Extract from the Zooxanthellate Jellyfish Cotylorhiza tuberculata Modulates Gap Junction Intercellular Communication in Human Cell Cultures




Abstract
:1. Introduction
2. Results and Discussion
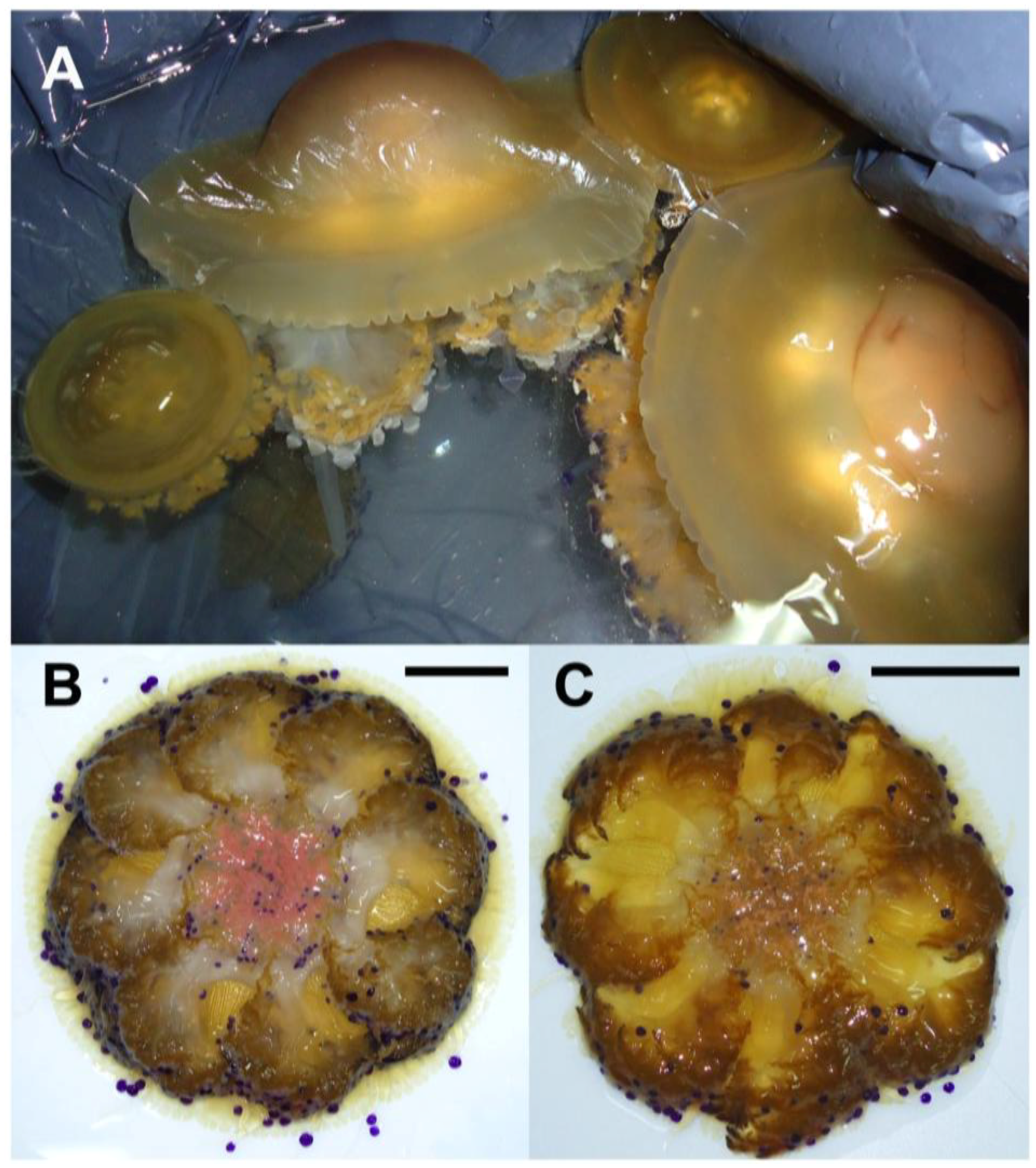
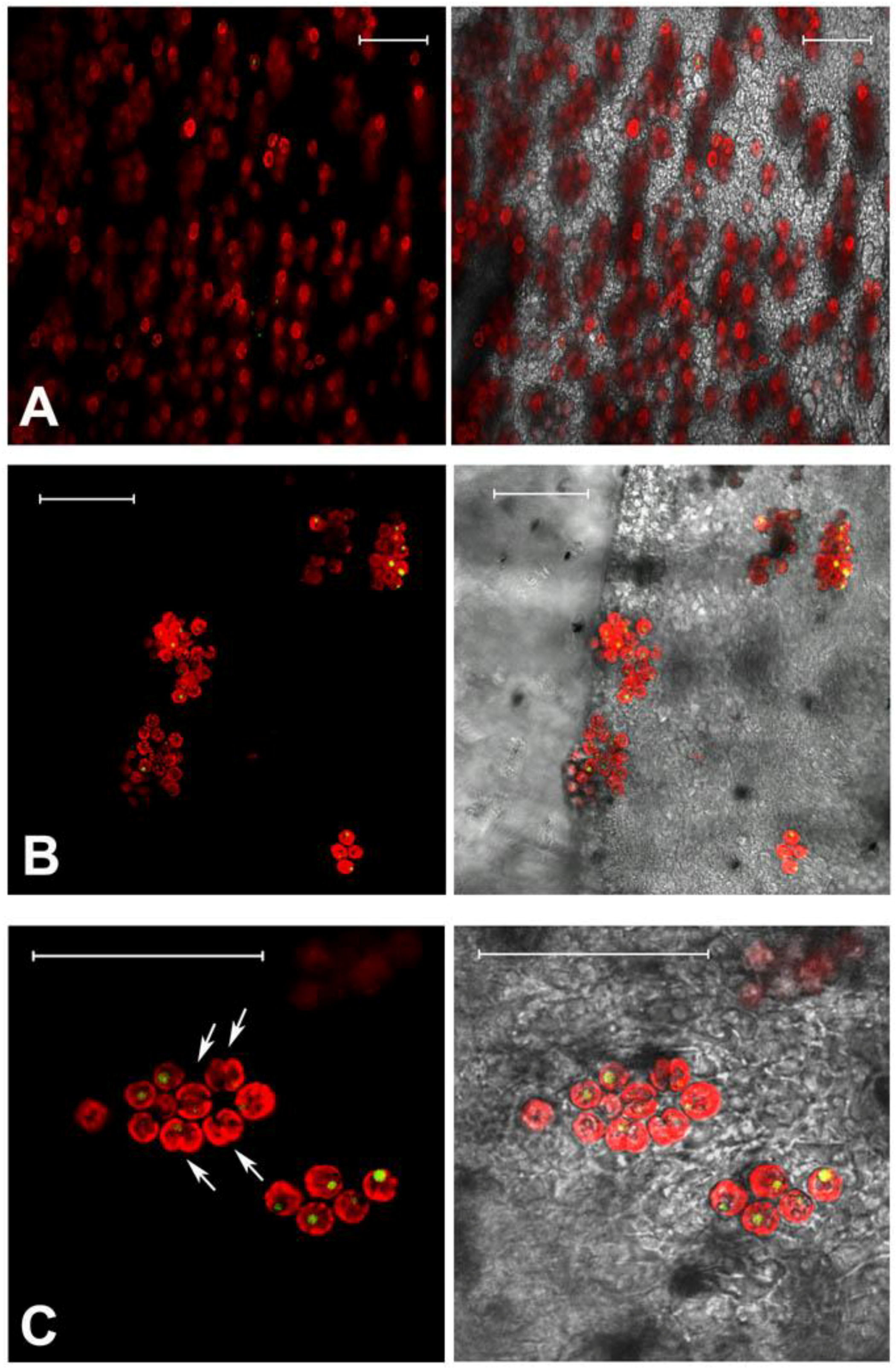
2.1. Jellyfish Sample Features
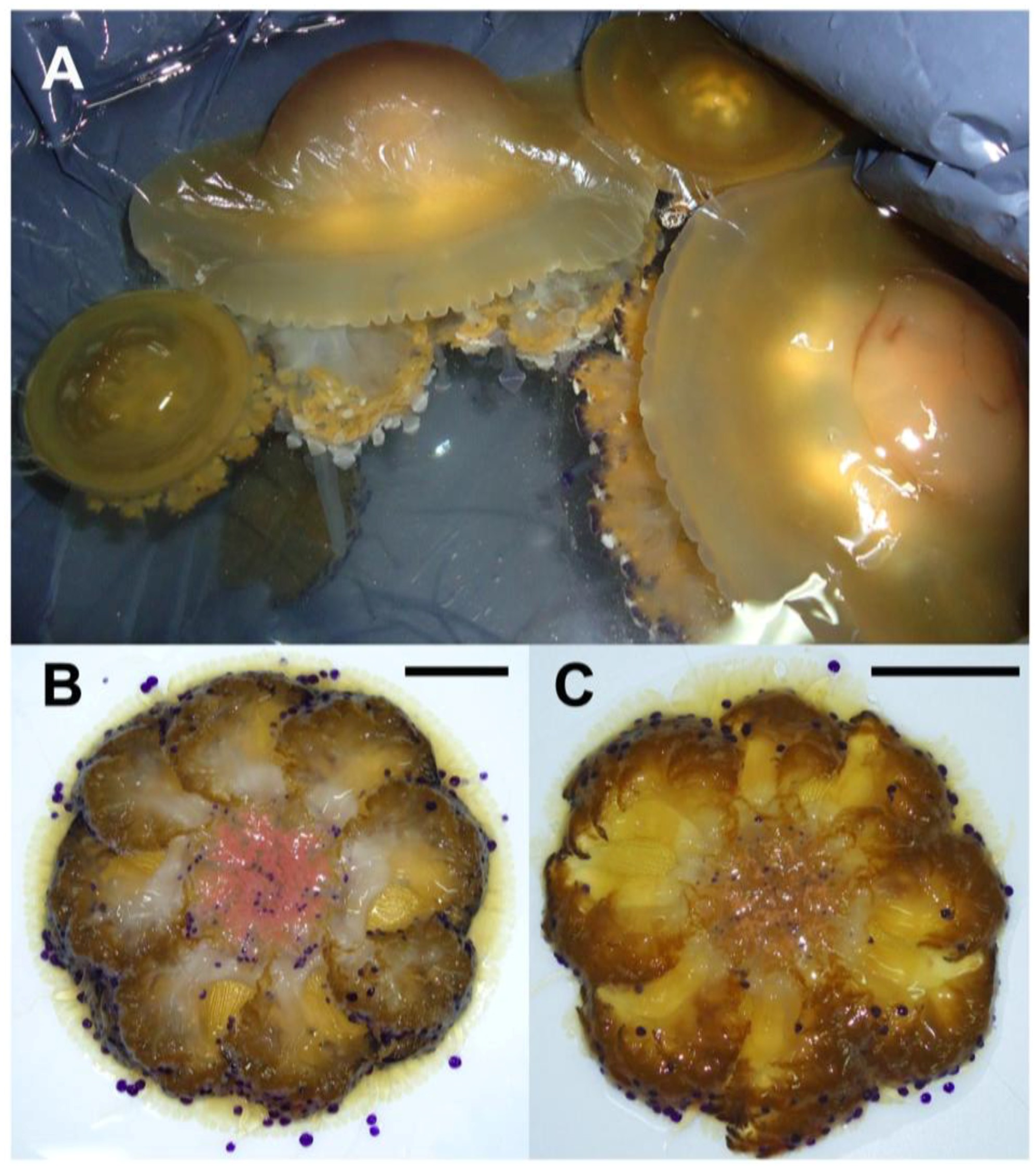
2.2. Hydro-Alcoholic Extraction and Fractioning

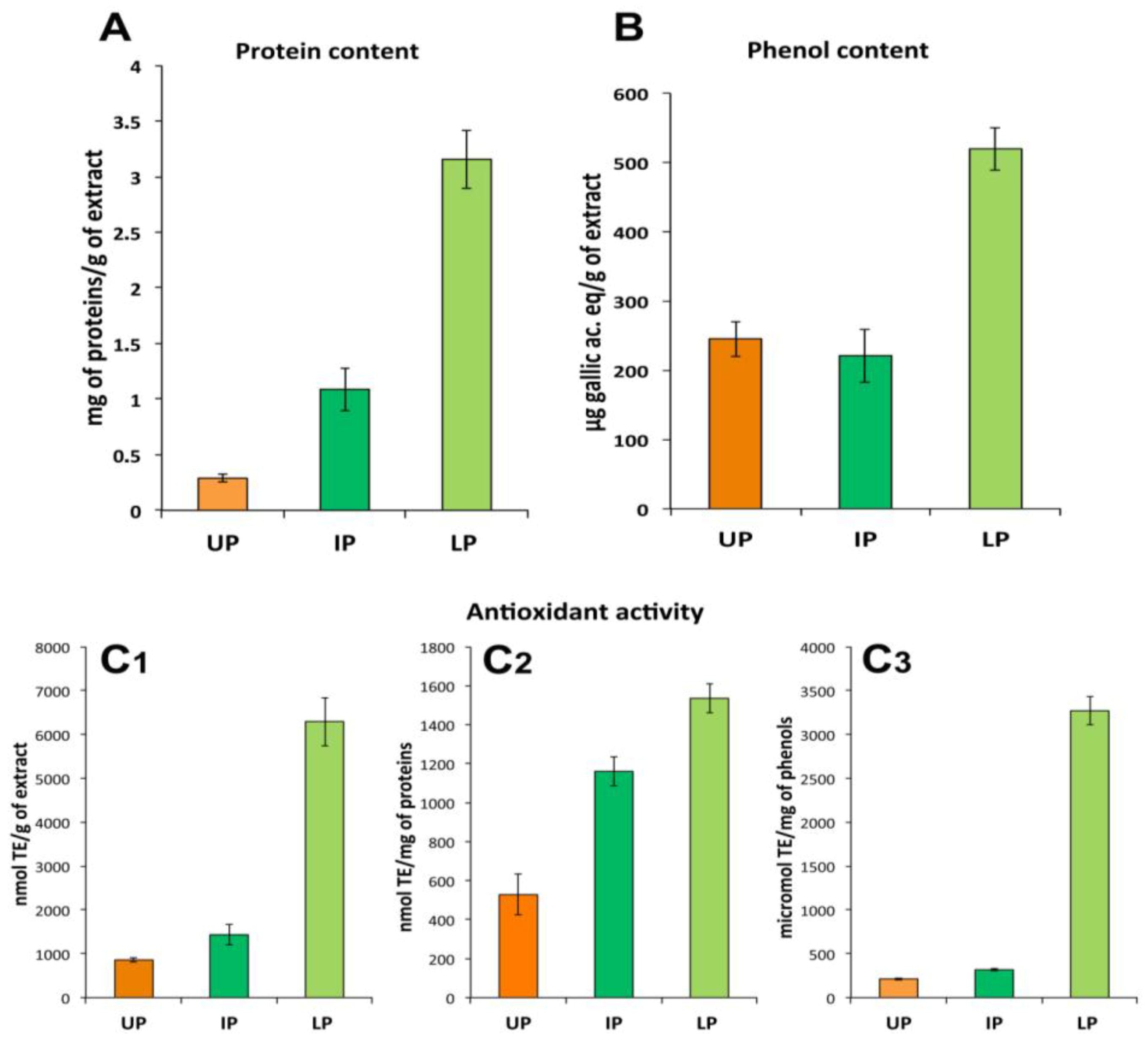
2.3. Fraction Characterization
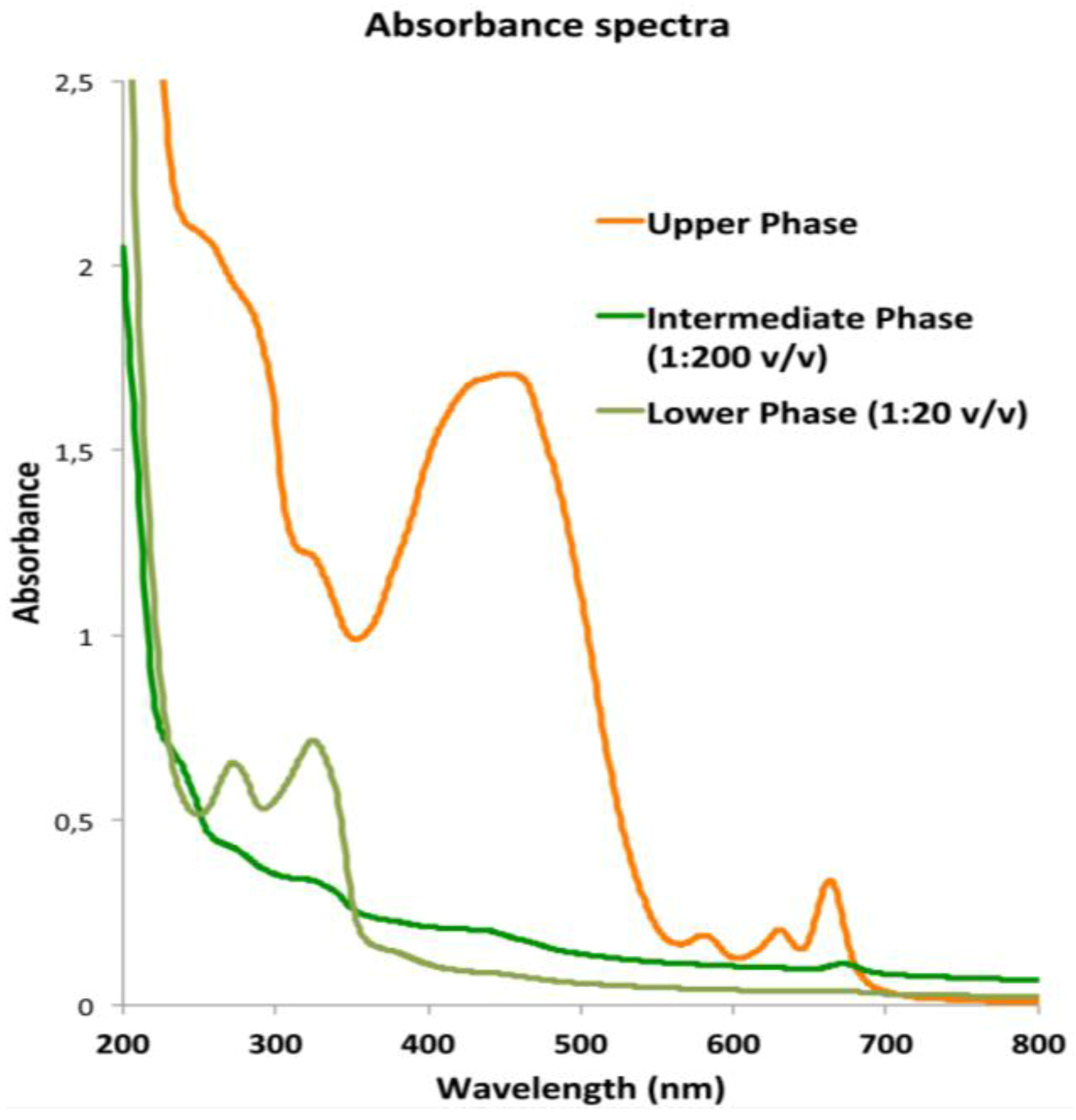
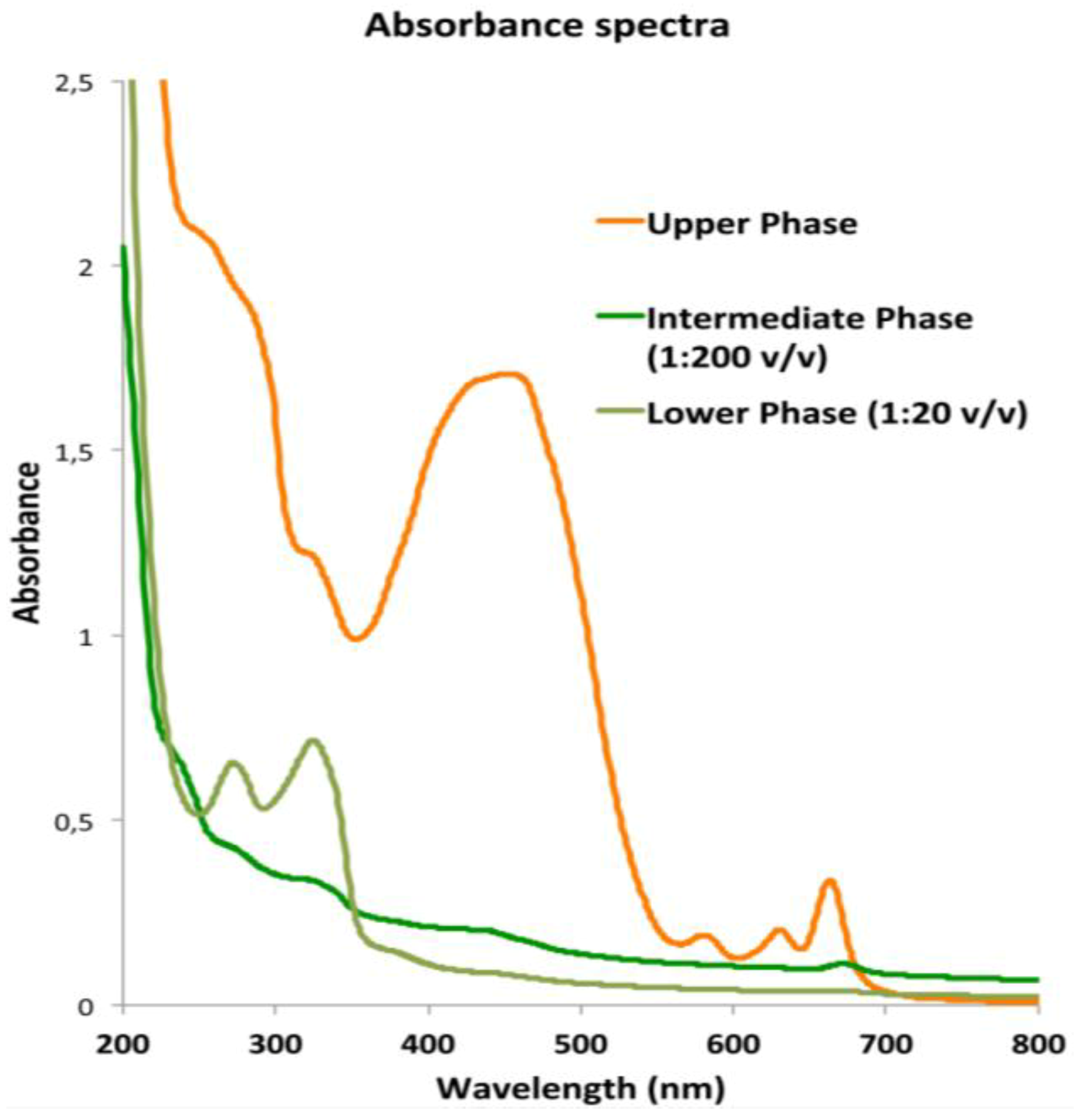
2.3.1. Lipophilic Fraction: Upper Phase
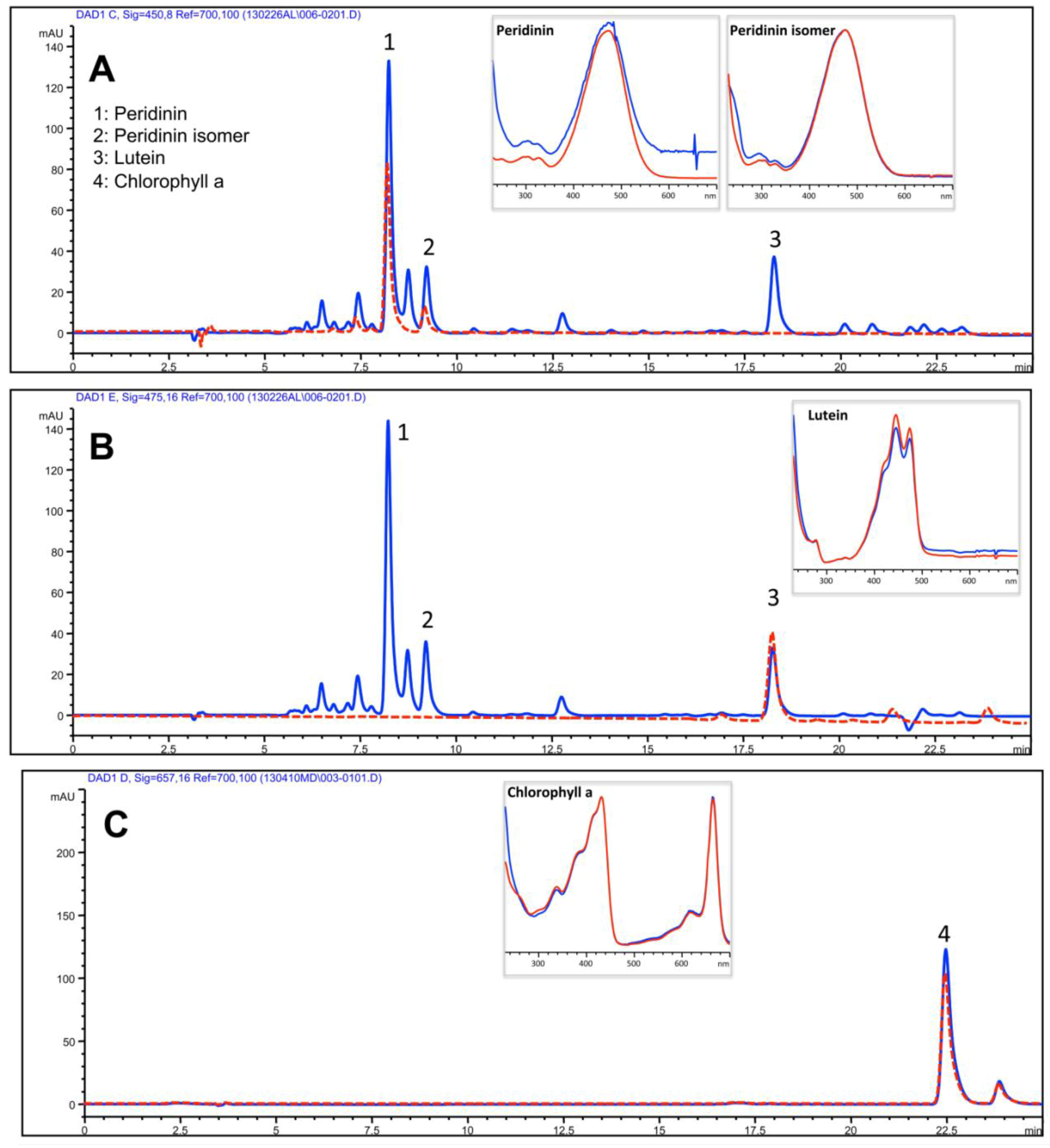

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
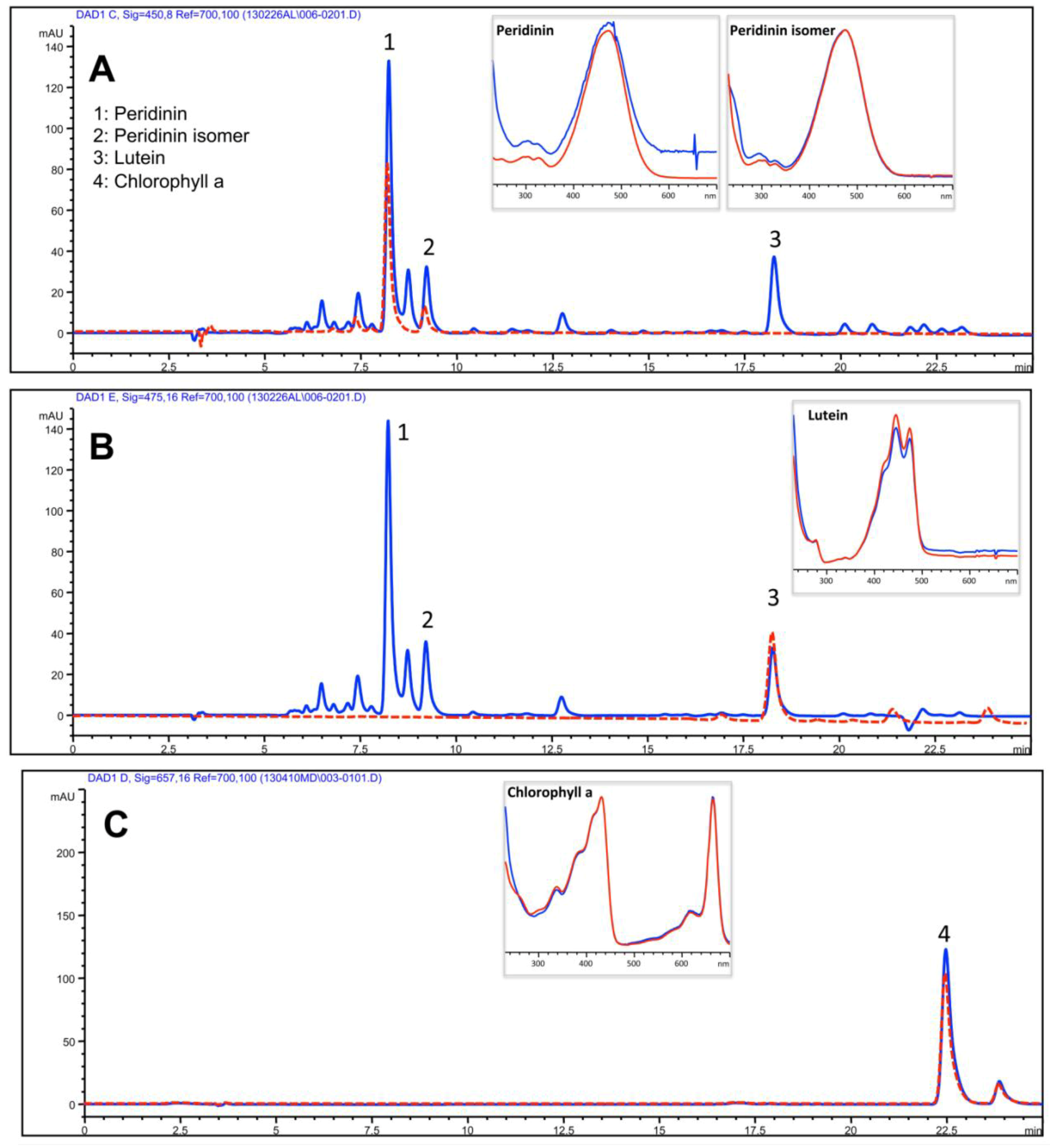
| Isoprenoids | Upper Phase of hydro-alcoholic extract |
|---|---|
| μg/g DW | |
| Peridinin | 385.1 ± 49.6 |
| Peridinin isomer | 186.5 ± 17.3 |
| Lutein | 108.7 ± 9.5 |
| Chlorophyll a | 20.3 ± 9.5 |
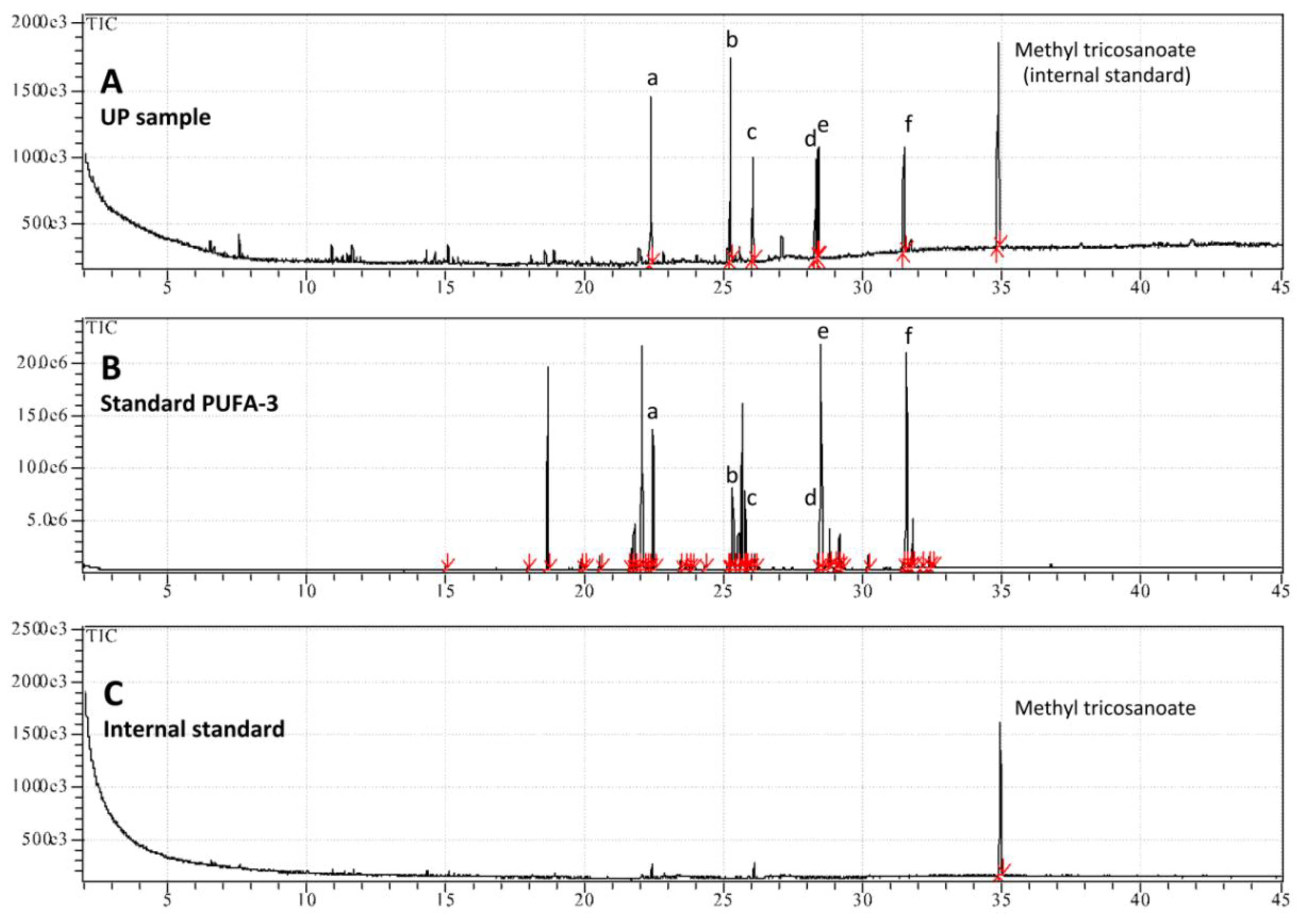
| Fatty acids | Upper Phase of hydro-alcoholic extract |
