1. Introduction
Natural sources that can be used for the treatment or prevention of diseases have been of much interest during the past decades. In this context, extracts and compounds from plants are among the main targets of studies [
1,
2]. Several studies in the past reported various biologically active plants with marine plants allocating a large part [
3]. Due to the harsh conditions of marine environments and the need for strong protection, marine organisms produce unique substances. Plant extracts and compounds have been intensely studied in order to fully understand their action mechanism against common complications such as oxidative stress and inflammation [
4,
5,
6], as well as diseases with high morbidity and mortality rates including diabetes, cancer, obesity and AIDS [
7,
8,
9]. The halophyte
Salicornia herbacea is endemic to the western coast of the Korean peninsula and a part of folk medicine due to its effect on constipation, diabetes, obesity,
etc., [
10]. As expected, a number of experiments have credited
S. herbacea for various bioactivities including antioxidative, antiinflamatory, antihyperglycemic and antihyperlidemic [
11,
12,
13]. It was proposed that
S. herbacea contained active flavonoids [
14,
15,
16]. However, very few investigations have been carried out in order to evaluate the effect of
S. herbacea on osteoporosis.
Bone-related diseases, namely osteoporosis and age-related osteopenia are reported to be associated with bone mass loss due to lack of osteoblastogenesis [
17]. Studies showed that glucocorticoid treatment also caused an increase in bone adipocytes, which finally resulted in fractures and osteoporosis [
18]. Several mechanisms have been suggested for possible cause of bone mass imbalance. Peroxisome proliferator activated receptor (PPAR)γ is confirmed to play crucial roles in the outcome of bone marrow mesenchymal cell differentiation [
19]. Both
in vitro and
