newman, D.J.; Hill, R.T. New drugs from marine microbes: The tide is turning. J. Ind. Microbiol. Biotechnol. 2006, 33, 539–544. [Google Scholar] [CrossRef]
- Martens, T.; Gram, L.; Grossart, H.P.; Kessler, D.; Muller, R.; Simon, M.; Wenzel, S.C.; Brinkhoff, T. Bacteria of the Roseobacter clade show potential for secondary metabolite production. Microb. Ecol. 2007, 54, 31–42. [Google Scholar] [CrossRef]
- Piel, J. Bacterial symbionts: Prospects for the sustainable production of invertebrate-derived pharmaceuticals. Curr. Med. Chem. 2006, 13, 39–50. [Google Scholar] [CrossRef]
- Sudek, S.; Lopanik, N.B.; Waggoner, L.E.; Hildebrand, M.; Anderson, C.; Liu, H.; Patel, A.; Sherman, D.H.; Haygood, M.G. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J. Nat. Prod. 2007, 70, 67–74. [Google Scholar] [CrossRef]
- Hentschel, U.; Schmid, M.; Wagner, M.; Fieseler, L.; Gernert, C.; Hacker, J. Isolation and phylogenetic analysis of bacteria with antimicrobial activities from the Mediterranean sponges Aplysina aerophoba and Aplysina cavernicola. FEMS Microbiol. Ecol. 2001, 35, 305–312. [Google Scholar] [CrossRef]
- Hentschel, U.; Usher, K.M.; Taylor, M.W. Marine sponges as microbial fermenters. FEMS Microbiol. Ecol. 2006, 55, 167–177. [Google Scholar] [CrossRef]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Ritacco, F.V.; Haltli, B.; Janso, J.E.; Greenstein, M.; Bernan, V.S. Dereplication of Streptomyces soil isolates and detection of specific biosynthetic genes using an automated ribotyping instrument. J. Ind. Microbiol. Biotechnol. 2003, 30, 472–479. [Google Scholar] [CrossRef]
- Jensen, P.R.; Williams, P.G.; Oh, D.C.; Zeigler, L.; Fenical, W. Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 2007, 73, 1146–1152. [Google Scholar] [CrossRef]
- Tawfike, A.F.; Viegelmann, C.; Edrada-Ebel, R. Metabolomics and dereplication strategies in natural products. Methods Mol. Biol. 2013, 1055, 227–244. [Google Scholar] [CrossRef]
- Bobzin, S.C.; Yang, S.; Kasten, T.P. LC-NMR: A new tool to expedite the dereplication and identification of natural products. J. Ind. Microbiol. Biotechnol. 2000, 25, 342–345. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication strategies for targeted isolation of new antitrypanosomal actinosporins A and B from a marine sponge associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220–1244. [Google Scholar] [CrossRef]
- Blunt, J.W. MarinLit; University of Canterbury: Christchurch, New Zealand, 2012. [Google Scholar]
- Laatsch, H. Antibase Version 4.0—The Natural Compound Identifier; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Nielsen, K.F.; Mansson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Klitgaard, A.; Iversen, A.; Andersen, M.R.; Larsen, T.O.; Frisvad, J.C.; Nielsen, K.F. Aggressive dereplication using UHPLC-DAD-QTOF: Screening extracts for up to 3000 fungal secondary metabolites. Anal. Bioanal. Chem. 2014, 406, 1933–1943. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef]
- Erve, J.C.; Gu, M.; Wang, Y.; DeMaio, W.; Talaat, R.E. Spectral accuracy of molecular ions in an LTQ/Orbitrap mass spectrometer and implications for elemental composition determination. J. Am. Soc. Mass Spectrom. 2009, 20, 2058–2069. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2001, 2, 155–168. [Google Scholar] [CrossRef]
- Van der Werf, M.J.; Jellema, R.H.; Hankemeier, T. Microbial metabolomics: Replacing trial-and-error by the unbiased selection and ranking of targets. J. Ind. Microbiol. Biotechnol. 2005, 32, 234–252. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar]
- Robinette, S.L.; Bruschweiler, R.; Schroeder, F.C.; Edison, A.S. NMR in metabolomics and natural products research: Two sides of the same coin. Acc. Chem. Res. 2012, 45, 288–297. [Google Scholar] [CrossRef]