|---|---|
| % of total FA | |
| Saturated Fatty Acids (SFA) | |
| Palmitic acid (C16:0) | 18.9 ± 0.7 |
| Stearic acid (C18:0) | 9.5 ± 0.4 |
| Total SFA | 28.4 |
| Monounsaturated Fatty Acids (MUFA) | n.d. 1 |
| Polyunsaturated Fatty Acids (PUFA) | |
| cis-8,11,14,17-Eicosatetraenoic acid C20:4 (ω-3) | 28.2 ± 2.6 |
| cis-5,8,11,14-Eicosatetraenoic acid C20:4 (ω-6) | 12.5 ± 0.4 |
| cis-5,8,11,14,17-Eicosapentaenoic acid C20:5 (ω-3) | 14.9 ± 1.4 |
| cis-4,7,10,13,16,19-Docosahexaenoic acid methyl ester C22:6 (ω-3) | 15.9 ± 0.9 |
| Total PUFA | 71.6 |
| TOTAL (%) | 100.0 |
| Total (mg/g DW of the extract) | 2.450 ± 0.289 |
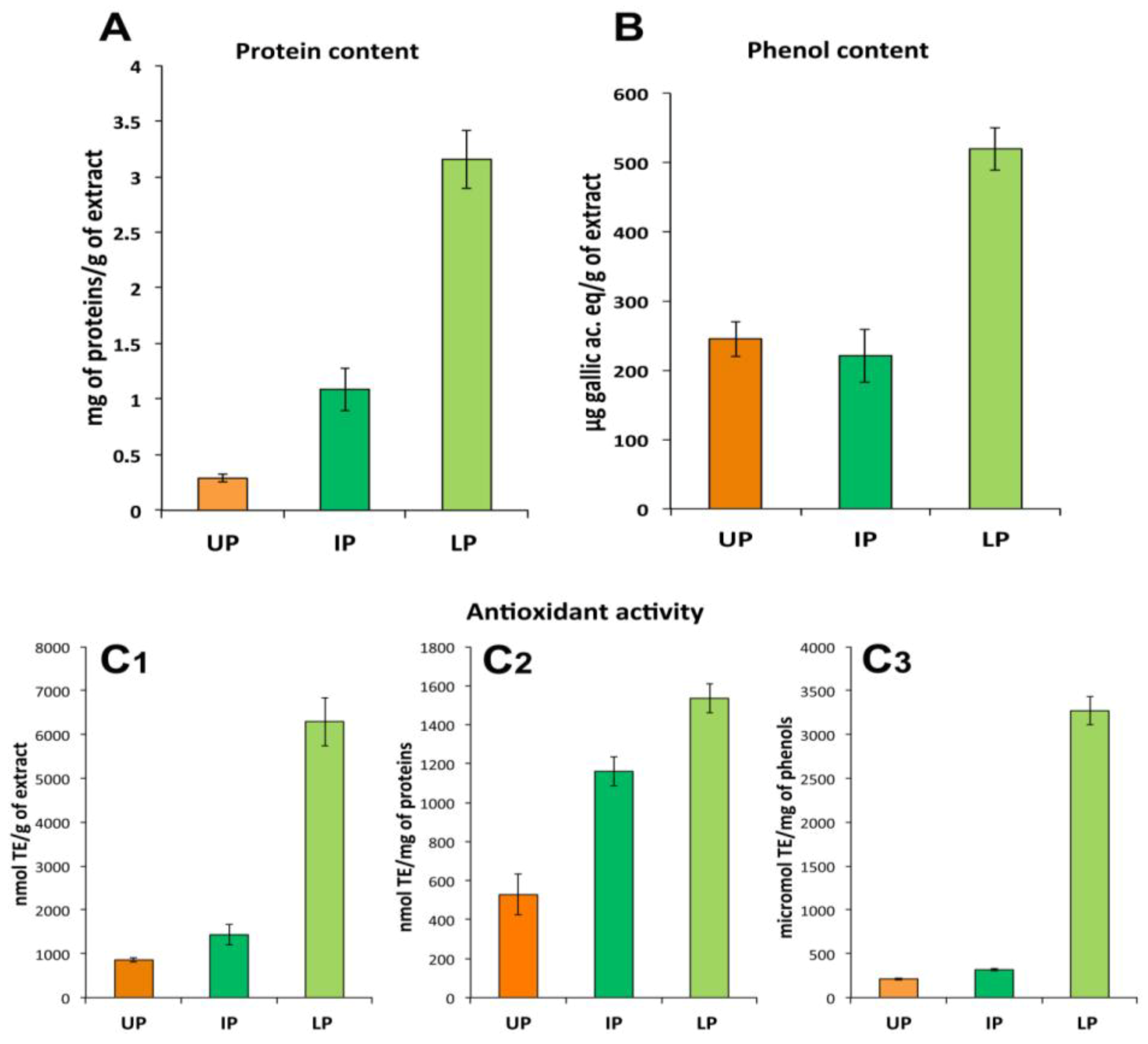
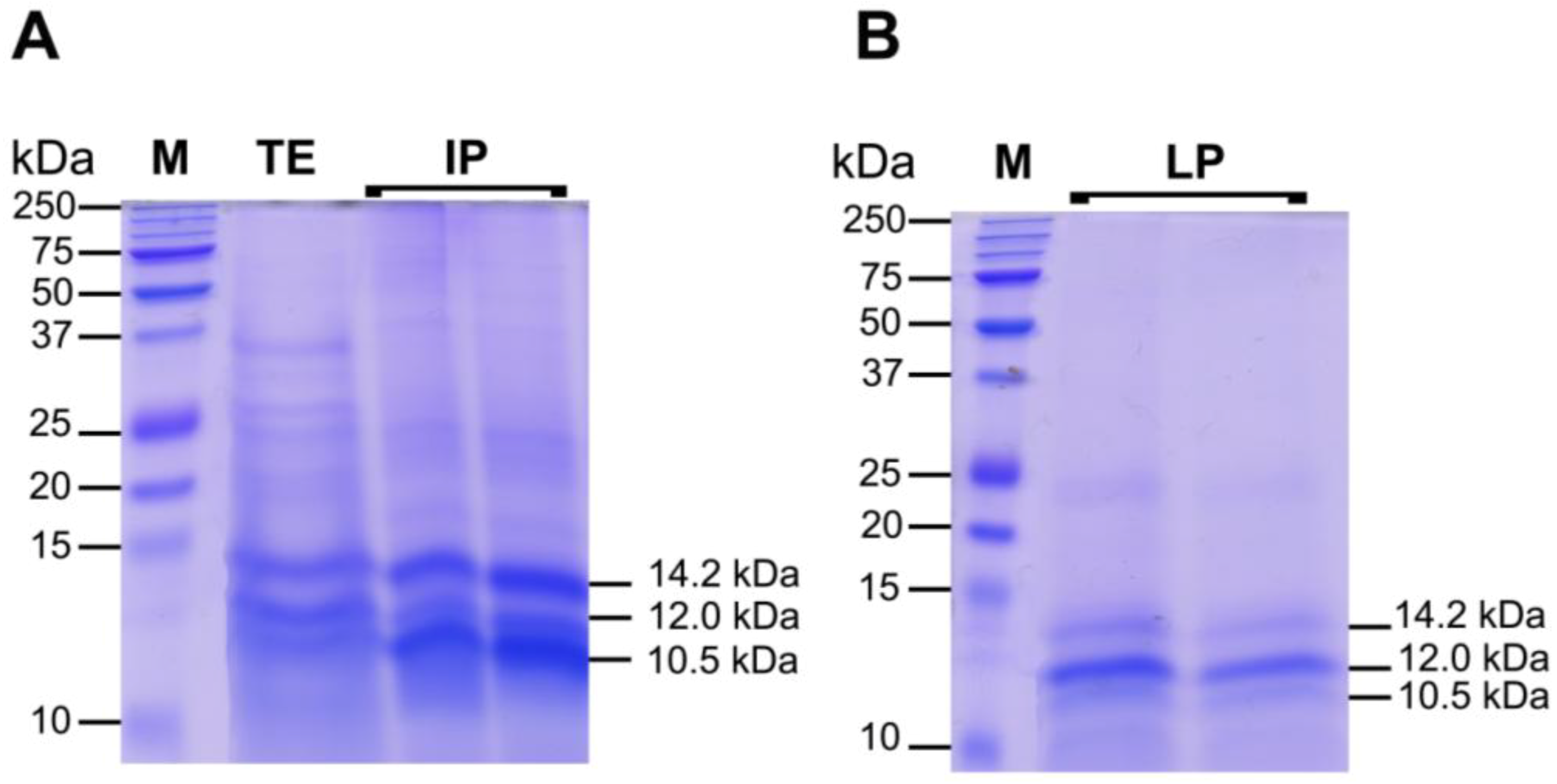
2.3.2. Protein Fractions: Intermediate and Lower Phases
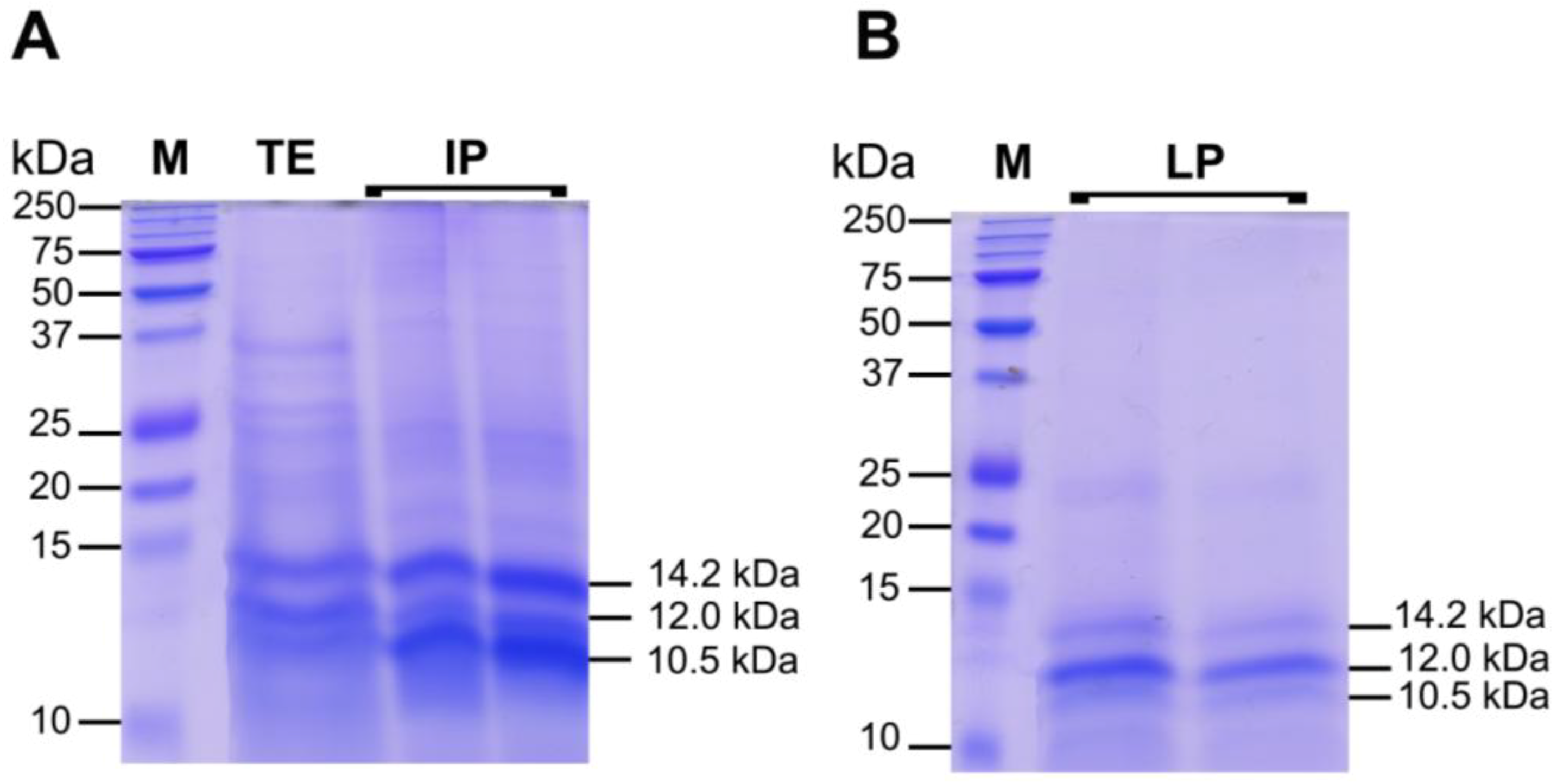
2.4. Biological Activities of Jellyfish Extract Fractions
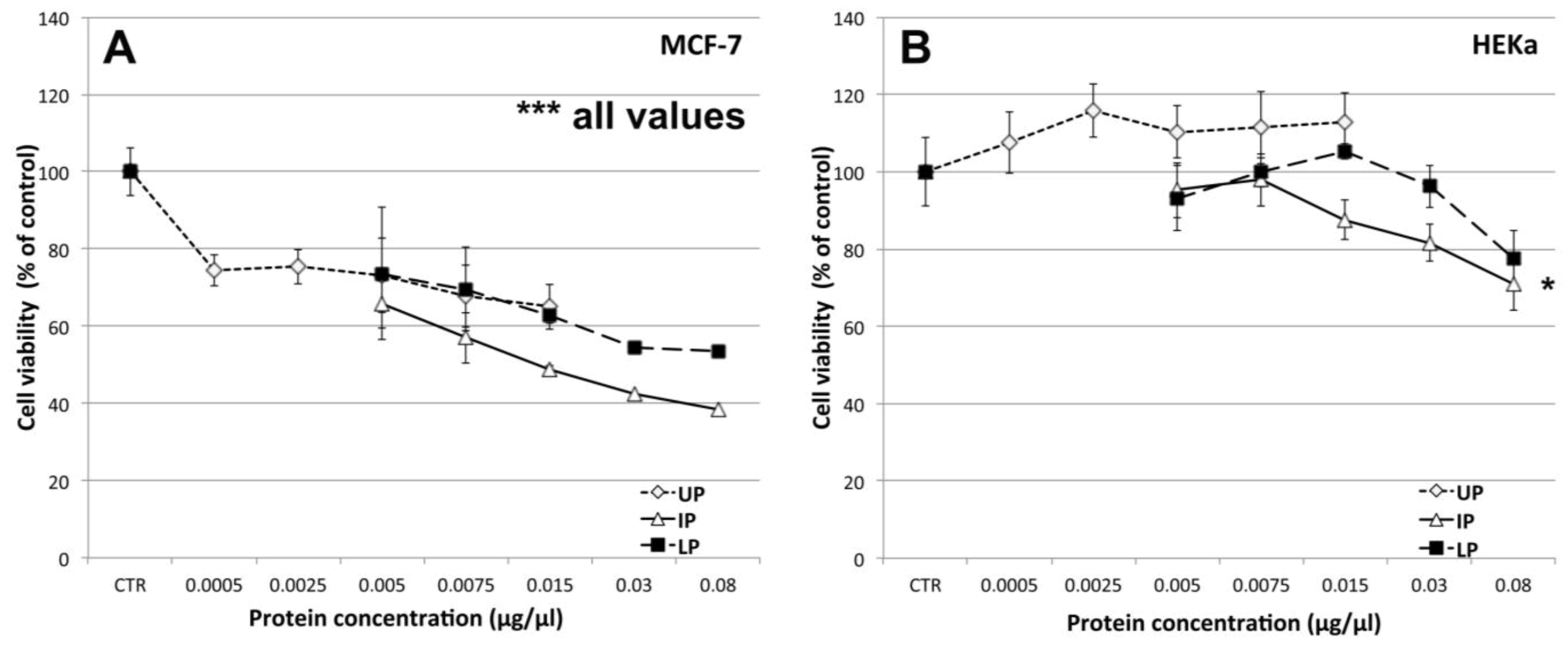
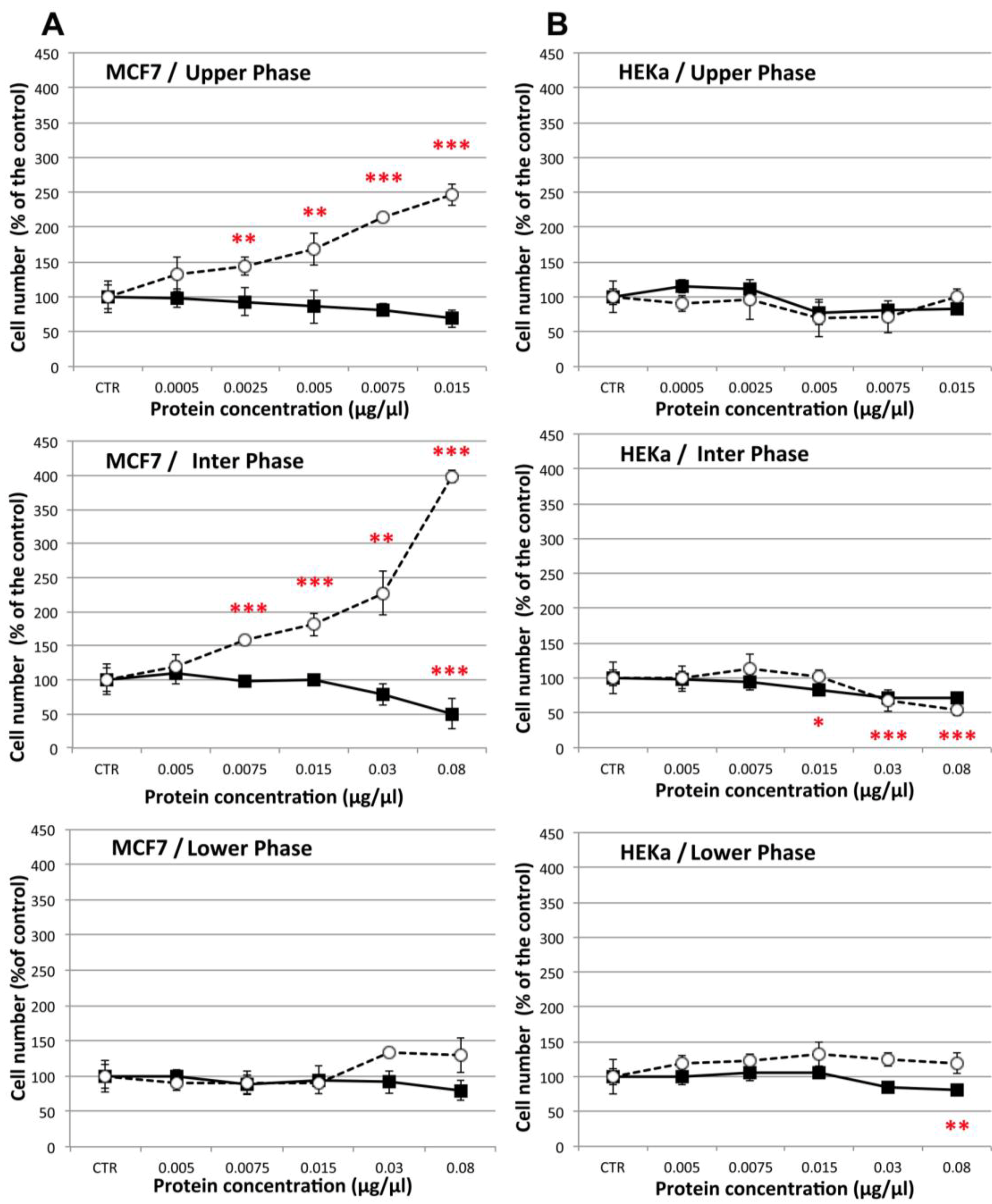
2.4.1. Effect on Cell Viability
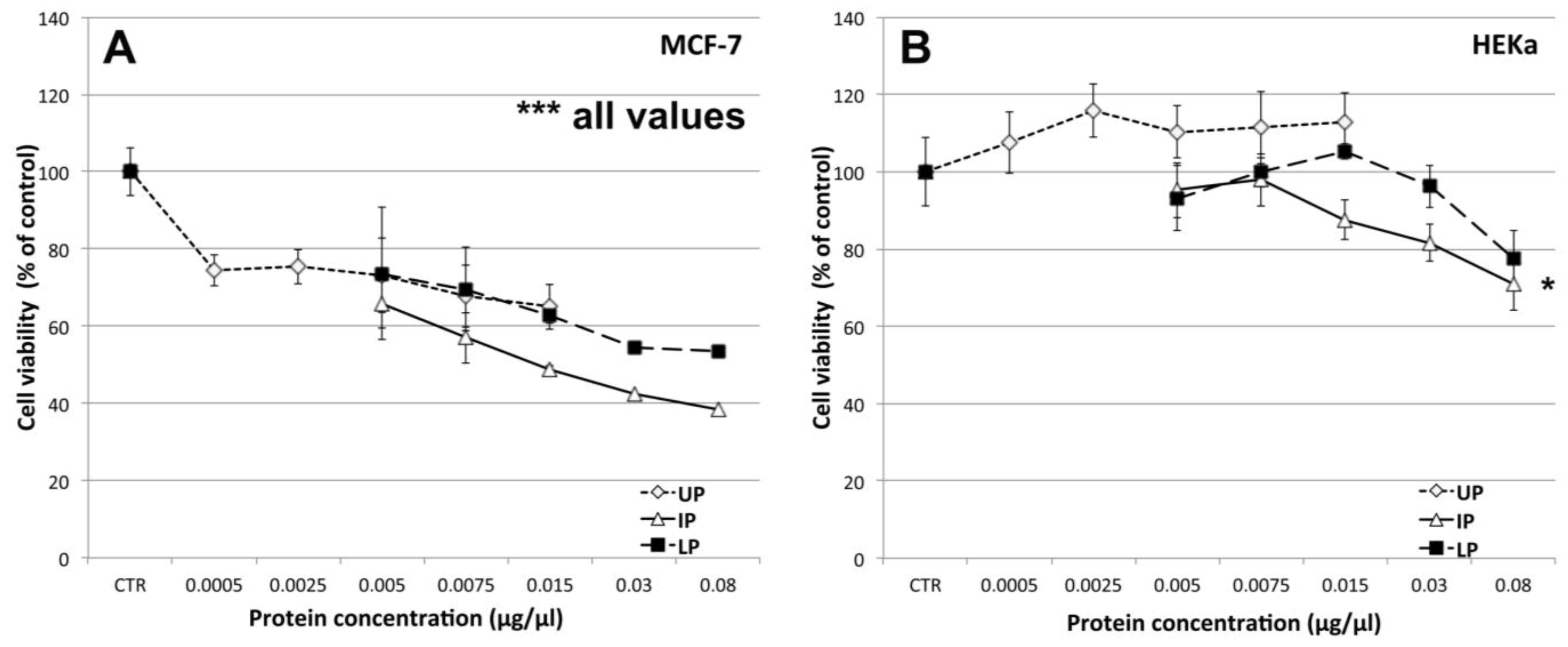
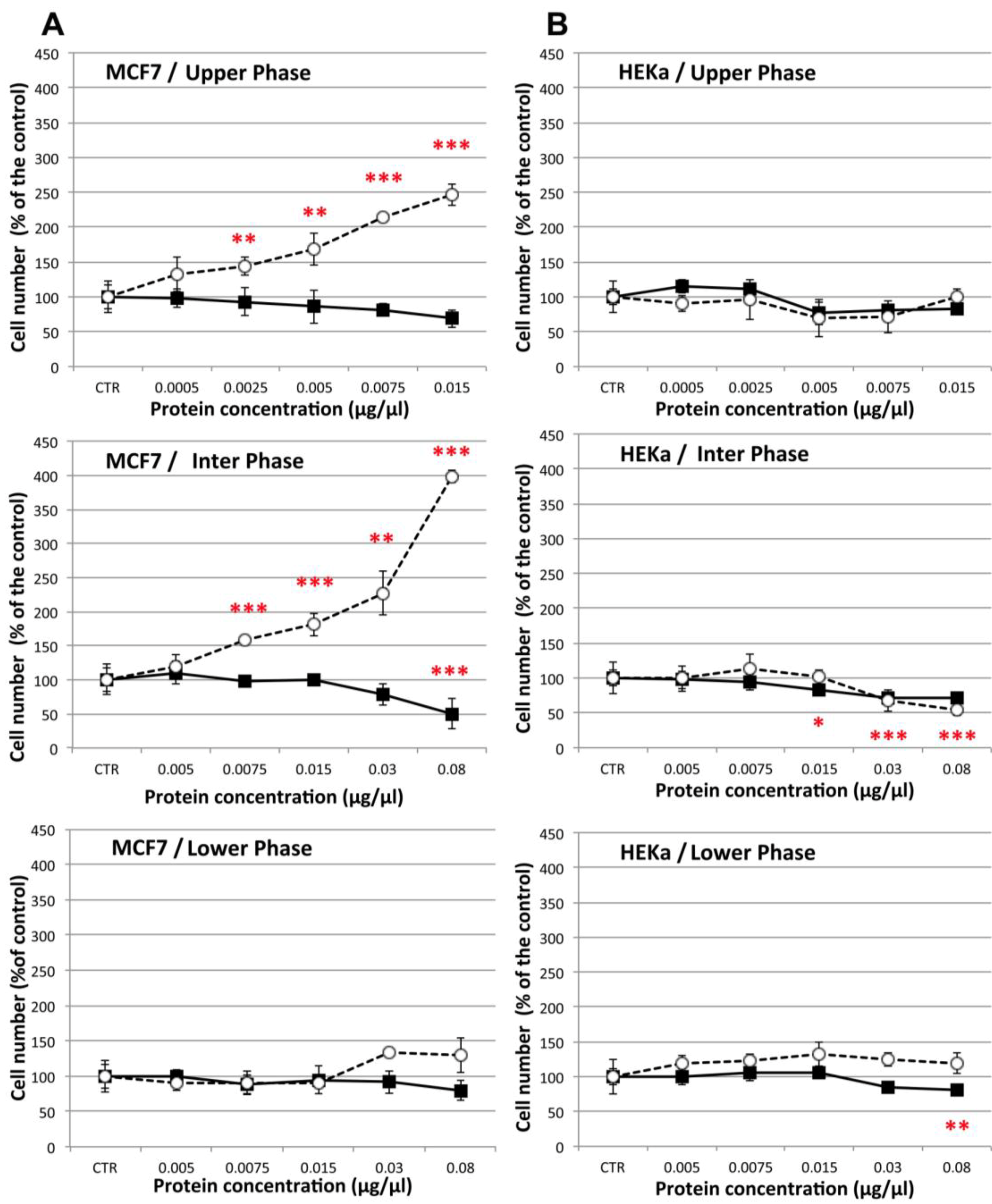
