Chromomycin A2 Induces Autophagy in Melanoma Cells


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Cytotoxicity of Chromomycin A2
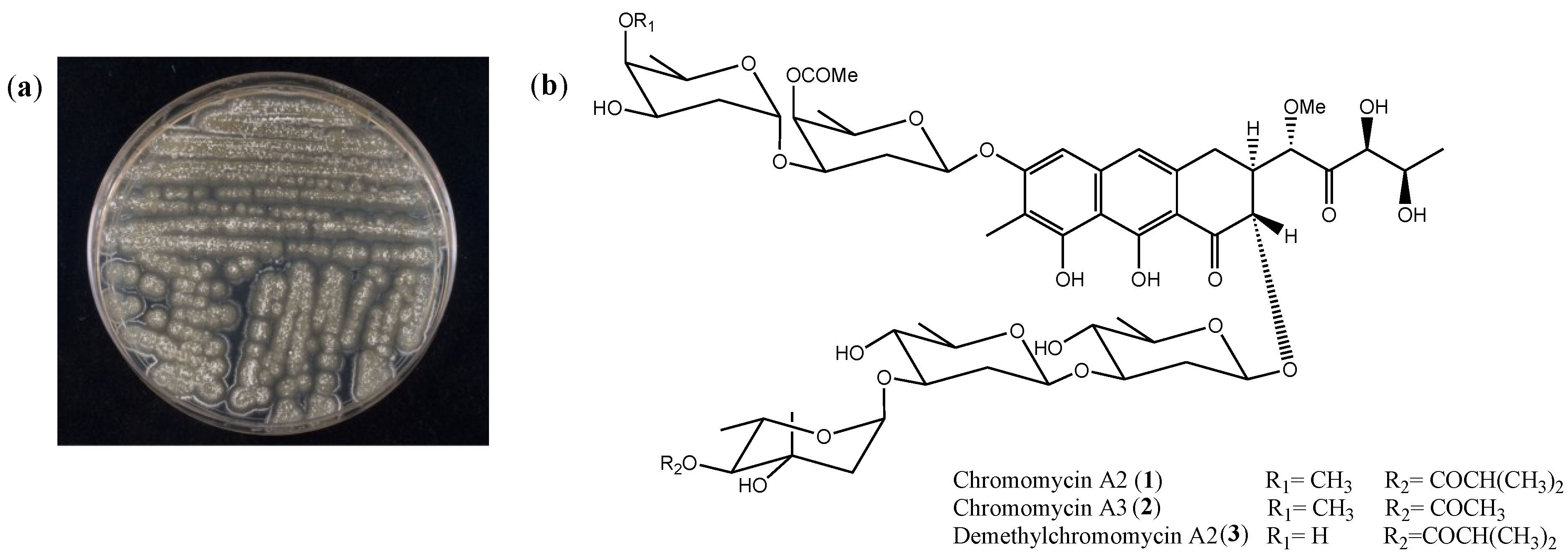

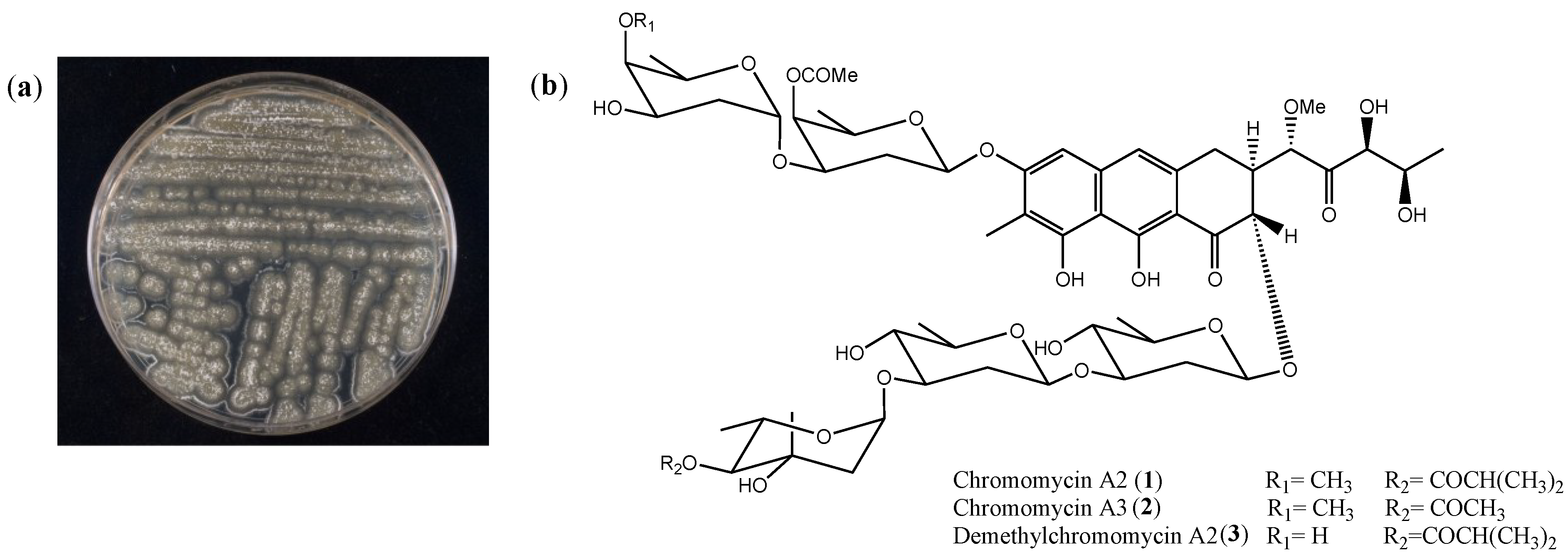{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Histological Origin (Duplication Time) | IC50 [nM] (CI 95%) | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| HCT-116 | Colon adenocarcinoma (17.4 h) | >400 | 21.6 (12.1–38.5) | 6.3 (4.6–8.7) |
| HL-60 | Promyelocitic leukemia (28.6 h) | >200 | 22.3 (19.7–25.3) | 10.1 (8.13–12.46) |
| MALME-3M | Metastatic melanoma (46.2 h) | >400 | 16.7 (7.2–38.5) | 18.7 (14.6–24.3) |
| OVCAR-8 | Ovarian adenocarcinoma (26.1 h) | >200 | 33.8 (28.2–40.5) | 8.8 (7.7–9.9) |
| PC-3M | Metastatic prostate carcinoma (20 h) | >400 | 209.8 (122.5–359.5) | 49.0 (27.5–87.3) |
| SF-295 | Glioblastoma (29.5 h) | >200 | n.d. | 20.8 (18.8–23.0) |
| MRC-5 | Lung fibroblast (27 h) | > 400 | 131.7 (86.6–200.3) | 109.0 (82.0–144.8) |
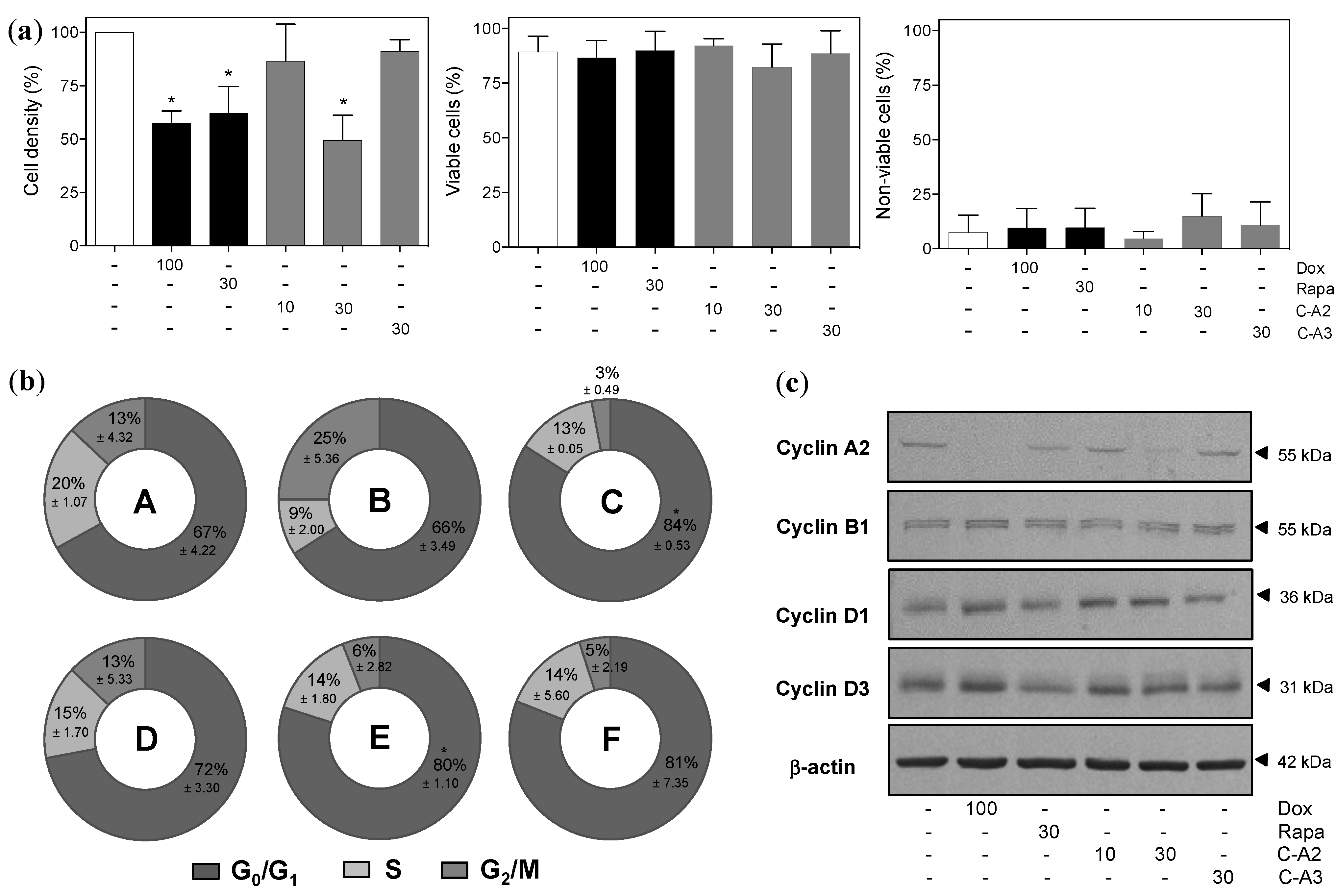
2.2. Chromomycin A2 Alters Cell Cycle of Metastatic Melanoma Cells MALME-3M
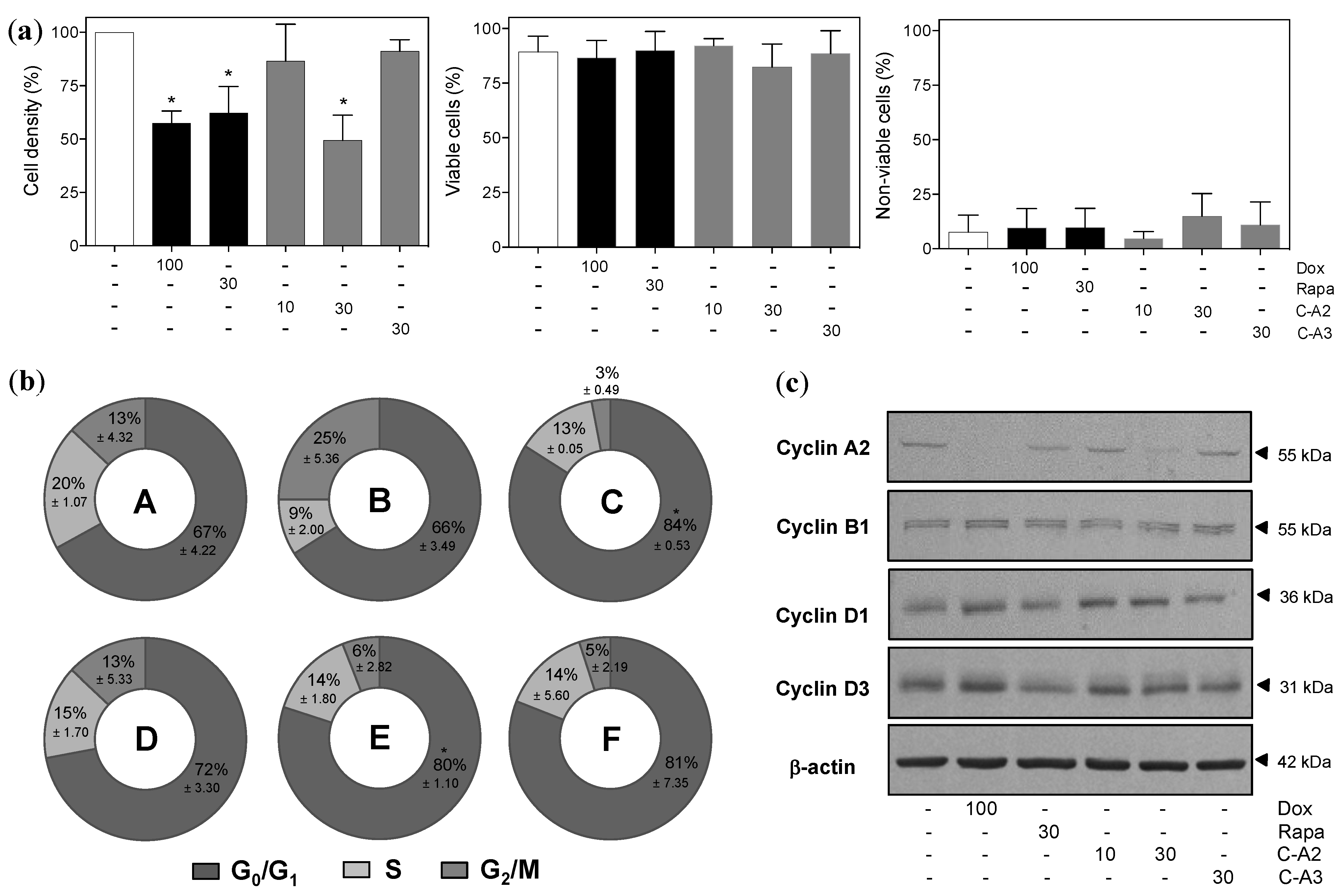
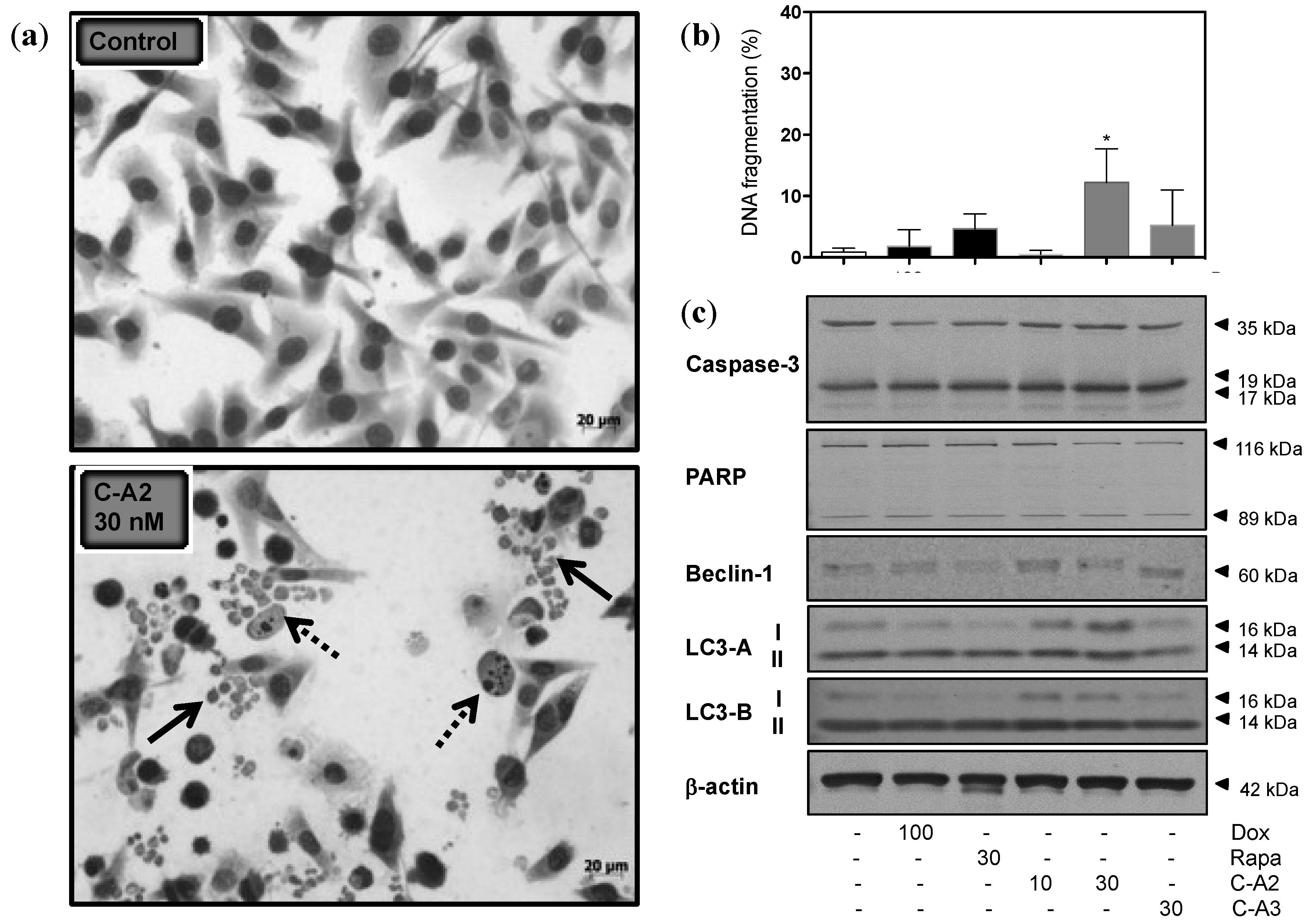
2.3. Alterations in Morphology and Expression of Autophagic Proteins by Chromomycin A2
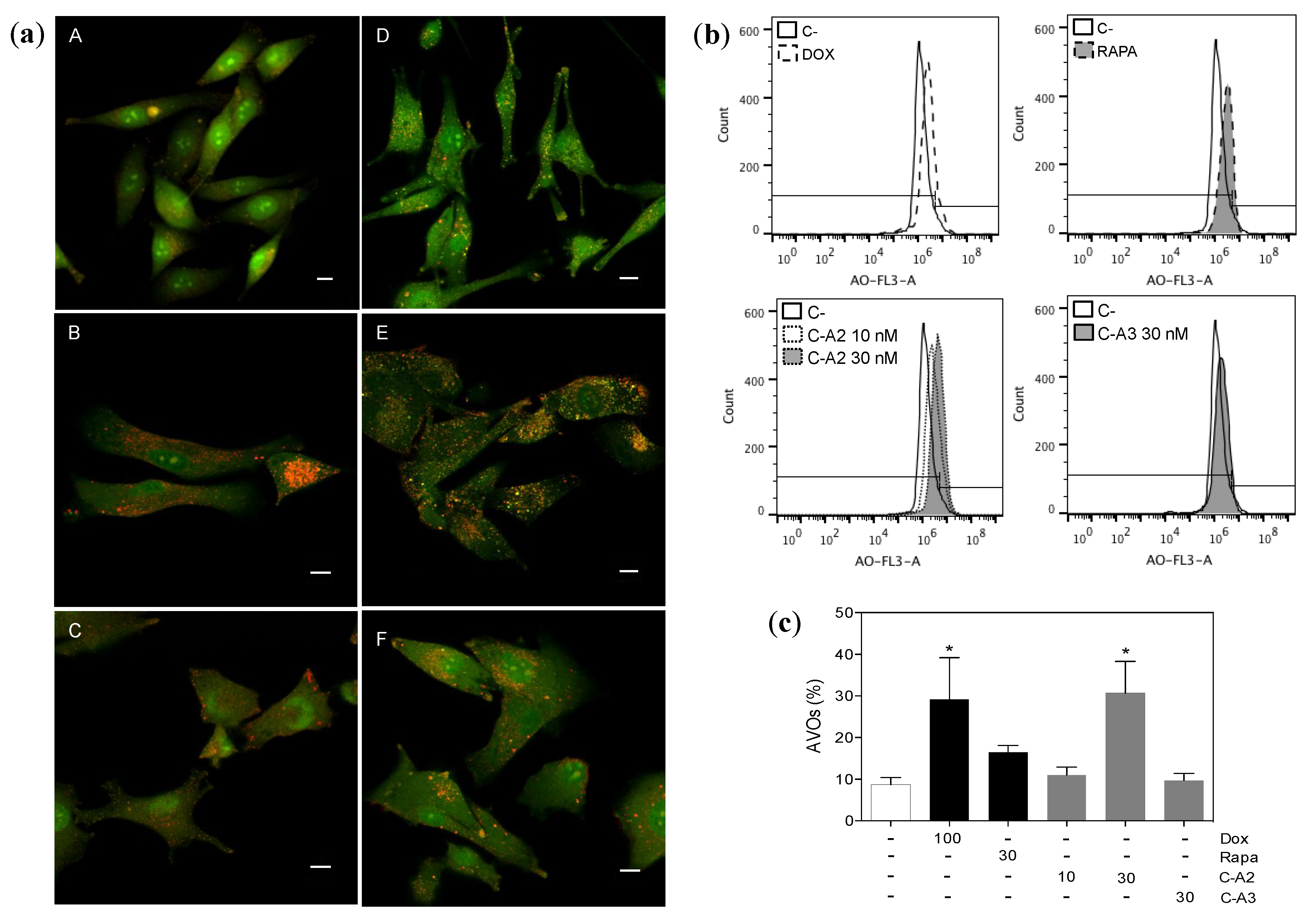
2.4. Induction of Autophagy by Chromomycin A2
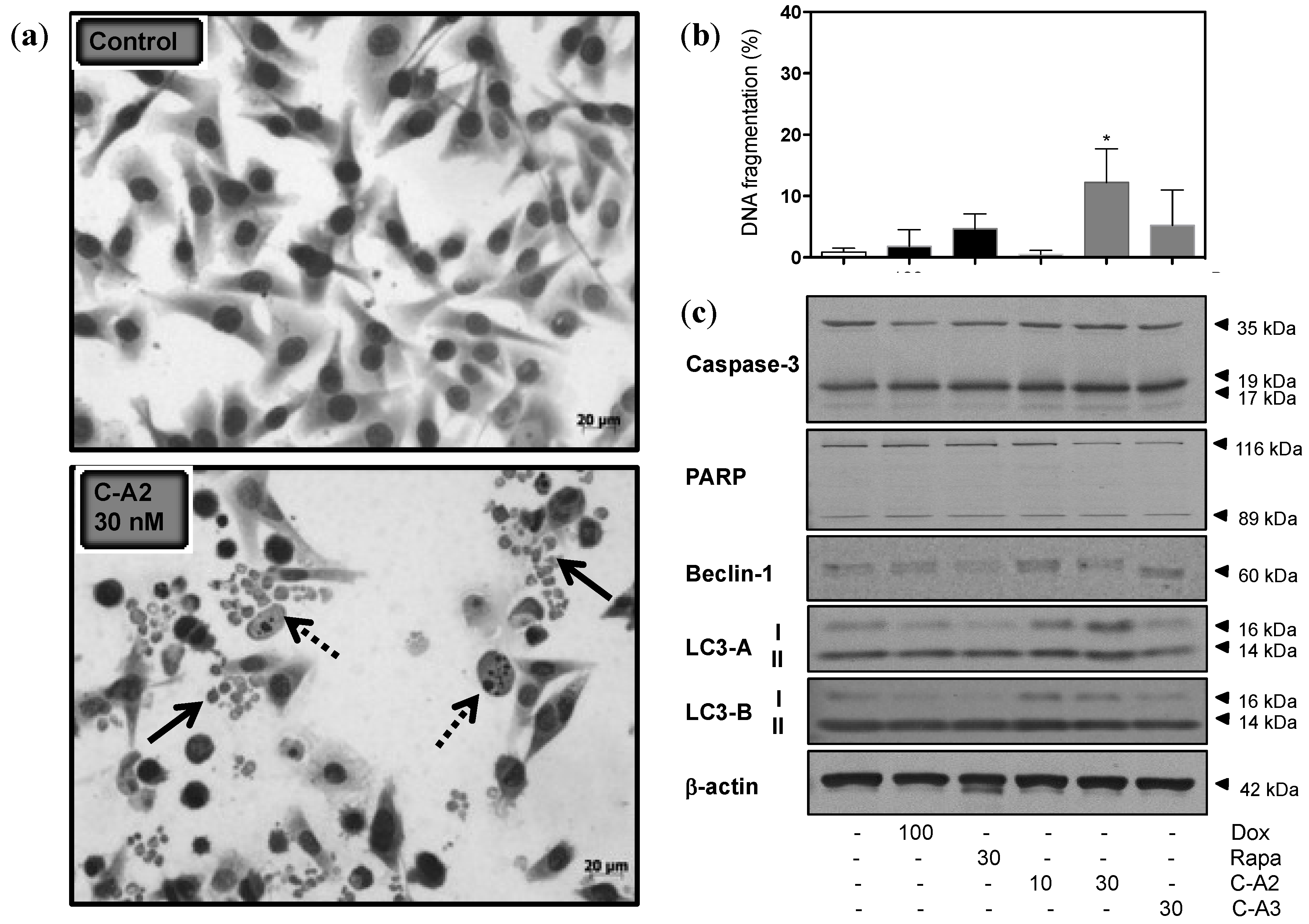
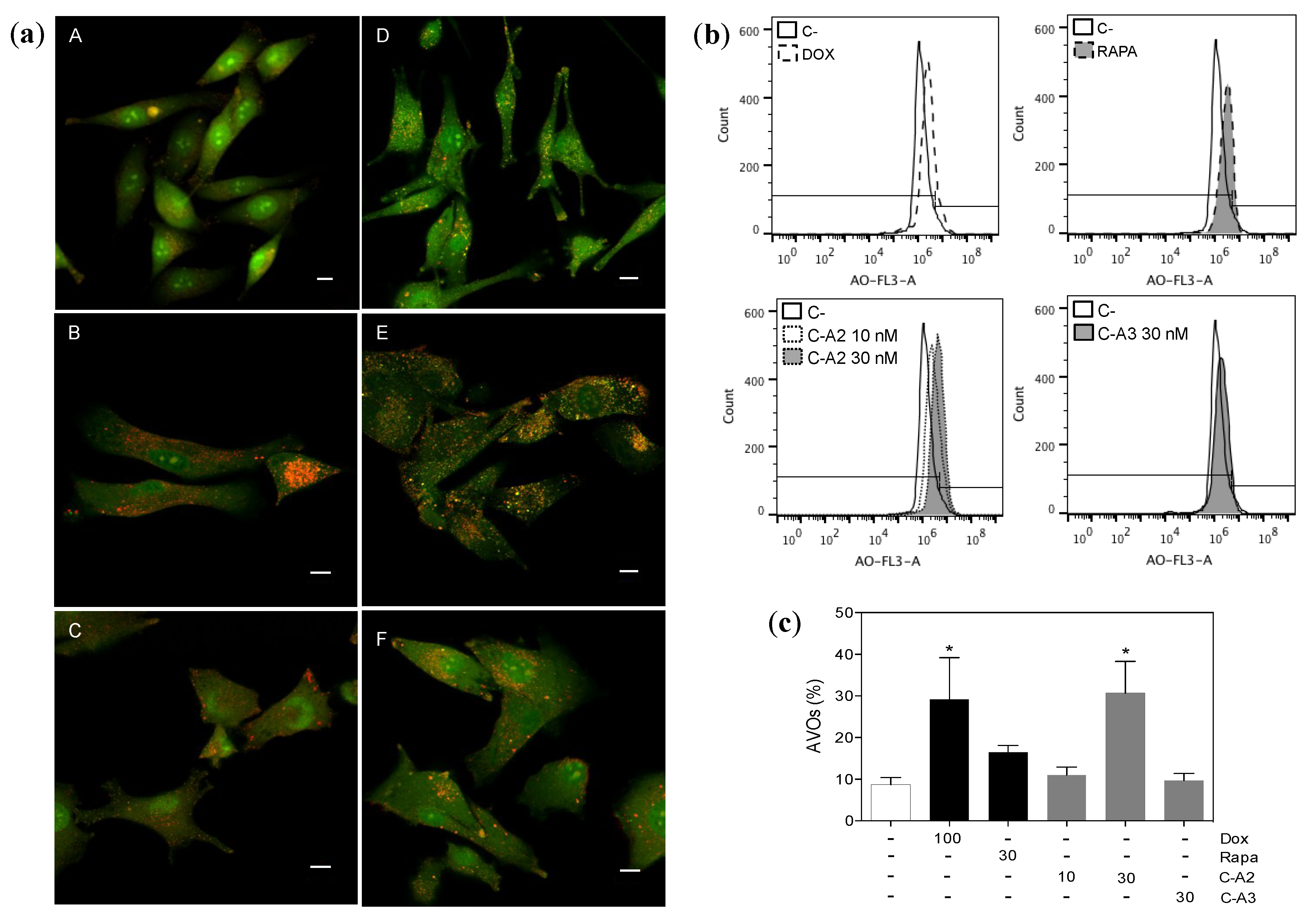