in vivo mechanism studies demonstrated that the activation of PPARγ promotes adipogenesis. Likewise, suppression of PPARγ pathway was shown to inhibit adipogenesis and stimulate osteoblast differentiation depending on the binding ligand [
20]. Several diabetic drugs as ligands of PPARγ activate adipogenesis and lower blood glucose. However, this activation also causes problems in bone mass by favoring adipogenesis of bone mesenchymal cells and deteriorating the bone mass balance. Diabetic drugs, obesity-related factors and long chain fatty acids were confirmed to be activating ligands for PPARγ [
21]. Overall, in this study
S. herbacea was tested for its potential effect on adipogenesis of pre-adipocytes and pre-osteoblast differentiation with a possible intervention in PPARγ pathway.
3. Discussion
The main age-related metabolic diseases are the major causes of diminished life quality for the elderly. Additionally, recent studies indicate an increasing trend for the younger generation to be diagnosed with obesity, type-2 diabetes and osteoporosis [
16,
22,
23]. Scientists are turning their attention to natural products to use to intervene with the progression of the aforementioned diseases due to bioavailability, biodegradation and fewer side effects. In this context, marine-based organisms are of high interest to pharmaceutical researchers as most marine plants and animals live in extreme conditions, which results in the need for unique compounds for survival. By means of this promising situation, past decades have been fruitful in ways of natural bioactive substance development. Numerous marine plants have been studied intensively in order to discover bioactive chemicals that can act against cancer, tumor growth, oxidative stress, diabetes, obesity and osteoporosis [
5,
24,
25]. Halophytes are also plants that survive under extreme conditions such as high concentrations of salt and harsh climate changes. In this context, the halophyte