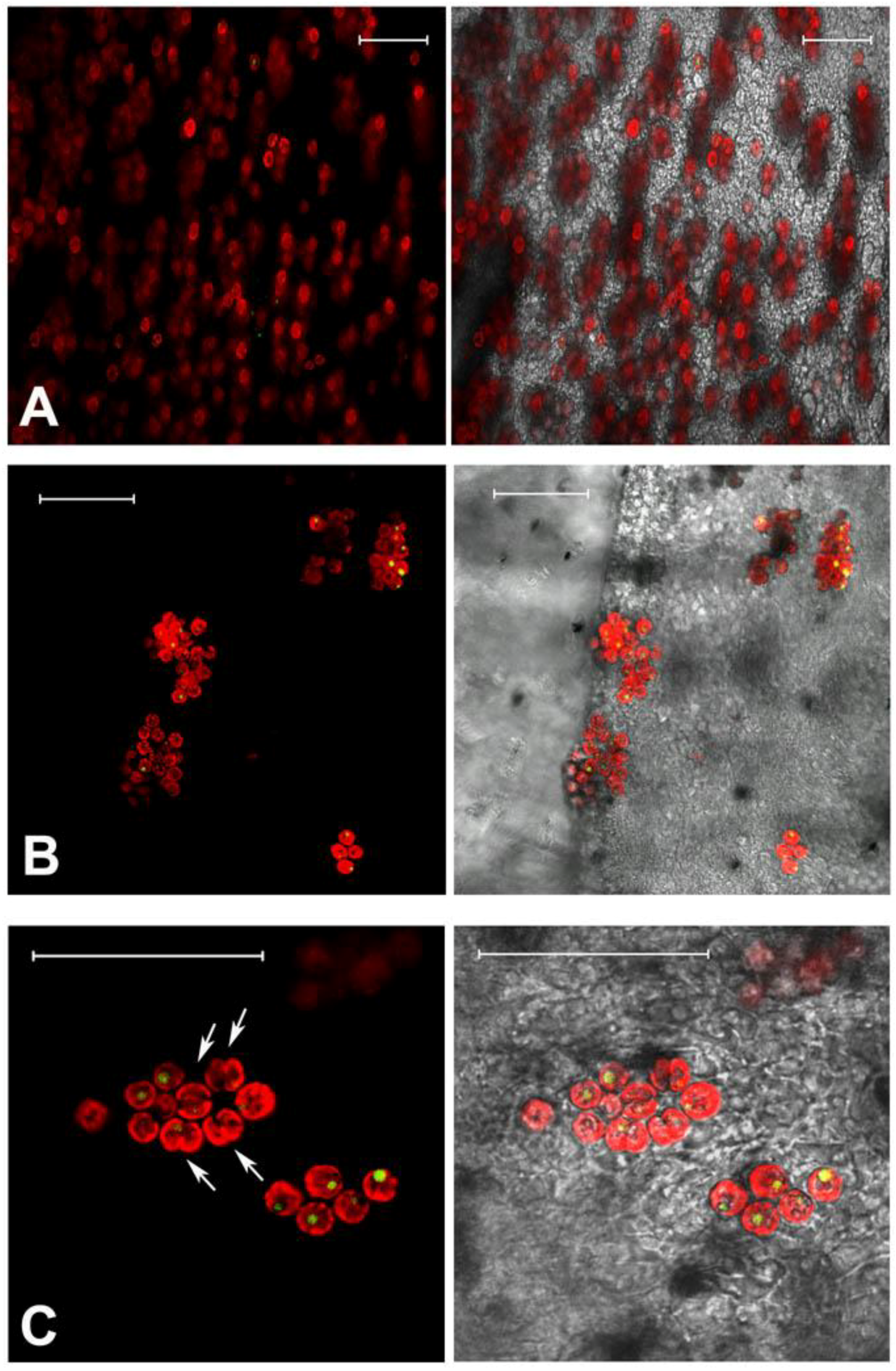
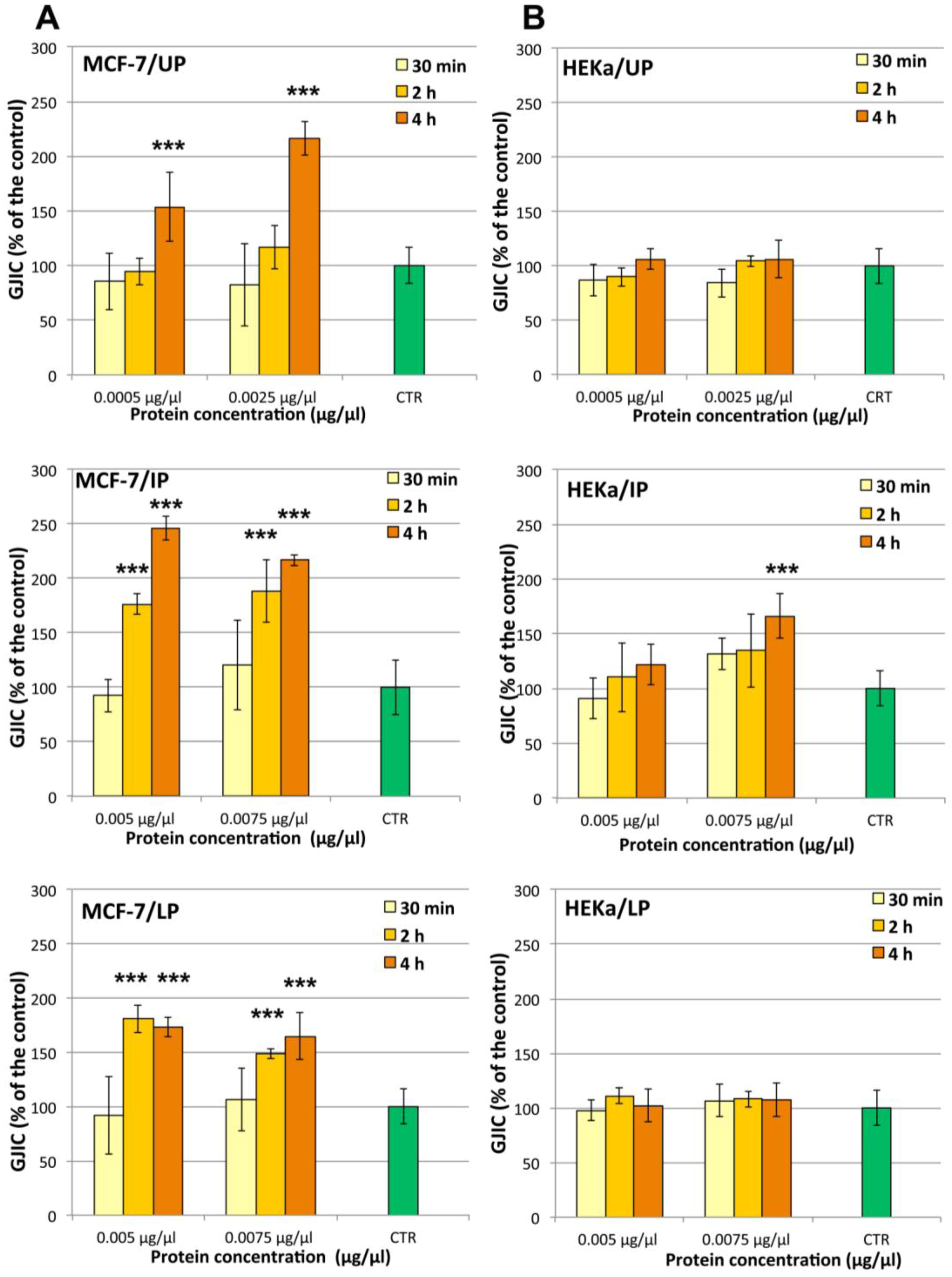
2.4.2. Effect on Cell-Cell Communication
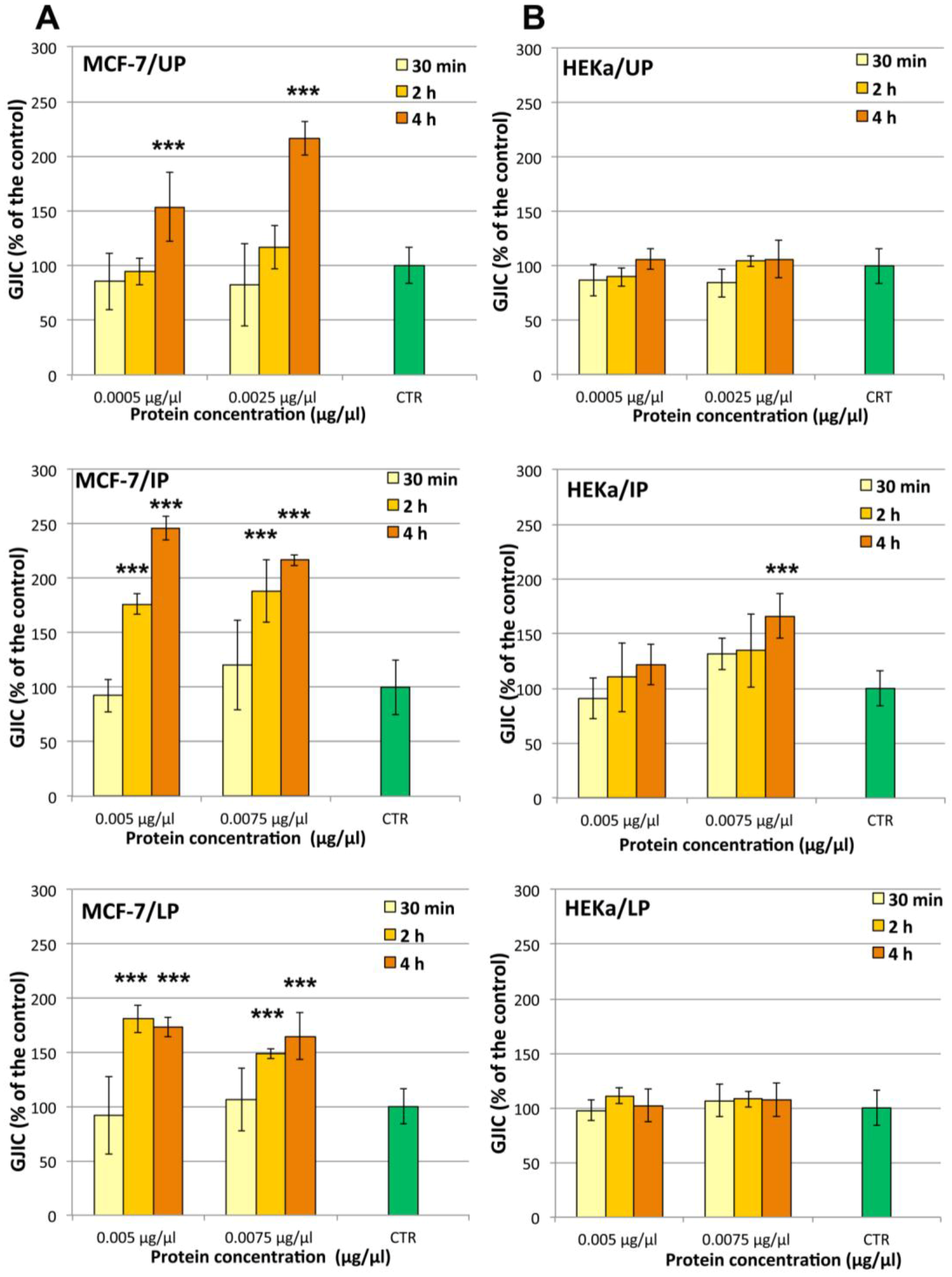
3. Experimental Section
3.1. Chemicals and Media
3.2. Jellyfish Samples
3.3. Preparation and Fractionation of the Jellyfish Extract
3.4. Protein Concentration
3.5. Determination of Phenol Content
3.6. In Vitro Antioxidant Activity Analysis
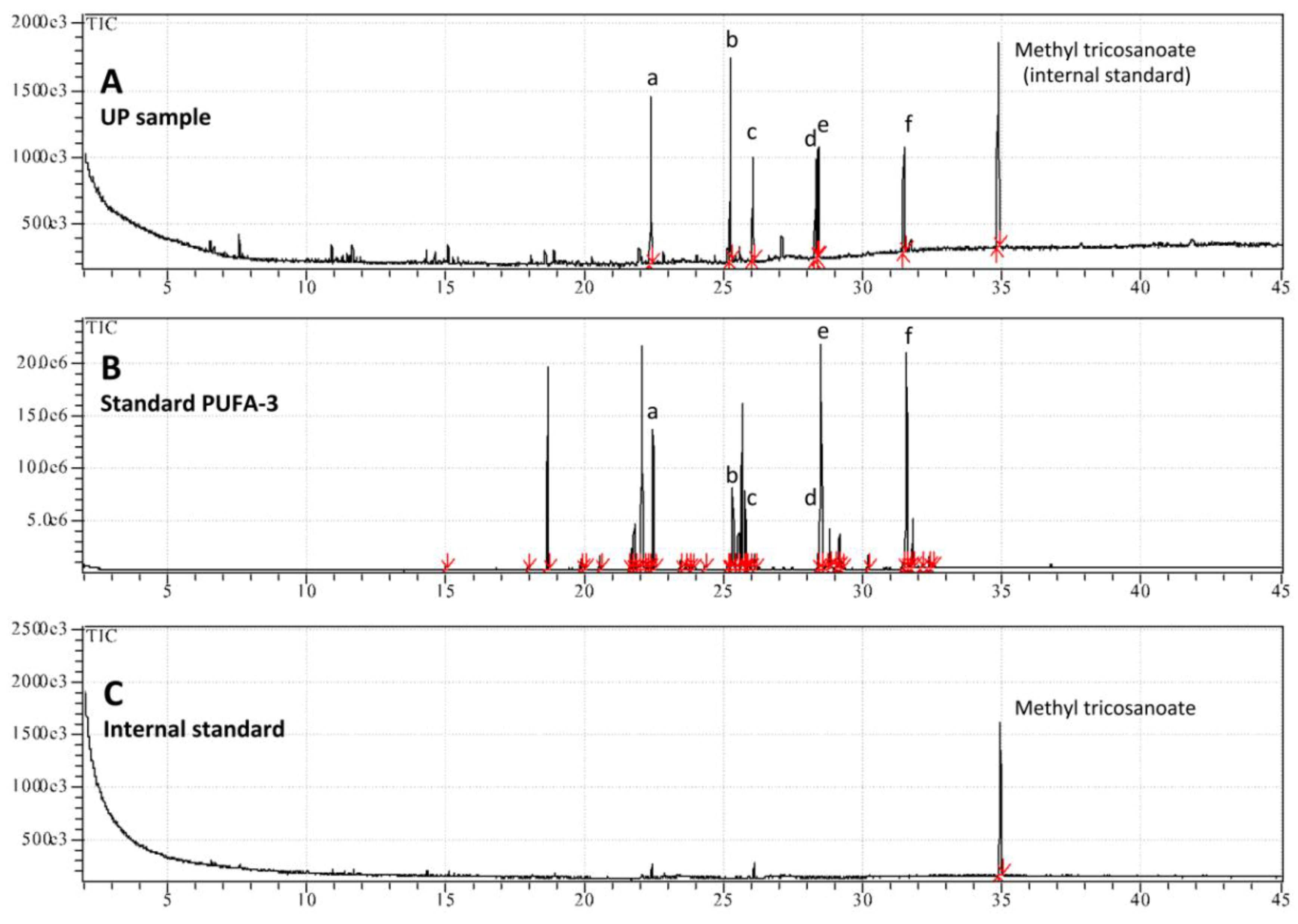
3.7. Fatty Acid Profiles Determination
3.8. HPLC Analysis of the UP Fraction
3.9. GC-MS Analysis
3.10. Protein Analysis by SDS-PAGE
3.11. Cell Cultures
3.12. Cell Viability and Treatments
3.12.1. MTS Assay
3.12.2. Trypan Blue Dye Exclusion Assay
3.13. Estimation of GJIC by Scrape-Loading Dye Transfer (SL/DT) Assay
3.14. Laser Scanning Confocal Microscopy
3.15. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Kijjoa, A.; Sawangwong, P. Drugs and cosmetics from the sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar]