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation and Identification of Bacterial Strain
3.3. Up-Scale Bacterial Growth and Isolation of Chromomycin A2
3.4. Cytotoxicity Assay
3.5. Cell Viability Analyses
3.6. Cell Cycle and DNA Fragmentation Analyses
3.7. Morphological Analysis
3.8. Detection and Quantification of Acidic Vesicular Organelles (AVOs)
3.9. Western Blot
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lombó, F.; Menéndez, N.; Salas, J.A.; Méndez, C. The aureolic acid family of antitumor compounds: Structure, mode of action, biosynthesis, and novel derivatives. Appl. Microbiol. Biotechnol. 2006, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Patel, D.J. Solution structure of the mithramycin dimer-DNA complex. Biochemistry 1993, 32, 6588–6604. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.J. Metabolic and toxic effects of mithramycin during tumor therapy. Am. J. Med. 1970, 49, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Ryan, W.G. Experiences in the treatment of paget’s disease of bone with mithramycin. J. Am. Vet. Med. Assoc. 1970, 213, 1153. [Google Scholar] [CrossRef]
- Rohr, J.; Méndez, C.; Salas, J.A. The biosynthesis of aureolic acid group antibiotics. Bioorg. Chem. 1999, 27, 41–54. [Google Scholar] [CrossRef]
- Sabín, J.G. Exploring novel opportunities for aureolic acids as anticancer drugs. Biochem. Pharmacol. 2013, 2, 1–4. [Google Scholar] [CrossRef]