S. herbacea was examined here for possible compounds with health beneficiary effects. Reports had already stated that
S. herbacea contains flavonoid glycosides and polysaccharides, which are natural antioxidants [
15].
In this study, we examined the ability of
S. herbacea to relieve osteoporosis conditions with a regulatory mechanism towards adipogenesis/osteoblastogenesis imbalance. Uncontrolled inducement of adipogenesis was shown to create a bone mass imbalance which resulted in elevated fragility and susceptibility. Differentiation of both pre-adipocyte and mesenchymal cells is partly regulated by PPARγ signaling [
26]. Obesity conditions were recently linked to increased osteoporosis progression in proportional to elevated adipokines [
27]. Adipokines and triglycerides trigger in a similar way an increase in adipogenesis, which is required to be in balance with osteoblastogenesis for healthy bones. Regulation of this imbalance is crucial to prevent and treat osteoporosis, especially when developed under obesity.
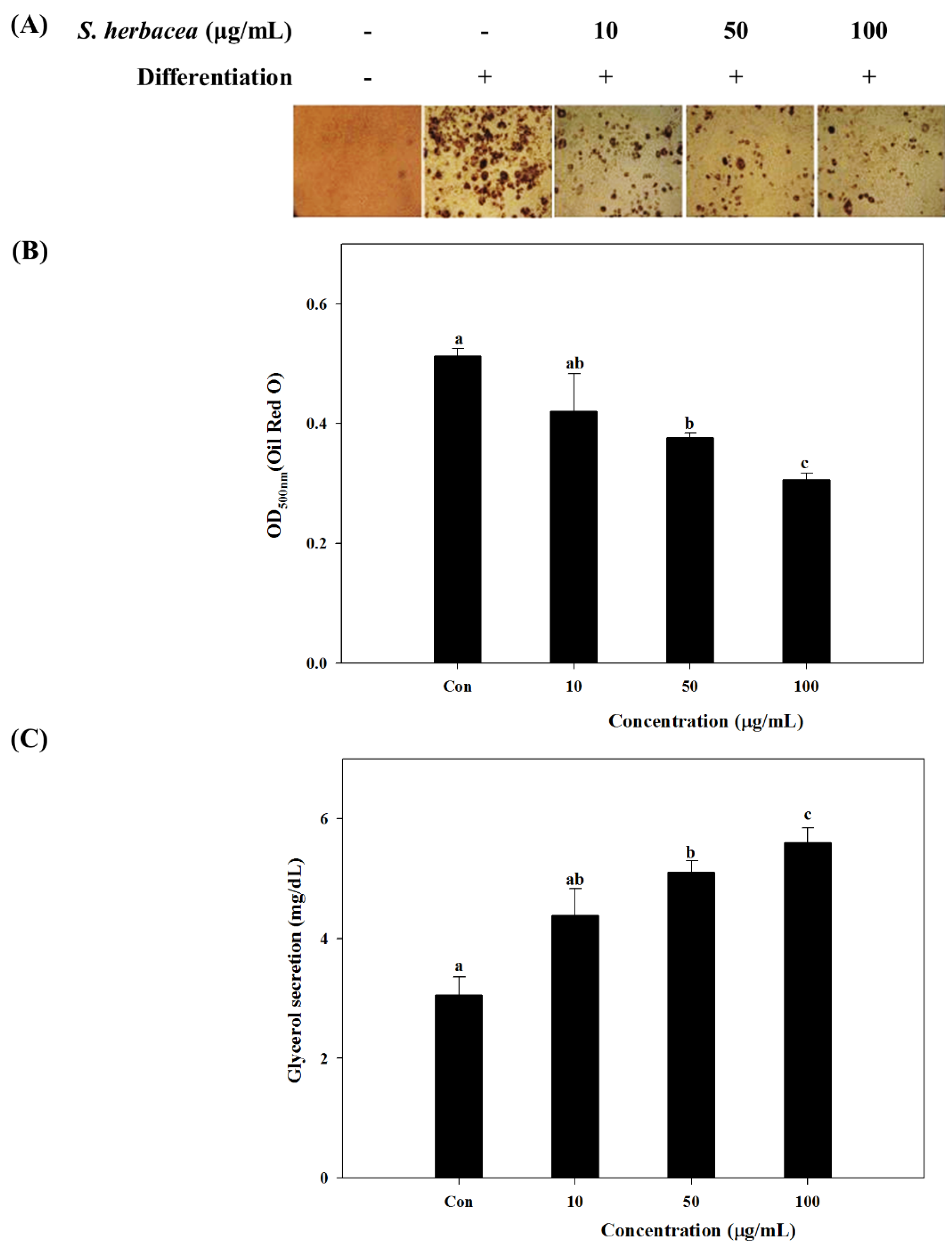
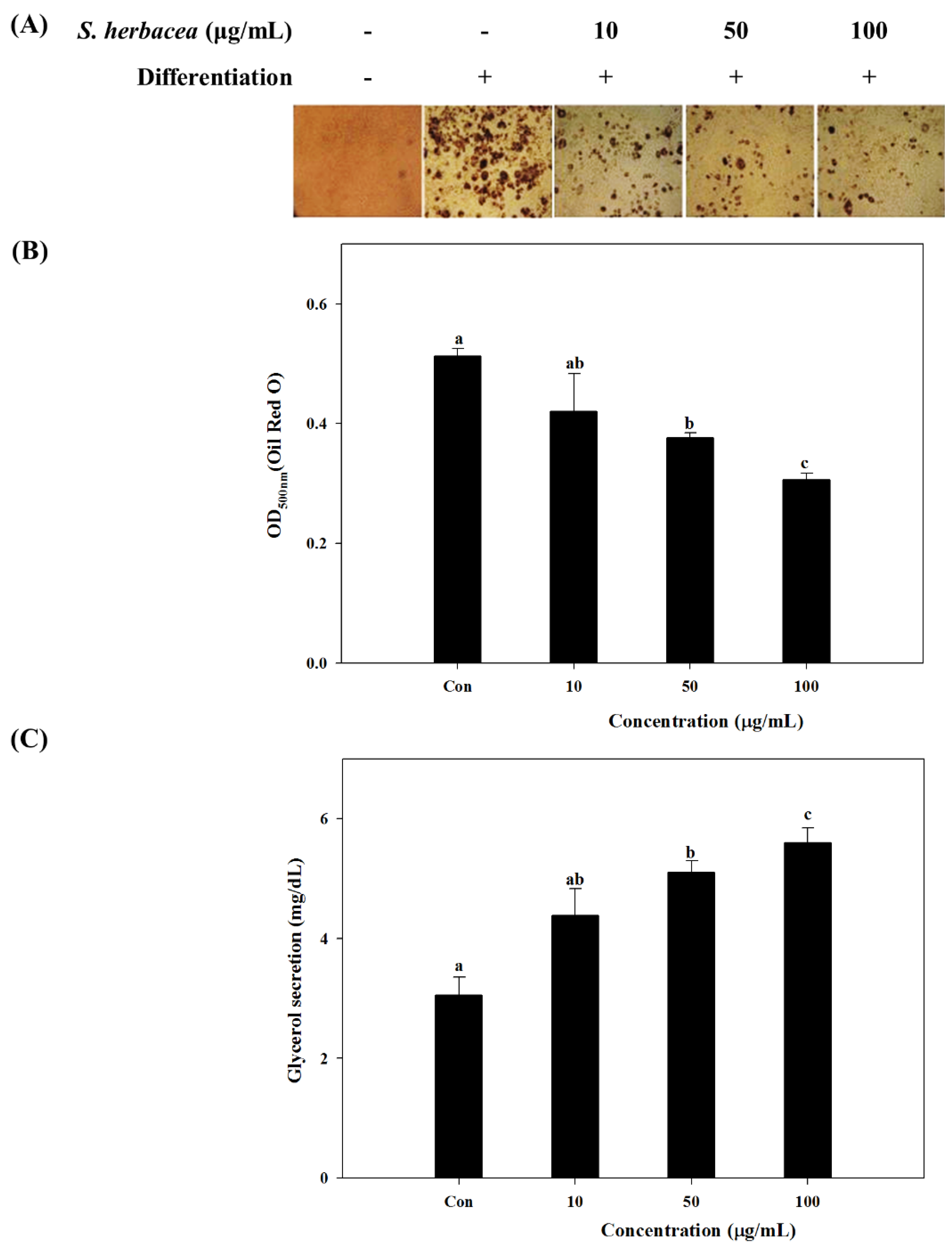
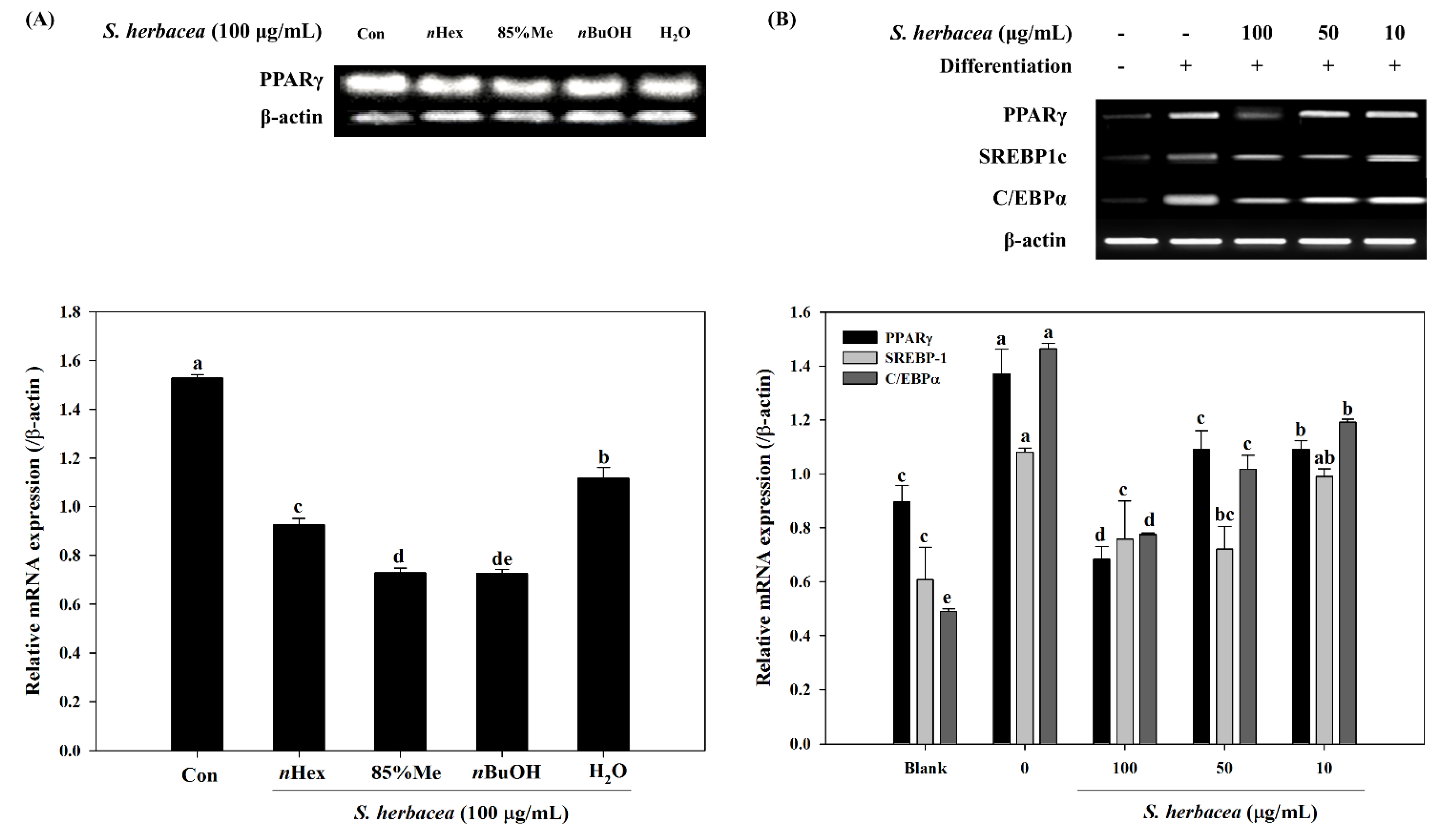
Our results showed a strong inhibition on lipid accumulation of differentiated 3T3-L1 adipocytes by
S. herbacea treatment. Glycerol assay results showed that cells treated with
S. herbacea released more glycerol to the culture medium. Furthermore, a possible mechanism behind the anti-adipogenic effect of
S. herbacea was evaluated by assessing expression levels of adipogenesis regulator factors, PPARγ, SREBP1c and C/EBPα. In addition to lowering lipid accumulation,
S. herbacea also lowered the expression of the aforementioned adipogenesis factors. Results indicated that
S. herbacea inhibited not only lipid accumulation, but also adipogenesis. In order to be used for further bioactivity-directed isolation, comparison of
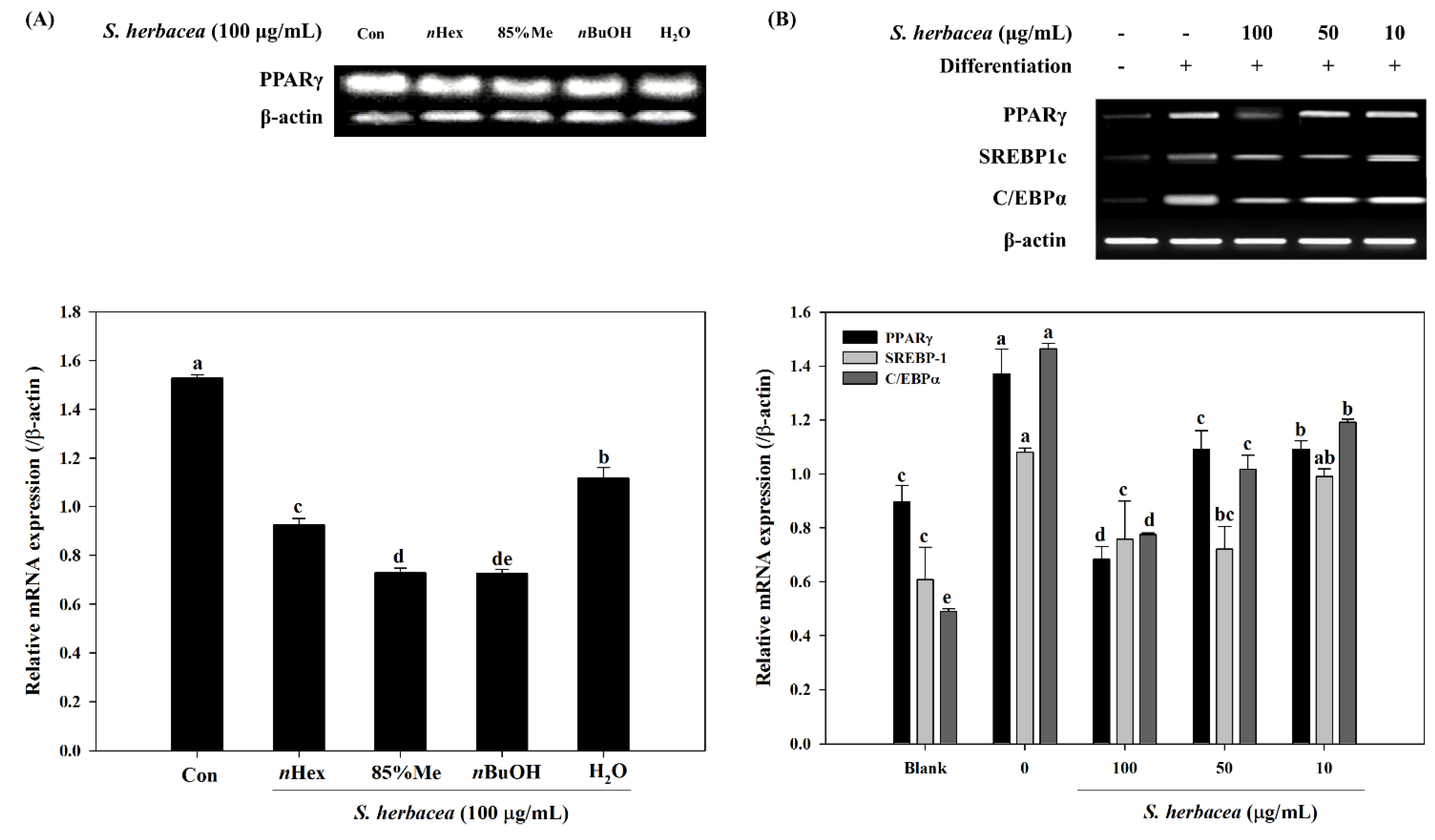