- Jha, R.K.; Zi-rong, X. Biomedical compounds from marine organisms. Mar. Drugs 2004, 2, 123–146. [Google Scholar] [CrossRef]
- Kuramoto, M.; Arimoto, H.; Uemura, D. Bioactive alkaloids from the sea: A review. Mar. Drugs 2004, 1, 39–54. [Google Scholar]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Piggott, A.M.; Karuso, P. Quality not quantity: The role of marine natural products in drug discovery and reverse chemical proteomics. Mar. Drugs 2005, 3, 36–63. [Google Scholar] [CrossRef]
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar]
- Ismail, H.; Lemriss, S.; Ben Aoun, Z.; Mhadhebi, L.; Dellai, A.; Kacem, Y.; Boiron, P.; Bouraoui, A. Antifungal activity of aqueous and methanolic extracts from the Mediterranean sea cucumber, Holoturia polii. J. Med. Mycol. 2008, 18, 23–26. [Google Scholar] [CrossRef]
- Kang, C.; Munawir, A.; Cha, M.; Sohn, E.T.; Lee, H.; Kim, J.S.; Yoon, W.D.; Lim, D.; Kim, E. Cytotoxicity and hemolytic activity of jellyfish Nemopilema nomurai (Scyphozoa: Rhizostomeae) venom. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 150, 85–90. [Google Scholar]