- Tatsuoka, S.; Nakazawa, K.; Miyake, A.; Kaziwara, K.; Aramaki, M.; Shibata, M.; Tanabe, K.; Hamada, Y.; Hitomi, H.; Myiamoto, M.; et al. Isolation, anticancer activity and pharmacology of new antibiotic chromomycin A3. Gann. 1958, 49, 23–24. [Google Scholar] [PubMed]
- Tutsuoka, S.; Tanaka, K.; Miyamoto, M.; Morita, K.; Kawamatsu, Y.; Nakanishi, K.; Nakadaira, Y.; Bhacca, N. The structure of chromomycin A3, a cancerostatic antibiotic. Proc. Jpn. Acad. 1964, 40, 236. [Google Scholar]
- Chatterjee, S.; Zaman, K.; Ryu, H.; Conforto, A.; Ratan, R.R. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann. Neurol. 2001, 49, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, N.; Osti, F.; Rutigliano, C.; Corradini, F.G.; Borsetti, E.; Tomassetti, M.; Mischiati, C.; Feriotto, G.; Gambari, R. The DNA-binding drugs mithramycin and chromomycin are powerful inducers of erythroid differentiation of human K562 cells. Br. J. Haematol. 1999, 104, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Espindola, A.P.D.M.; Stewart, N.A.; Wei, S.; Posner, B.A.; MacMillan, J.B. Chromomycin SA analogs from a marine-derived Streptomyces sp. Bioorg. Med. Chem. 2011, 19, 5183–5189. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Bhattacharyya, D.; Dasgupta, D. Structural basis of DNA recognition by anticancer antibiotics, chromomycin A(3), and mithramycin: Roles of minor groove width and ligand flexibility. Biopolymers 2001, 56, 85–95. [Google Scholar] [CrossRef]