S. herbacea solvent fractions was carried out. Among all fractions,
nBuOH was found to be the most active to inhibit PPARγ mRNA expression. In addition, Kong
et al. [
14] also reported that two glycosides isolated from
S. herbacea were shown to possess chemoprotective effect against cancer through matrix metalloproteinase inhibition.
Increase in bone adipocytes is accompanied by severe fragility that defines osteoporosis. In order to relieve the deteriorated stem cell differentiation, inducing differentiating cells towards osteoblastogenesis is considered to be a crucial treatment step. Our results showed that S. herbacea inhibited adipogenesis by possible interaction with the PPAR-γ pathway and lipolysis. Results also indicated that S. herbacea might attenuate the imbalance of bone cell differentiation towards osteoblastogenesis.
In osteoporosis, mesenchymal cell perseverance towards adipogenesis is also accompanied by diminished osteoblastogenesis [
28,
29]. If an increase in adipogenesis is coupled with a decrease in osteoblastogenesis, bone loses its sturdiness and tends to be more fragile.
We examined the ability of
S. herbacea extract to enhance osteoblast differentiation in MC3T3-E1 pre-osteoblasts. Differentiating MC3T3-E1 pre-osteoblasts were introduced to
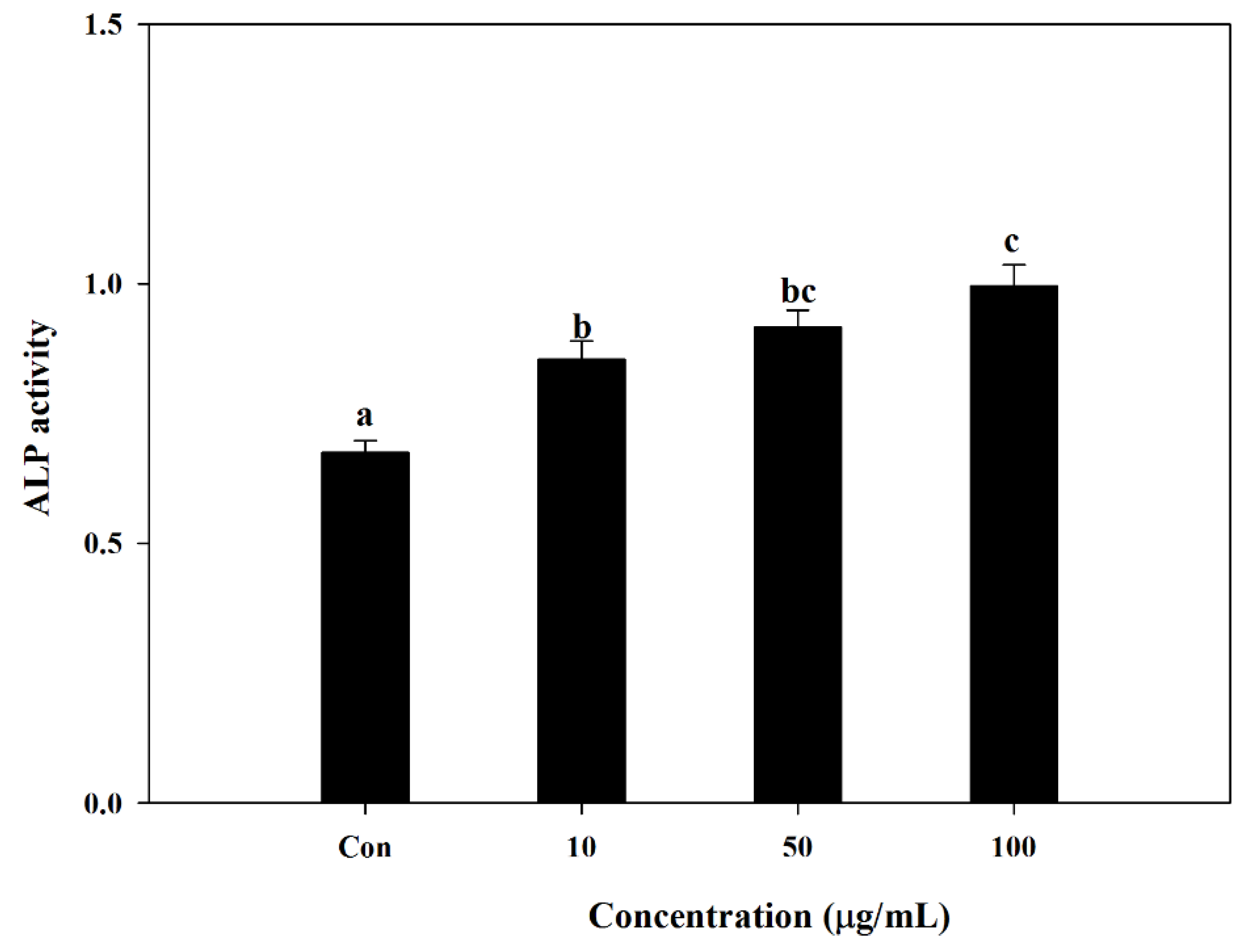
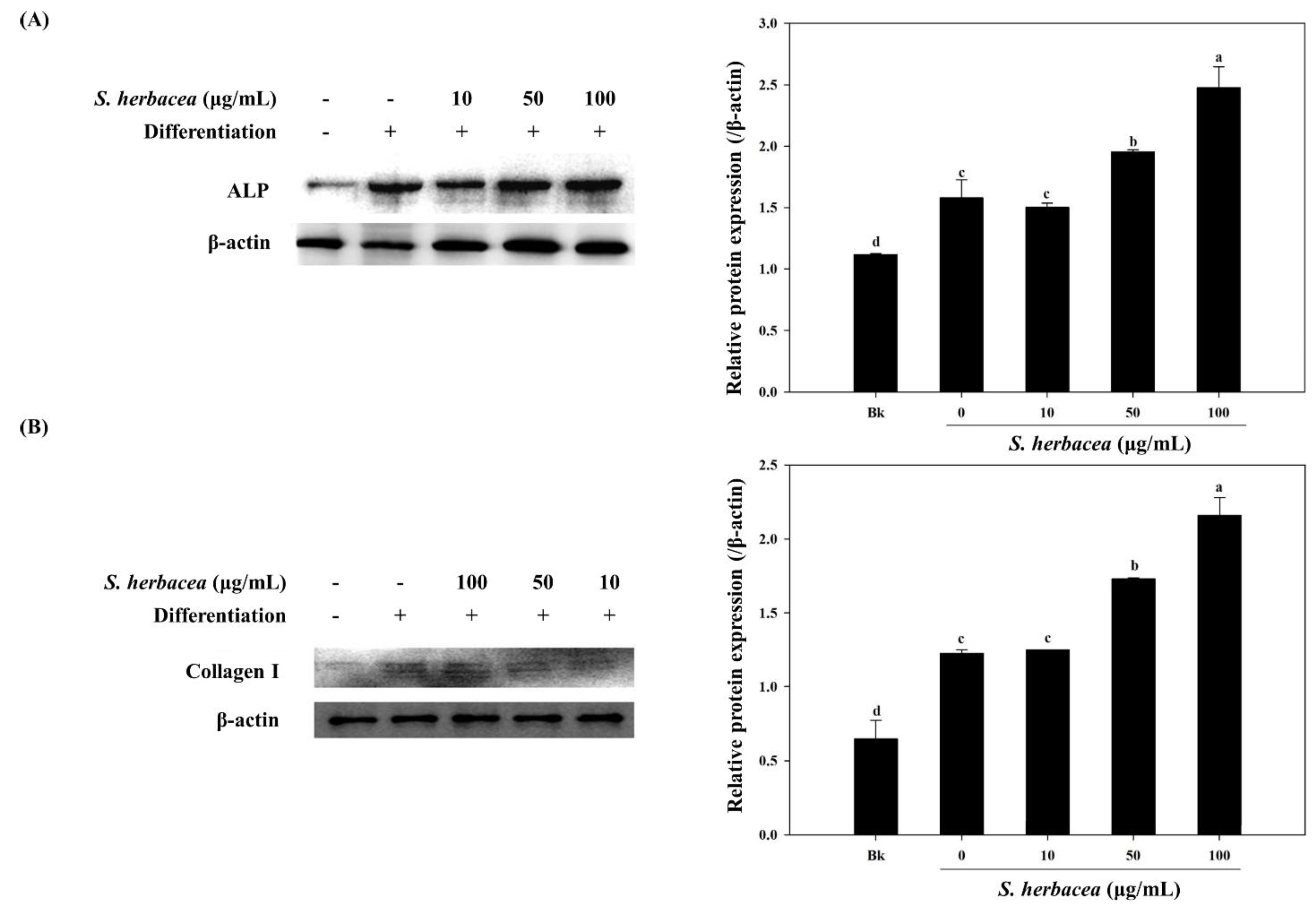
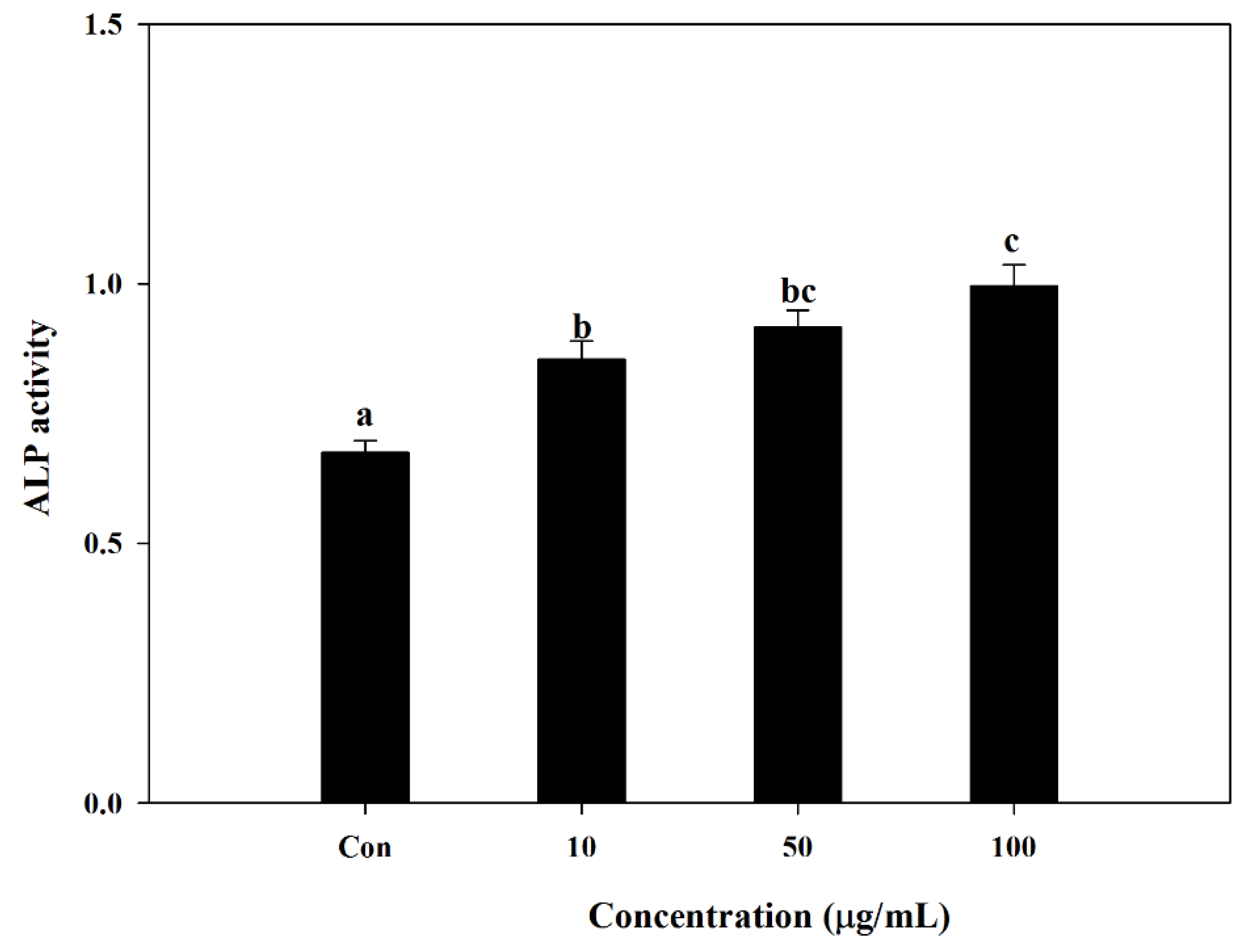
S. herbacea in different concentrations. Following a full differentiation, alkaline phosphatase (ALP) activity of cell lysates was evaluated. The alkaline phosphatase level is known to be an indicator of successful osteoblast differentiation, as it plays a crucial role in the mineralization of bone [
30]. In this regard, results indicated that the presence of
S. herbacea elevated the ALP activity suggesting a role in the mineralization of osteoblasts at all treated concentrations, indicating an enhancement towards bone formation.