- Yu, H.; Liu, X.; Dong, X.; Li, C.; Xing, R.; Liu, S.; Li, P. Insecticidal activity of proteinous venom from tentacle of jellyfish Rhopilema esculenta Kishinouye. Bioorg. Med. Chem. Lett. 2005, 15, 4949–4952. [Google Scholar]
- Ramasamy, S.; Isbister, G.K.; Seymour, J.E.; Hodgson, W.C. Pharmacologically distinct cardiovascular effects of box jellyfish (Chironex leckeri) venom and a tentacle-only extract in rats. Toxicol. Lett. 2005, 155, 219–226. [Google Scholar] [CrossRef]
- Yu, H.; Liu, X.; Xing, R.; Liu, S.; Li, C.; Li, P. Radical scavenging activity of protein from tentacles of jellyfish Rhopilema esculenta. Bioorg. Med. Chem. Lett. 2005, 15, 2659–2664. [Google Scholar]
- Ovchinnikova, T.V.; Balandin, S.V.; Aleshina, G.M.; Tagaev, A.A.; Leonova, Y.F.; Krasnodembsky, E.D.; Men’shenin, A.V.; Kokryakov, V.N. Aurelin, a novel antimicrobial peptide from jellyfish Aurelia aurita with structural features of defensins and channel-blocking toxins. Biochem. Biophys. Res. Commun. 2006, 348, 514–523. [Google Scholar] [CrossRef]