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef]
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar] [CrossRef]
- Boroujerdi, A.F.; Vizcaino, M.I.; Meyers, A.; Pollock, E.C.; Huynh, S.L.; Schock, T.B.; Morris, P.J.; Bearden, D.W. NMR-based microbial metabolomics and the temperature-dependent coral pathogen Vibrio coralliilyticus. Environ. Sci. Technol. 2009, 43, 7658–7664. [Google Scholar] [CrossRef]
- Tang, J. Microbial metabolomics. Curr. Genomics 2011, 12, 391–403. [Google Scholar] [CrossRef]
- Vynne, N.G.; Mansson, M.; Nielsen, K.F.; Gram, L. Bioactivity, chemical profiling, and 16S rRNA-based phylogeny of Pseudoalteromonas strains collected on a global research cruise. Mar. Biotechnol. 2011, 13, 1062–1073. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jin, Y.; Jin, M.; Yu, X. A comparative study on the phylogenetic diversity of culturable actinobacteria isolated from five marine sponge species. Antonie Van Leeuwenhoek 2008, 93, 241–248. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Pimentel-Elardo, S.M.; Hanora, A.; Radwan, M.; Abou-El-Ela, S.H.; Ahmed, S.; Hentschel, U. Isolation, phylogenetic analysis and anti-infective activity screening of marine sponge-associated actinomycetes. Mar. Drugs 2010, 8, 399–412. [Google Scholar] [CrossRef]
- Joint, I.; Muhling, M.; Querellou, J. Culturing marine bacteria—An essential prerequisite for biodiscovery. Microb. Biotechnol. 2010, 3, 564–575. [Google Scholar] [CrossRef]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef]
- Pluskal, T.; Uehara, T.; Yanagida, M. Highly accurate chemical formula prediction tool utilizing high-resolution mass spectra, MS/MS fragmentation, heuristic rules, and isotope pattern matching. Anal. Chem. 2012, 84, 4396–4403. [Google Scholar] [CrossRef]
- Naruse, N.; Tenmyo, O.; Kobaru, S.; Kamei, H.; Miyaki, T.; Konishi, M.; Oki, T. Pumilacidin, a complex of new antiviral antibiotics. Production, isolation, chemical properties, structure and biological activity. J. Antibiot. 1990, 43, 267–280. [Google Scholar] [CrossRef]
- Stein, T. Bacillus subtilis antibiotics: Structures, syntheses and specific functions. Mol. Microbiol. 2005, 56, 845–857. [Google Scholar] [CrossRef]
- Goldstein, B.J. Protein-tyrosine phosphatase 1B (PTP1B): A novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets Immune Endocr. Metab. Disord. 2001, 1, 265–275. [Google Scholar] [CrossRef]
- Shiomi, K.; Haneda, K.; Tomoda, H.; Iwai, Y.; Omura, S. Cytosaminomycins, new anticoccidial agents produced by Streptomyces sp. KO-8119. II. Structure elucidation of cytosaminomycins A, B, C and D. J. Antibiot. 1994, 47, 782–786. [Google Scholar] [CrossRef]
- Haneda, K.; Shinose, M.; Seino, A.; Tabata, N.; Tomoda, H.; Iwai, Y.; Omura, S. Cytosaminomycins, new anticoccidial agents produced by Streptomyces sp. KO-8119. I. Taxonomy, production, isolation and physico-chemical and biological properties. J. Antibiot. 1994, 47, 774–781. [Google Scholar] [CrossRef]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef]
- Shirling, E.B.; Gottlieb, D. Methods for characterization of Streptomyces species. Int. J. Syst. Bacteriol. 1966, 16, 313–340. [Google Scholar] [CrossRef]
- Olson, J.B.; Lord, C.C.; McCarthy, P.J. Improved recoverability of microbial colonies from marine sponge samples. Microb. Ecol. 2000, 40, 139–147. [Google Scholar]
- Weiner, R.M.; Segall, A.M.; Colwell, R.R. Characterization of a Marine Bacterium Associated with Crassostrea virginica (the Eastern Oyster). Appl. Environ. Microbiol. 1985, 49, 83–90. [Google Scholar]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 1985, 49, 1–7. [Google Scholar]
- ProteoWizard Software Foundation. ProteoWizard Homepage. Available online: http://proteowizard.sourceforge.net/ (accessed on 15 January 2014).