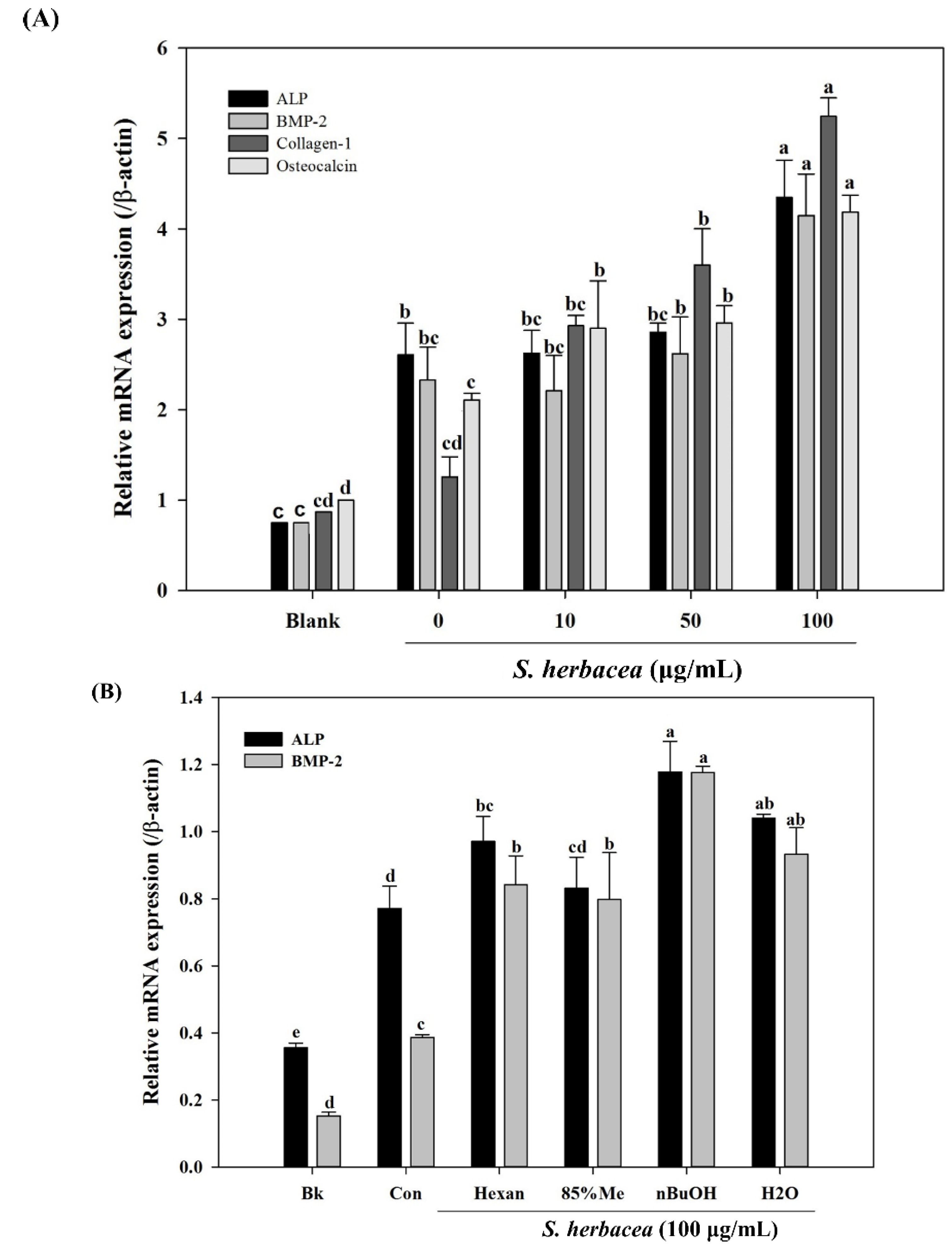
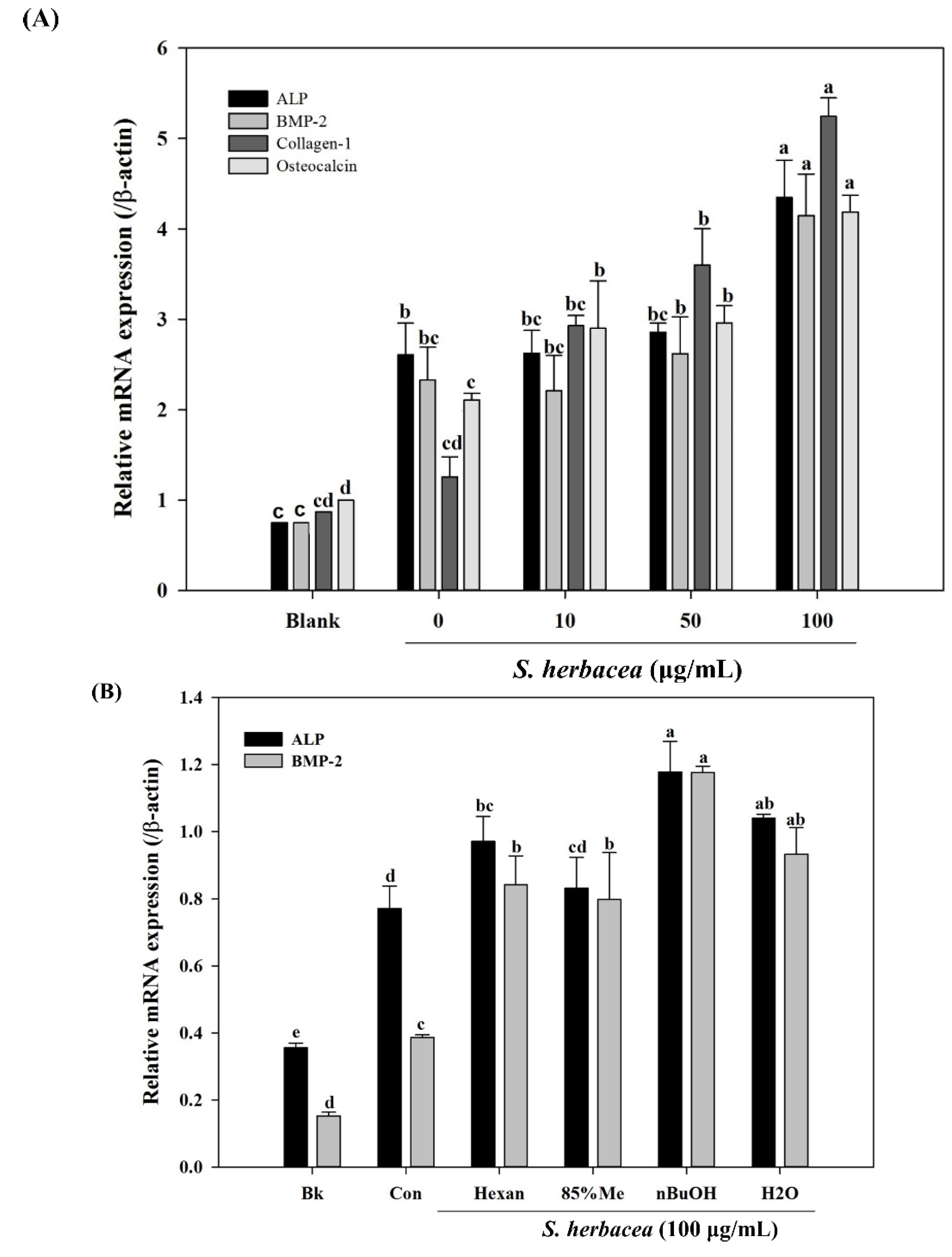
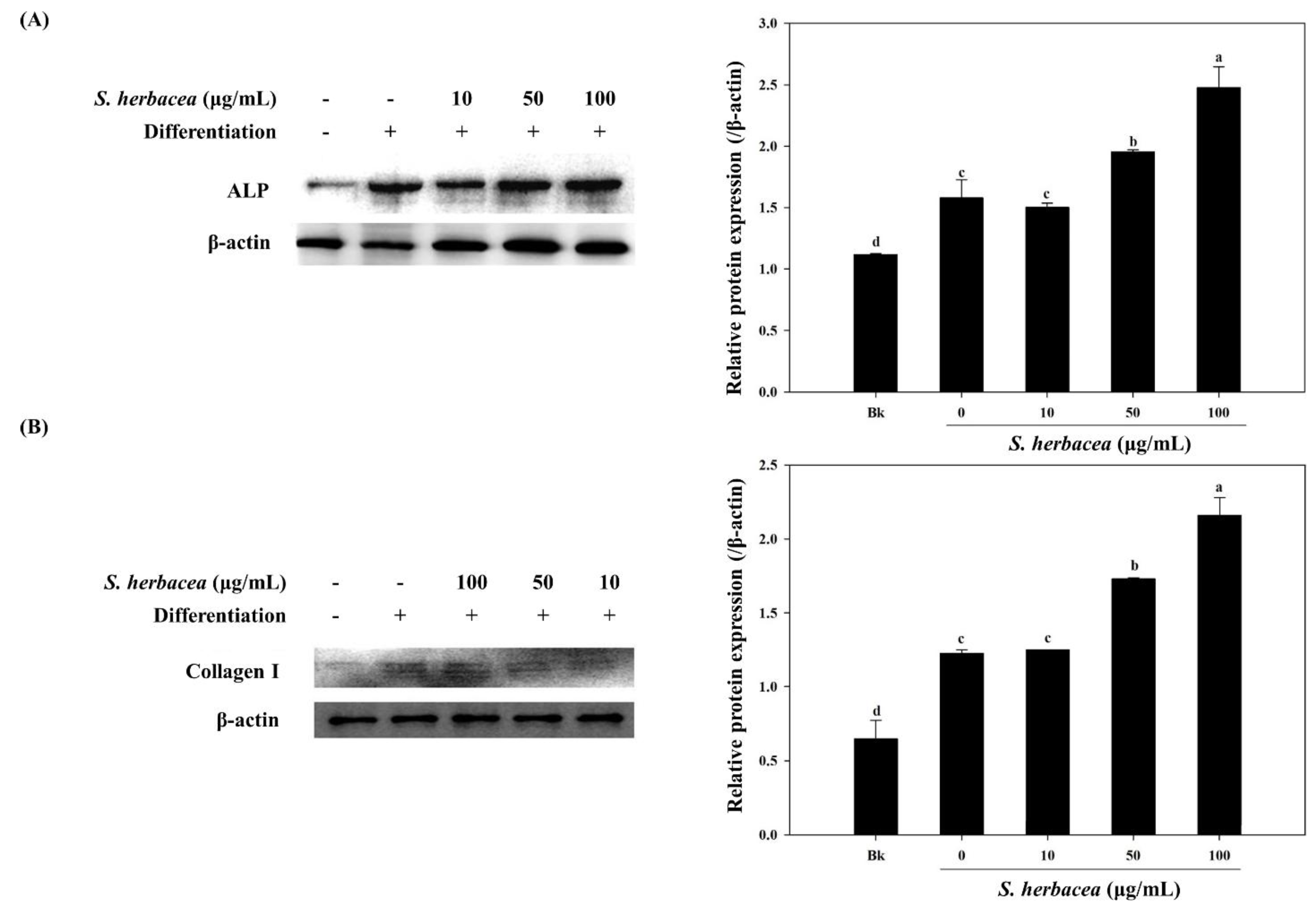
Bone formation is carried out by a distinctive and well-studied pathway of factors and proteins. In this cascade of signaling, bone morphogenetic protein (BMP) 2, 4 and 7 and osteocalcin are some of the key factors elevated at gene expression levels [
31]. During osteoblastogenesis, osteocalcin has also been reported to be a cell marker for the final differentiation state. On the other hand, BMPs are known to enhance expression of alkaline phosphatase (ALP), type I collagen (collagen-I) and other non-collagenous bone proteins as indicators for successful osteoblastogenesis [
30]. Therefore, mRNA expression of ALP, BMP-2, osteocalcin and collagen-I were assessed in the absence and presence of
S. herbacea. After full maturation into osteoblasts, RT-PCR experiments suggested an increasing trend towards expression of the aforementioned markers in cells treated with
S. herbacea, a result confirmed for ALP and collagen-I by western blot. In the light of these results, it was suggested that
S. herbacea enhanced osteoblast differentiation while inhibiting adipogenesis in pre-adipocytes. Ha
et al. [
32] also reported that
S. herbacea might be a potential source of antioxidant agents because of its effect on ovariectomy-induced oxidative stress, which is considered to cause age-related diseases including osteoporosis. Taken together, our results on
S. herbacea extracts provide evidence for an inhibitory effect on adipogenesis while enhancing osteoblast differentiation. Therefore,
S. herbacea is proposed as a promising source of bioactive agents for the effective prevention and treatment of osteoporosis.