- Rastogi, A.; Biswas, S.; Sarkar, A.; Chakrabarty, D. Anticoagulant activity of Moon jellyfish (Aurelia aurita) tentacle extract. Toxicon 2012, 60, 719–723. [Google Scholar] [CrossRef]
- Ayed, Y.; Bousabbeh, M.; Mabrouk, H.B.; Morjen, M.; Marrakchi, N.; Bacha, H. Impairment of the cell-to-matrix adhesion and cytotoxicity induced by the Mediterranean jellyfish Pelagia noctiluca venom and its fractions in cultured glioblastoma cells. Lipids Health Dis. 2012, 28, 11–84. [Google Scholar]
- Zhuang, Y.; Hou, H.; Zhao, X.; Zhang, Z.; Li, B. Effects of collagen and collagen hydrolysate from jellyfish (Rhopilema esculentum) on mice skin photoaging induced by UV irradiation. J. Food Sci. 2009, 74, 183–138. [Google Scholar]
- Nishimoto, S.; Goto, Y.; Morishige, H.; Shiraishi, R.; Doi, M.; Akiyama, K.; Yamauchi, S.; Sugahara, T. Mode of action of the immunostimulatory effect of collagen from jellyfish. Biosci. Biotechnol. Biochem. 2008, 72, 2806–2828. [Google Scholar] [CrossRef]