- Hsu, C.-W.; Chuang, S.-M.; Wu, W.-L.; Hou, M.-H. The crucial role of divalent metal ions in the DNA-acting efficacy and inhibition of the transcription of dimeric chromomycin A3. PLoS One 2012, 7, e43792. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, M.; Williams, S.T. Ecology of actinomycetes. Annu. Rev. Microbiol. 1983, 37, 189–216. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Lyla, P.S.; Ajmal Khan, S. Distribution and generic composition of culturable marine actinomycetes from the sediments of Indian continental slope of Bay of Bengal. Chin. J. Oceanol. Limnol. 2008, 26, 166–177. [Google Scholar] [CrossRef]
- Bérdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Kawamatsu, Y.; Kawashima, K.; Shinohara, M.; Tanaka, K.; Tatsuoka, S.; Nakanishi, K. Chromomycin A2, A3 and A4. Tetrahedron 1967, 23, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Koenuma, M.; Matsumoto, K.; Tori, K.; Terui, Y. NMR studies of chromomycins, olivomycins, and their derivatives. J. Antibiot. 1988, 41, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Toume, K.; Tsukahara, K.; Ito, H.; Arai, M.A.; Ishibashi, M. Chromomycins A2 and A3 from marine actinomycetes with TRAIL resistance-overcoming and Wnt signal inhibitory activities. Mar. Drugs 2014, 12, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Kimura, T.; Tawara, K.; Sasaki, K.; Nakajima, K.; Shimaoka, N.; Okamodo, S.; Shimohira, M.; Isono, J. Aburamycin, a new antibiotic. J. Antibiot. 1957, 10, 205–212. [Google Scholar] [PubMed]