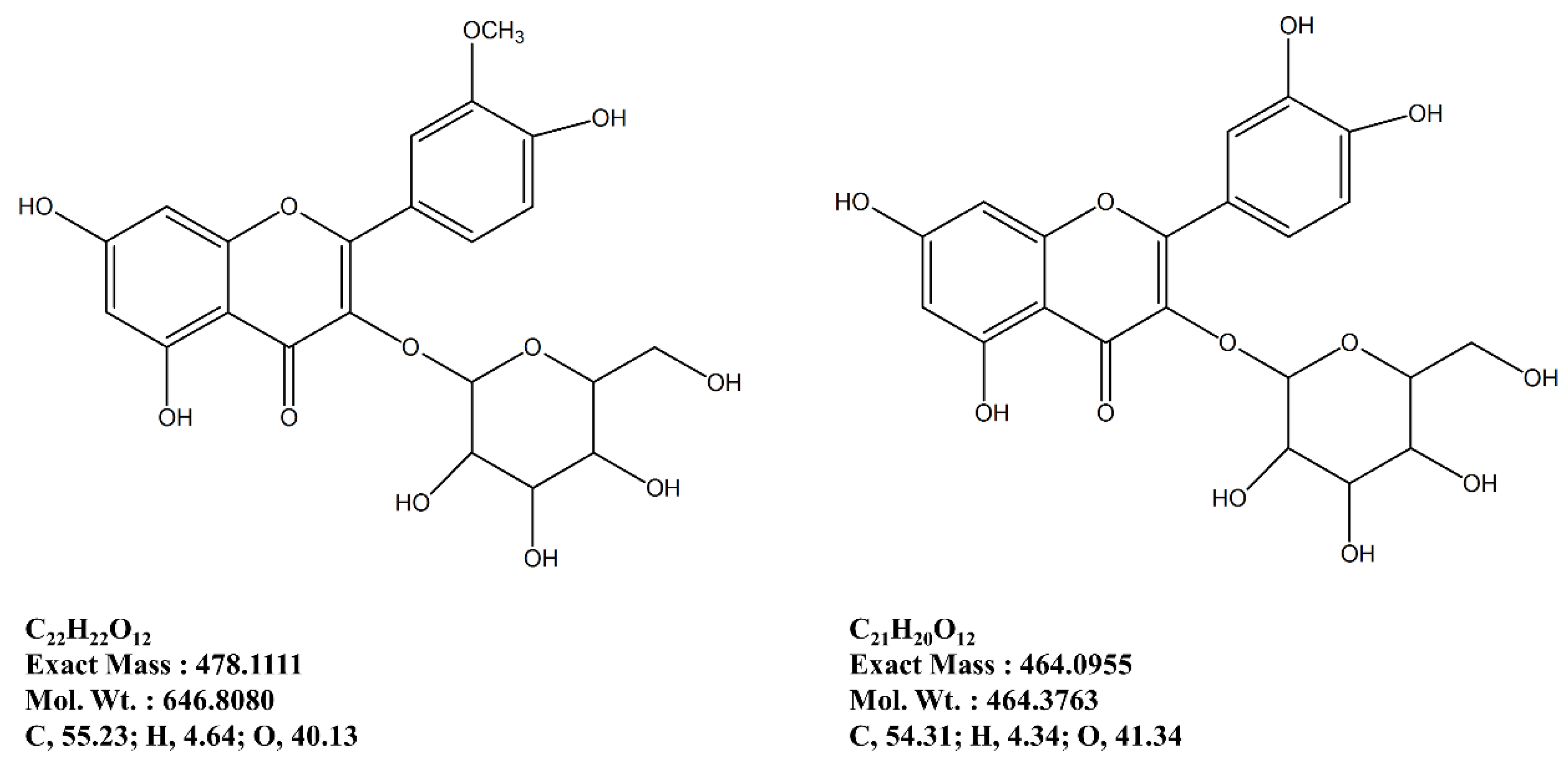
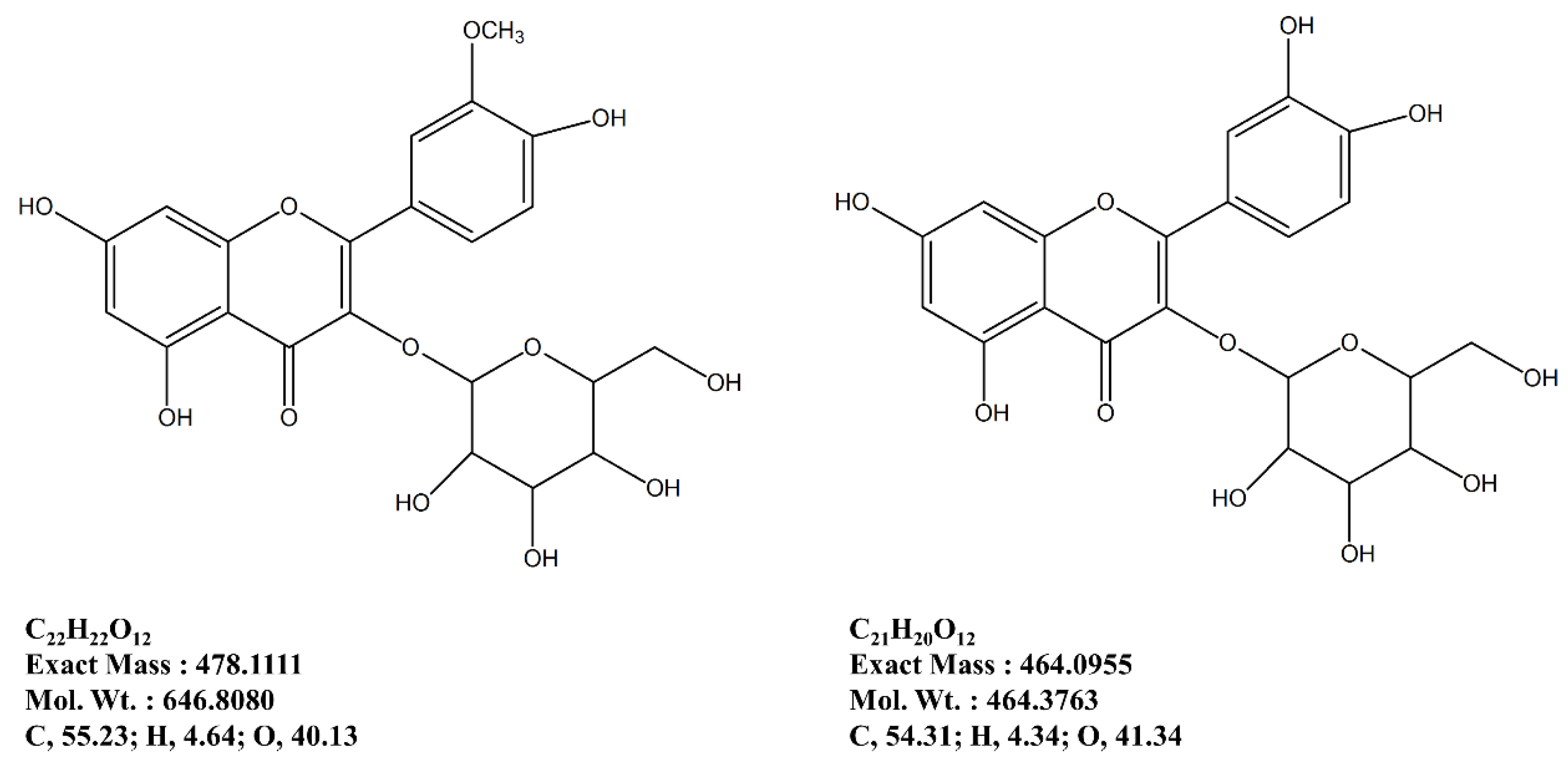
In this regard, following an earlier reported isolation method adapted for anti-osteoporosis bioactivity-directed flow, two formerly known flavonoid glycosides were isolated [
14]. These glycosides, namely isorhamnetin 3-
O-β-
d-glucoside and quercetin 3-
O-β-
d-glucoside are suggested to be possible bioactive reagents of
S. herbacea, responsible for relieving the effect of osteoporosis through regulation of adipogenesis/osteoblastogenesis imbalance by inhibiting PPARγ pathway and enhancing bone formation. Isolation of glycosides from the
nBuOH fraction was expected as this fraction was the most active for inhibiting adipogenesis and enhancing osteoblastogenesis. Several other flavonoid glycosides were isolated and evaluated for their anti-adipogenic activities. Studies also showed the possible absorption and bioactivity mechanisms of dietary flavonoids through the small intestine. Our previous results [
14,
15] were also in accordance with current assays and suggested that isorhamnetin 3-
O-β-
d-glucoside and quercetin 3-
O-β-
d-glucoside are strong bioactive substituents of
S. herbacea. On the other hand, isorhamnetin 3-
O-β-
d-glucoside and quercetin 3-
O-β-
d-glucoside were also shown to act on inhibition of differentiation of 3T3-L1 cells through AMPK/MAPK pathways. Notoya
et al. [
33] suggested an inhibitory effect of the flavonoid, quercetin, on proliferation, differentiation and mineralization of osteoblasts. However, Kim
et al. [
34] also stated that quercetin was able to inhibit proliferation while elevating the osteogenic differentiation of adipose stromal cells. In such a case, quercetin derivatives might also show the same distinct bioactivity on adipogenesis and osteoblastogenesis as our preliminary results indicated. The isolated derivative of quercetin, quercetin 3-
O-β-
d-glucoside has the quercetin backbone with a glucoside side-chain containing -OH branches similar to phloroglucinol derivatives. Considering that phloroglucinol derivatives have been reported to have anti-adipogenesis and pro-osteoblastogenesis activities, it could be suggested that these flavonoids might be responsible for the reported bioactivity of S. herbacea. Nonetheless, the
nBuOH fraction of
S. herbacea showed strong effects on adipogenesis and osteoblastogenesis and two flavonoid glycosides were isolated from the fraction. However, further studies to reveal the real action mechanisms of isolated compounds and their efficiency on adipogenic and osteogenic differentiation are needed. According to the results, the
nBuOH fraction of
S. herbacea might contain other bioactive constituents that are responsible for anti-adipogenic and pro-osteoblastogenic effects. In the future, coupled with
in vivo assays, results of this study will help to evaluate the true potential of
S. herbacea, as a potential source of compounds against obesity-related osteoporosis.
4. Experimental Section
4.1. Plant Materials
The whole plant of S. herbacea was briefly dried under shade and kept at −25 °C until use. The air-dried sample of S. herbacea was chopped into small pieces and extracted for 24 h with CH2Cl2 (3 L × 2) at room temperature. After removal of the solvent, the residue was re-extracted for 24 h with MeOH (3 L × 2) at room temperature.
The combined crude extracts (50 g) were suspended between CH2Cl2 and water. The organic layer was further partitioned between 85% aqueous MeOH and n-hexane and then the aqueous layer was fractioned with n-BuOH and H2O, respectively, to afford the n-hexane (3.2 g), 85% aq. MeOH (12.1 g), n-BuOH (16.3 g) and water (15.5 g) fractions.
4.2. Cell Culture and Adipocyte/Osteoblast Differentiations
Murine 3T3-L1 pre-adipocytes were seeded in 6-well plates at a density of 2 × 105 cells/well prior to experiments and grown to confluence in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere of 5% CO2. At 1 day postconfluence (designated “day 0”), a mixture of 3-isobutyl-1-methylxanthine (0.5 mM), dexamethasone (0.25 M) and insulin (5 µg/mL) in DMEM containing 10% FBS was introduced into cells in order to induce cell differentiation. After 48 h (day 2), DMEM containing 10% FBS supplemented with insulin (5 µg/mL) was introduced to cells following removal of induction medium. While replacing this medium with a fresh one every two days, S. herbacea extract was administered to the culture medium from day 0 to day 6 for Oil Red O and RT-PCR experiments and day 6 to day 8 for the glycerol secretion assay.
Murine osteoblast-like MC3T3-E1 cells were seeded in 6-well plates at a density of 1 × 10
5 cells/well and grown to confluence in α-Modified minimal essential medium (αMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1 mM sodium pyruvate, 100 units/L penicillin and 100 mg/L streptomycin at 37 °C in a humidified atmosphere of 5% CO
2. The cells were induced into osteoblastogenesis by adding of ascorbic acid and β-glycerophosphate into medium for five days under the conditions of the earlier report [
35]. Following confluence, the cell differentiation was initiated with culture medium containing 50 μg/mL ascorbic acid and 10 mM β-glycerophosphate for three days. Then, the induction medium was removed and the cell monolayer was washed twice with phosphate buffered saline (PBS).