- Morishige, H.; Sugahara, T.; Nishimoto, S.; Muranaka, A.; Ohno, F.; Shiraishi, R.; Doi, M. Immunostimulatory effects of collagen from jellyfish in vivo. Cytotechnology 2011, 63, 481–492. [Google Scholar]
- Brotz, L.; Cheung, W.W.L.; Kleisner, K.; Pakhomov, E.; Pauly, D. Increasing jellyfish populations: Trends in large marine ecosystems. Hydrobiologia 2012, 690, 3–20. [Google Scholar]
- Boero, F.; Bouillon, J.; Gravili, C.; Miglietta, M.P.; Parsons, T.; Piraino, S. Gelatinous plankton: Irregularities rule the world (sometimes). Mar. Ecol. Prog. Ser. 2008, 356, 299–310. [Google Scholar] [CrossRef]
- Purcell, J.E. Jellyfish and ctenophore blooms coincide with human proliferations and environmental perturbations. Ann. Rev. Mar. Sci. 2012, 4, 209–235. [Google Scholar] [CrossRef]
- Rottini-Sandrini, L.; Avian, M.; Franchi, A.; Troian, A.; Vio, E. Dommagesà la Peche en Correlation Avec la Presence de Grandes Quantites de Meduses. In Proceeding of Workshop on Jellyfish Blooms in the Mediterranean, UNEP, Athens, Greece, 31 October–4 November 1983.