- Miyamoto, M.; Kawamatsu, Y.; Kawashima, K.; Shinohara, M.; Nakanishi, K. The full structures of three chromomycins, A2, A3 and A4. Tetrahedron Lett. 1966, 7, 545–552. [Google Scholar] [CrossRef]
- Koenuma, M.; Uchida, N.; Yamagughi, K.; Kawamura, Y.; Matsumoto, K. New aureolic acid antibiotics I. Screening, isolation, characterization and biological properties. J. Antibiot. 1988, 41, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Koenuma, M.; Yoshimura, Y.; Matsumoto, K.; Terui, Y. New aureolic acid antibiotics II. Structure determination. J. Antibiot. 1988, 41, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, N.; Nur-e-Alam, M.; Fischer, C.; Braña, A.F.; Salas, J.A.; Rohr, J.; Méndez, C. Deoxysugar transfer during chromomycin A3 biosynthesis in Streptomyces griseus subsp. griseus: New derivatives with antitumor activity. Appl. Environ. Microbiol. 2006, 72, 167–177. [Google Scholar]
- Xiong, Z.-Q.; Zhang, Z.-P.; Li, J.-H.; Wei, S.-J.; Tu, G.-Q. Characterization of Streptomyces padanus JAU4234, a producer of actinomycin X2, fungichromin, and a new polyene macrolide antibiotic. Appl. Environ. Microbiol. 2012, 78, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wen, Y.; Qian, C.; Li, O.; Fang, H.; Chen, W. Taxonomic study of a chromomycin-producing strain and reclassification of Streptomyces cavourensis subsp. washingtonensis as a later synonym of Streptomyces griseus. Int. J. Syst. Evol. Microbiol. 2008, 58, 2783–2787. [Google Scholar] [CrossRef]