- Lane, D.J. 16S/23S rRNA Sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley: Chichester, UK, 1991; pp. 115–175. [Google Scholar]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2014, 42, D32–D37. [Google Scholar] [CrossRef]
- Viegelmann, C.; Parker, J.; Ooi, T.; Clements, C.; Abbott, G.; Young, L.; Kennedy, J.; Dobson, A.D.W.; Edrada-Ebel, R. Isolation and Identification of Antitrypanosomal and Antimycobacterial Active Steroids from the Sponge Haliclona simulans. Mar. Drugs 2014, 12, 2937–2952. [Google Scholar] [CrossRef]
- Inderlied, C.B.; Salfinger, M. Antimicrobial agents and susceptibility tests: Mycobacteria. In Manual of Clinical Microbiology, 6th ed.; Murray, P.R., Baron, E.J., Pfaller, M.A., Tenover, F.C., Yolken, R.H., Eds.; American Society for Microbiology: Washington, DC, USA, 1995; pp. 1385–1404. [Google Scholar]
- Molecular Devices Home Page. Available online: http://info.moleculardevices.com/acton/attachment/2560/f-0bc0/0/-/-/-/-/file.pdf (accessed on 1 March 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Macintyre, L.; Zhang, T.; Viegelmann, C.; Martinez, I.J.; Cheng, C.; Dowdells, C.; Abdelmohsen, U.R.; Gernert, C.; Hentschel, U.; Edrada-Ebel, R. Metabolomic Tools for Secondary Metabolite Discovery from Marine Microbial Symbionts. Mar. Drugs 2014, 12, 3416-3448. https://doi.org/10.3390/md12063416
Macintyre L, Zhang T, Viegelmann C, Martinez IJ, Cheng C, Dowdells C, Abdelmohsen UR, Gernert C, Hentschel U, Edrada-Ebel R. Metabolomic Tools for Secondary Metabolite Discovery from Marine Microbial Symbionts. Marine Drugs. 2014; 12(6):3416-3448. https://doi.org/10.3390/md12063416
Chicago/Turabian StyleMacintyre, Lynsey, Tong Zhang, Christina Viegelmann, Ignacio Juarez Martinez, Cheng Cheng, Catherine Dowdells, Usama Ramadan Abdelmohsen, Christine Gernert, Ute Hentschel, and RuAngelie Edrada-Ebel. 2014. "Metabolomic Tools for Secondary Metabolite Discovery from Marine Microbial Symbionts" Marine Drugs 12, no. 6: 3416-3448. https://doi.org/10.3390/md12063416
APA StyleMacintyre, L., Zhang, T., Viegelmann, C., Martinez, I. J., Cheng, C., Dowdells, C., Abdelmohsen, U. R., Gernert, C., Hentschel, U., & Edrada-Ebel, R. (2014). Metabolomic Tools for Secondary Metabolite Discovery from Marine Microbial Symbionts. Marine Drugs, 12(6), 3416-3448. https://doi.org/10.3390/md12063416