S. herbacea extract was administered to the culture medium prior to further incubation of 48 h.
4.3. Cytotoxicity Determination Using MTT Assay
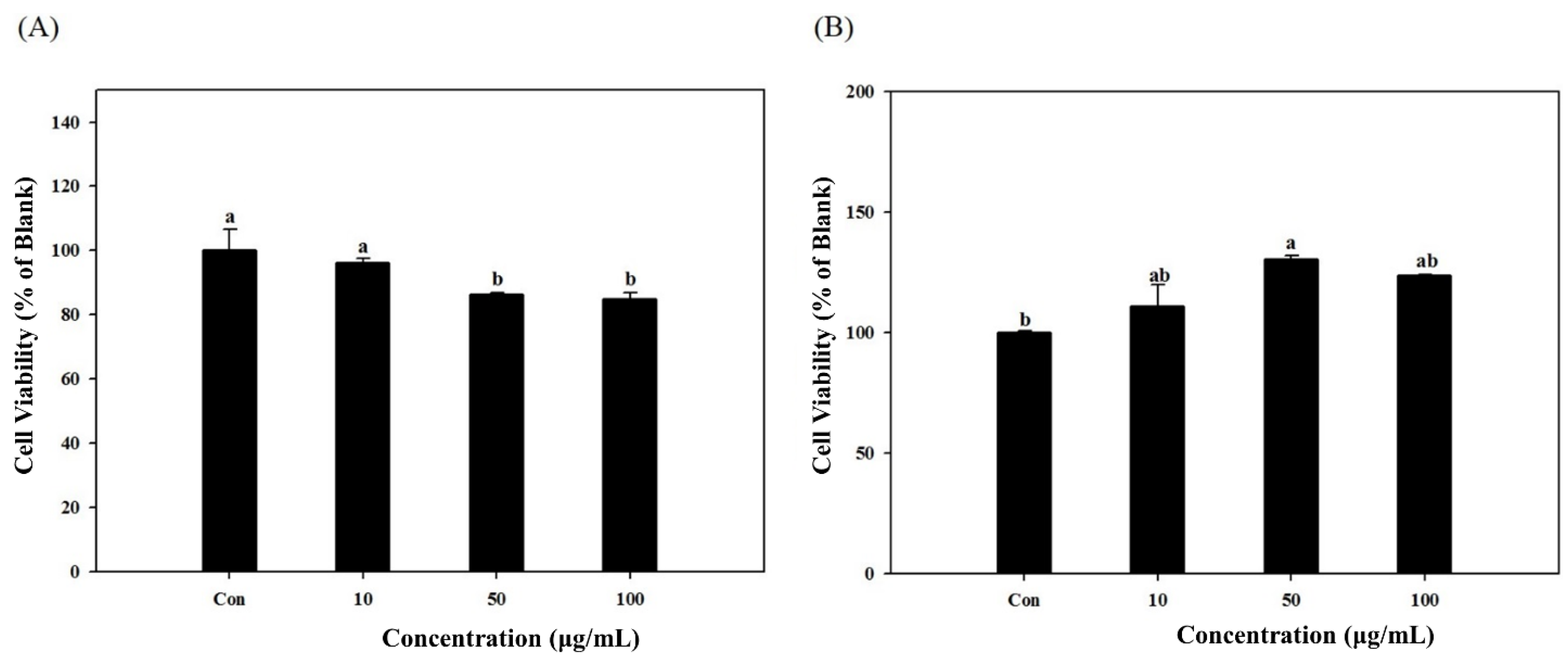
Cytotoxic levels of the S. herbacea on cultured cells were measured using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, which is based on the conversion of MTT to MTT-formazan by mitochondrial enzyme. The cells were grown in 96-well plates at a density of 5 × 103 cells/well. After 24 h, the cells were washed with fresh medium and were treated with control medium or the medium supplemented with S. herbacea. After incubation for seven days while changing the medium and re-treating the samples every two days, cells were rewashed and 100 μL of MTT solution (1 mg/mL) was added and incubated for 4 h. Finally, 100 μL of DMSO was added to solubilize the formed formazan crystals and the amount of formazan crystal was determined by measuring the absorbance at 540 nm using a GENios® microplate reader (Tecan Austria GmbH, Grödig, Austria). Relative cell viability was determined by the amount of MTT converted into formazan crystal. Viability of cells was quantified as a percentage compared to the control and dose response curves were developed.
4.4. Oil-Red O Staining and Glycerol Release Assay
Fully differentiated cells were fixed with 10% fresh formaldehyde in PBS for 1 h at room temperature and stained with filtered Oil-Red O solution (60% isopropanol and 40% water) for at least 1 h. After incubation, the wells were emptied of Oil-Red O staining solution, washed with distilled water and air dried. Images of lipid droplets in 3T3-L1 adipocytes were collected by an Olympus microscope (Tokyo, Japan). Finally, dye retained in the cells was eluted with isopropanol and quantified by measuring the optical absorbance at 500 nm using a microplate reader (Tecan Qustria GmbH, Grödig, Austria).
The glycerol levels were determined using the enzymatic reagent, free glycerol reagent (Sigma, St. Louis, MO, USA), directed by the protocol of GPO-TRINDER (Sigma, St. Louis, MO, USA).
4.5. Cellular ALP Activity
Cellular ALP activity of S. herbacea-treated and control cells was measured following incubation of 14 days. The cell monolayer was gently washed twice with PBS and lysed using 0.1% Triton X-100 and 25 mM carbonate buffer. The lysates were centrifuged at 4 °C 12,000× g for 15 min. Enzyme assay buffer (15 mM ρ-nitrophenyl phosphate, 1.5 mM MgCl2 and 200 mM carbonate buffer) was used to measure the ALP activity of the supernatants. The absorbance of reactive solution was measured at 405 nm.
4.6. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction Analysis
Total RNA was isolated from 3T3-L1 and D1 adipocytes and MC3T3-E1 osteoblasts in the presence/absence of S. herbacea using Trizol reagent (Invitrogen Co., Carlsbad, CA, USA). For synthesis of cDNA, RNA (2 μg) was added to RNase-free water and oligo (dT), denaturated at 70 °C for 5 min and cooled immediately. RNA was reverse transcribed in a master mix containing 1× RT buffer, 1 mM dNTPs, 500 ng oligo (dT), 140 U M-MLV reserve transcriptase and 40 U RNase inhibitor at 42 °C for 60 min and at 72 °C for 5 min using an automatic T100 Thermo Cycler (Bio-Rad, Hertfordshire, UK). The target cDNA was amplified using the following sense and antisense primers: forward 5′-TTT-TCA-AGG-GTG-CCA-GTT-TC-3′ and reverse 5′-AAT-CCT-TGG-CCC-TCT-GAG-AT-3′ for PPARγ; forward 5′-TGT-TGG-CAT-CCT-GCT-ATC-TG-3′ and reverse 5′-AGG-GAA-AGC-TTT-GGG-GTC-TA-3′ for SREBP1c; forward 5′-TTA-CAA-CAG-GCC-AGG-TTT-CC-3′ and reverse 5′-GGC-TGG-CGA-CAT-ACA-GTA-CA-3′ for C/EBPα; forward 5′-CCA-CAG-CTG-AGA-GGG-AAA-TC-3′ and reverse 5′-AAG-GAA-GGC-TGG-AAA-AGA-GC-3′ for β-actin. The amplification cycles were carried out at 95 °C for 45 s, 60 °C for 1 min and 72 °C for 45 s. Final PCR products were separated by electrophoresis on 1.5% agarose gel for 30 min at 100 V after 30 cycles. Gels were then stained with 1 mg/mL ethidium bromide visualized by UV light using Davinch-Chemi imager™ (CAS-400SM, Wako Co., Osaka, Japan).
4.7. Real-Time RT-PCR Analysis of mRNA Expression
Gene expression was measured by real time RT-PCR in a Thermal Cycler Dice® Real Time System TP800 (Takara Bio Inc., Ohtsu, Japan) following the manufacturer’s protocol. Briefly 1.0 μL of DNA sample and 12.5 μL of Maxima® SYBR Green qPCR Master Mix (Fermentas, Waltham, MA, USA) containing Taq DNA polymerase, dNTP and reaction buffer were mixed. The target cDNA was amplified using the following sense and antisense primers: forward 5′-CCA-GCA-GGT-TTC-TCT-CTT-GG-3′ and reverse 5′-CTG-GGA-GTC-TCA-TCC-TGA-GC-3′ for ALP; forward 5′-GGA-CCC-GCT-GTC-TTC-TAG-TG-3′ and reverse 5′-GCC-TGC-GGT-ACA-GAT-CTA-GC-3′ for BMP-2; forward 5′-GCT-GTG-TTG-GAA-ACG-GAG-TT-3′ and reverse 5′-CAT-GTG-GGT-TCT-GAC-TGG-TG-3′ for Osteocalcin; forward 5′-GAG-CGG-AGA-GTA-CTG-GAT-CG-3′ and reverse 5′-TAC-TCG-AAC-GGG-AAT-CCA-TC-3′ for Collagen I; forward 5′-CCA-CAG-CTG-AGA-GGG-AAA-TC-3′ and reverse 5′-AAG-GAA-GGC-TGG-AAA-AGA-GC-3′ for β-actin. The PCR amplification was carried out for an initial denaturation at 95 °C for 10 min, followed by 40 PCR cycles. Each cycle proceeded at 95 °C for 15 s, 60 °C for 60 s. Relative quantification was calculated using the 2-(ΔΔCT) method. β-Actin was used as an internal control.
4.8. Western Blot Analysis
Western blotting was performed according to standard procedures. Briefly, cells were lysed in RIPA lysis buffer (Sigma-Aldrich Corp., St. Louis, MO, USA) at 4 °C for 30 min. Cell lysates (35 μg) were separated by 12% SDS-polyacrylamide gel electrophoresis, transferred onto a polyvinylidene fluoride membrane (Amersham Pharmacia Biotech., Amersham, England, UK), blocked with 5% skimmed milk and hybridized with primary antibodies (diluted 1:1000) against ALP and collagen I. After incubation with horseradish-peroxidase-conjugated secondary antibody at room temperature, immunoreactive proteins were detected using a chemiluminescece ECL assay kit (Amersham Pharmacia Biosciences, England, UK) according to the manufacturer's instructions. Western blot bands were visualized using a Davinch-Chemi imager™ (CAS-400SM, Wako Co., Osaka, Japan).
4.9. Statistical Analysis
The data were presented as mean ± SD. Differences between the means of the individual groups were analyzed using the analysis of variance (ANOVA) procedure of Statistical Analysis System, SAS v9.1 (SAS Institute, Cary, NC, USA) with Duncan’s multiple range tests. The significance of differences was defined at the p < 0.05 level.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}