- Lu, J.; Ma, Y.; Liang, J.; Xing, Y.; Xi, T.; Lu, Y. Aureolic acids from a marine-derived Streptomyces sp. WBF16. Microbiol. Res. 2012, 167, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Korman, J.B.; Fisher, D.E. Developing melanoma therapeutics: Overview and update. Wiley Interdiscip. Rev. Syst. Biol .Med. 2013, 5, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Battie, C.; Gohara, M.; Verschoore, M.; Roberts, W. Skin cancer in skin of color: An update on current facts, trends, and misconceptions. J. Drugs Dermatol. 2013, 12, 194–198. [Google Scholar] [PubMed]
- Hartman, K.G.; McKnight, L.E.; Liriano, M.A.; Weber, D.J. The evolution of S100B inhibitors for the treatment of malignant melanoma. Future Med. Chem. 2013, 5, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Fiala, R.; Patel, D.J. Solution structure of mithramycin dimers bound to partially overlapping sites on DNA. J. Mol. Biol. 1995, 251, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.A.; Owen, E.A.; Shafer, R.H. The three-dimensional structure of the 4:1 mithramycin:d(ACCCGGGT)(2) complex: Evidence for an interaction between the E saccharides. Biopolymers 2000, 54, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, N.; Nur-E-Alam, M.; Braña, A.F.; Rohr, J.; Salas, J.A.; Méndez, C. Tailoring modification of deoxysugars during biosynthesis of the antitumour drug chromomycin A by Streptomyces griseus ssp. griseus. Mol. Microbiol. 2004, 53, 903–915. [Google Scholar] [CrossRef]
- Hou, M.-H.; Lu, W.-J.; Lin, H.-Y.; Yuann, J.-M.P. Studies of sequence-specific DNA binding, DNA cleavage, and topoisomerase I inhibition by the dimeric chromomycin A3 complexed with Fe(II). Biochemistry 2008, 47, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Hou, M.-H. The binding of the Co(II) complex of dimeric chromomycin A3 to GC sites with flanking G:G mismatches. J. Inorg. Biochem. 2013, 121, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Preobrazhenskaya, M.N.; Tevyashova, A.N.; Olsufyeva, E.N.; Huang, K.; Huang, H. Second generation drugs-derivatives of natural antitumor anthracycline antibiotics daunorubicin, doxorubicin and carminomycin. J. Med. Sci. 2006, 26, 119–128. [Google Scholar]
- Tanemura, M.; Ohmura, Y.; Deguchi, T.; Machida, T.; Tsukamoto, R.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Ito, T.; et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am. J. Transplant 2012, 12, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.; Schulze-Osthoff, K. Activation and caspase-mediated inhibition of PARP: A molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 2002, 13, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Garcia, G.D.; Gregoracci, G.B.; Santos, E.O.; Meirelles, P.M.; Silva, G.G.Z.; Edwards, R.; Sawabe, T.; Gotoh, K.; Nakamura, S.; Iida, T.; et al. Metagenomic analysis of healthy and white lague-affected Mussismilia braziliensis corals. Microb. Ecol. 2013, 65, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimarães, L.A.; Jimenez, P.C.; Sousa, T.D.S.; Freitas, H.P.S.; Rocha, D.D.; Wilke, D.V.; Martín, J.; Reyes, F.; Deusdênia Loiola Pessoa, O.; Costa-Lotufo, L.V. Chromomycin A2 Induces Autophagy in Melanoma Cells. Mar. Drugs 2014, 12, 5839-5855. https://doi.org/10.3390/md12125839
Guimarães LA, Jimenez PC, Sousa TDS, Freitas HPS, Rocha DD, Wilke DV, Martín J, Reyes F, Deusdênia Loiola Pessoa O, Costa-Lotufo LV. Chromomycin A2 Induces Autophagy in Melanoma Cells. Marine Drugs. 2014; 12(12):5839-5855. https://doi.org/10.3390/md12125839
Chicago/Turabian StyleGuimarães, Larissa Alves, Paula Christine Jimenez, Thiciana Da Silva Sousa, Hozana Patrícia S. Freitas, Danilo Damasceno Rocha, Diego Veras Wilke, Jesús Martín, Fernando Reyes, Otília Deusdênia Loiola Pessoa, and Letícia Veras Costa-Lotufo. 2014. "Chromomycin A2 Induces Autophagy in Melanoma Cells" Marine Drugs 12, no. 12: 5839-5855. https://doi.org/10.3390/md12125839
APA StyleGuimarães, L. A., Jimenez, P. C., Sousa, T. D. S., Freitas, H. P. S., Rocha, D. D., Wilke, D. V., Martín, J., Reyes, F., Deusdênia Loiola Pessoa, O., & Costa-Lotufo, L. V. (2014). Chromomycin A2 Induces Autophagy in Melanoma Cells. Marine Drugs, 12(12), 5839-5855. https://doi.org/10.3390/md12125839