- Bernard, P.; Couasnon, F.; Soubiran, J.P.; Goujon, J.F. Surveillance estivale de la méduse Pelagia noctiluca (Cnidaria, Scyphozoa) sur les côtes Méditerranéennes Françaises. Ann. Inst. Océanogr. 1988, 64, 115–125. [Google Scholar]
- Arai, M.N. A Functional Biology of Scyphozoa; Chapman & Hall: London, UK, 1997; pp. 1–316. [Google Scholar]
- Mills, C.E. Jellyfish blooms: Are populations increasing globally in response to changing ocean conditions? Hydrobiologia 2001, 451, 55–68. [Google Scholar] [CrossRef]
- Uye, S.; Ueta, Y. Recent increase of jellyfish populations and their nuisance to fisheries in the Inland Sea of Japan. Bull. Jpn. Soc. Fish. Oceanogr. 2004, 68, 9–19. [Google Scholar]
- Hay, S. Marine ecology: Gelatinous bells may ring change in marine ecosystems. Curr. Biol. 2006, 16, 679–682. [Google Scholar] [CrossRef]
- Purcell, J.E.; Uye, S.I.; Lo, W.T. Anthropogenic causes of jellyfish blooms and direct consequences for humans: A review. Mar. Ecol. Prog. Ser. 2007, 350, 153–174. [Google Scholar] [CrossRef]
- Marino, A.; Morabito, R.; Pizzata, T.; La Spada, G. Effect of various factors on Pelagia noctiluca (Cnidaria: Scyphozoa) crude venom-induced haemolysis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2008, 151, 144–149. [Google Scholar] [CrossRef]
- Senevirathne, M.; Kim, S.K. Utilization of seafood processing by-products: Medicinal applications. Adv. Food. Nutr. Res. 2012, 65, 495–512. [Google Scholar] [CrossRef]
- Macrì, D.S. Nuove osservazioni intorno la storia naturale del polmone marino degli antichi. Available online: http://books.google.it/books?id=wJw-AAAAcAAJ&printsec=frontcover&dq=inauthor:%22Saverio+MACRI%22&hl=it&sa=X&ei=vFiSUYrAJ8Ku7AbJk4H4BA&ved=0CEAQ6AEwAg#v=onepage&q&f=false (accessed on 16 May 2013).
- Kikinger, R. Cotylorhiza tuberculata (Cnidaria: Scyphozoa)-life history of a stationary population. Mar. Ecol. 1992, 13, 333–362. [Google Scholar] [CrossRef]
- Pérez-Ruzafa, A.; Gilabert, J.; Gutiérrez, J.M.; Fernández, A.I.; Marcos, C.; Sabah, S. Evidence of a planktonic food web response to changes in nutrient input dynamics in the Mar Menor coastal lagoon, Spain. Hydrobiologia 2002, 475–476, 359–369. [Google Scholar]
- Ruiz, J.; Prieto, L.; Astorga, D. A model for temperature control of jellyfish (Cotylorhiza tuberculata) outbreaks: A causal analysis in a Mediterranean coastal lagoon. Ecol. Model. 2012, 233, 59–69. [Google Scholar] [CrossRef]
- ECOS. Poblaciones de medusas en el Mar Menor. Available online: http://www.murcianatural.carm.es/c/document_library/get_file?uuid=1a94f256-80e9-442d-beec-5a2c3d102d84&groupId=14 (accessed on 16 May 2013).
- Mayer, A.G. Scyphomedusae. In Medusae of the World; Carnegie Institution: Washington, DC, USA, 1910; Volume 3, pp. 499–735. [Google Scholar]
- Astorga, D.; Ruiz, J.; Prieto, L. Ecological aspects of early life stages of Cotylorhiza tuberculata (Scyphozoa: Rhizostomae) affecting its pelagic population success. Hydrobiologia 2012, 690, 141–155. [Google Scholar] [CrossRef]
- LaJeunesse, T.C.; Loh, W.; Trench, R.K. Do introduced endosymbiotic dinoflagellates ‘take’ to new hosts? Biol. Invasions 2009, 11, 995–1003. [Google Scholar] [CrossRef]
- Prieto, L.; Astorga, D.; Navarro, G.; Ruiz, J. Environmental control of phase transition and polyp survival of a massive-outbreaker jellyfish. PLoS One 2010, 5, e13793. [Google Scholar]
- Pitt, K.A.; Welsh, D.T.; Condon, R.H. Influence of jellyfish blooms on carbon, nitrogen and phosphorus cycling and plankton production. Hydrobiologia 2009, 616, 133–149. [Google Scholar] [CrossRef]
- Kita, M.; Ohno, O.; Han, C.; Uemura, D. Bioactive secondary metabolites from symbiotic marine dinoflagellates: Symbiodinolide and durinskiols. Chem. Rec. 2010, 10, 57–69. [Google Scholar] [CrossRef]
- Rosic, N.N. Phylogenetic analysis of genes involved in mycosporine-like amino acid biosynthesis in symbiotic dinoflagellates. Appl. Microbiol. Biotechnol. 2012, 94, 29–37. [Google Scholar] [CrossRef]
- Iglesias-Prieto, R.; Trench, R.K. Acclimation and adaptation to irradiance in symbiotic dinoflagellates. II. Response of chlorophyll-protein complexes to different photon-flux densities. Mar. Biol. 1997, 130, 23–33. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, H.; Kang, Y.; Bina, D.; Lo, C.S.; Blankenship, R.E. Characterization of the peridinin-chlorophyll a-protein complex in the dinoflagellate Symbiodinium. Biochim. Biophys. Acta 2012, 1817, 983–989. [Google Scholar] [CrossRef]
- Niedzwiedzki, D.M.; Jiang, J.; Lo, C.S.; Blankenship, R.E. Spectroscopic properties of the Chlorophyll a-Chlorophyll c (2)-Peridinin-Protein-Complex (acpPC) from the coral symbiotic dinoflagellate Symbiodinium. Photosynth. Res. 2013. [Google Scholar] [CrossRef]
- Venn, A.A.; Wilson, M.A.; Trapido-Rosenthal, H.G.; Keely, B.J.; Douglas, A.E. The impact of coral bleaching on the pigment profile of the symbiotic alga, Symbiodinium. Plant Cell Environ. 2006, 29, 2133–2142. [Google Scholar] [CrossRef]
- Latyshev, N.A.; Naumenko, N.V.; Svetashev, V.I.; Latypov, Y.Y. Fatty acids of reef-building corals. Mar. Ecol. Prog. Ser. 1991, 76, 295–301. [Google Scholar] [CrossRef]
- Harland, A.D.; Navarro, J.C.; Spencer Davies, P.; Fixter, L.M. Lipids of some Caribbean and Red Sea corals: Total lipid, wax esters, triglycerides and fatty acids. Mar. Biol. 1993, 117, 113–117. [Google Scholar] [CrossRef]
- Dunn, S.R.; Thomas, M.C.; Nette, G.W.; Dove, S.G. A lipidomic approach to understanding free fatty acid lipogenesis derived from dissolved inorganic carbon within cnidarian-dinoflagellate symbiosis. PLoS One 2012, 7, e46801. [Google Scholar]
- Leblond, J.D.; Chapman, P.J. Lipid class distribution of highly unsaturated long chain fatty acids in marine dinoflagellates. J. Phycol. 2000, 36, 1103–1108. [Google Scholar] [CrossRef]
- Berquin, I.M.; Edwards, I.J.; Chen, Y.Q. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef]
- Kremmyda, L.S.; Tvrzicka, E.; Stankova, B.; Zak, A. Fatty acids as biocompounds: Their role in human metabolism, health and disease: A review. Part 2: Fatty acid physiological roles and applications in human health and disease. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2011, 155, 195–218. [Google Scholar] [CrossRef]
- Yongmanitchai, W.C.; Ward, O.P. Omega-3 fatty acids—Alternative sources of production. Process Biochem. 1989, 24, 117–125. [Google Scholar]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar]
- Phelan, P.; Bacon, J.P.; Davies, J.A.; Stebbings, L.A.; Todman, M.G.; Avery, L.; Baines, R.A.; Barnes, T.M.; Ford, C.; Hekimi, S.; et al. Innexins: A family of invertebrate gap junction proteins. Trends Genet. 1998, 14, 348–349. [Google Scholar] [CrossRef]
- Vinken, M.; Decrock, E.; de vuyst, E.; Ponsaerts, R.; D’hondt, C.; Bultynck, G.; Ceelen, L.; Vanhaecke, T.; Leybaert, L.; Rogiers, V. Connexins: Sensors and regulators of cell cycling. Biochim. Biophys. Acta 2011, 1815, 13–25. [Google Scholar]
- Bedner, P.; Steinhäuser, C.; Theis, M. Functional redundancy and compensation among members of gap junction protein families? Biochim. Biophys. Acta 2012, 1818, 1971–1984. [Google Scholar] [CrossRef]
- Evans, W.H.; Martin, P.E. Gap junctions: Structure and function (Review). Mol. Membr. Biol. 2002, 19, 121–136. [Google Scholar] [CrossRef]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harbor Perspect. Biol. 2009, 1, 2576. [Google Scholar] [CrossRef]
- Hesketh, G.G.; van Eyk, J.E.; Tomaselli, G.F. Mechanisms of gap junction traffic in health and disease. Cardiovasc. Pharmacol. 2009, 54, 263–272. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 1991, 115, 1357–1374. [Google Scholar] [CrossRef]
- Bruzzone, R.; White, T.W.; Paul, D.L. Connections with connexins: The molecular basis of intercellular signaling. Eur. J. Biochem. 1996, 238, 1–27. [Google Scholar]
- Laird, D.W. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 2005, 1711, 172–182. [Google Scholar] [CrossRef]
- Pahujaa, M.; Anikin, M.; Goldberg, G.S. Phosphorylation of connexin43 induced by Src: Regulation of gap junctional communication between transformed cells. Exp. Cell Res. 2007, 313, 4083–4090. [Google Scholar] [CrossRef]
- Trosko, J.E.; Ruch, R.J. Gap junctions as targets for cancer chemoprevention and chemotherapy. Curr. Drug Targets 2002, 3, 465–482. [Google Scholar] [CrossRef]
- Trosko, J.E.; Chang, C.C.; Upham, B.; Wilson, M. Epigenetic toxicology as toxicant-induced changes in intracellular signalling leading to altered gap junctional intercellular communication. Toxicol. Lett. 1998, 102–103, 71–78. [Google Scholar]
- Trosko, J.E. Stem cells and cell-cell communication in the understanding of the role of diet and nutrients in human diseases. J. Food Hyg. Saf. 2007, 22, 1–14. [Google Scholar]
- Trosko, J.E.; Chang, C.C. Mechanism of up-regulated gap junctional intercellular communication during chemoprevention and chemotherapy of cancer. Mutat. Res. 2001, 480–481, 219–229. [Google Scholar]
- Trosko, J.E.; Chang, C.C. Modulation of cell-cell communication in the cause and chemoprevention/chemotherapy of cancer. BioFactors 2000, 12, 259–263. [Google Scholar] [CrossRef]
- Leone, A.; Longo, C.; Trosko, J.E. The chemopreventive role of dietary phytochemicals through gap junctional intercellular communication. Phytochem. Rev. 2012, 11, 285–307. [Google Scholar] [CrossRef]
- Leone, A.; Zefferino, R.; Longo, C.; Leo, L.; Zacheo, G. Supercritical CO2-extracted tomato oleoresins enhance gap junction intercellular communications and recover from mercury chloride inhibition in keratinocytes. J. Agric. Food Chem. 2010, 58, 4769–4778. [Google Scholar] [CrossRef]
- Sai, K.; Kanno, J.; Hasegawa, R.; Trosko, J.E.; Inoue, T. Prevention of the down-regulation of gap junctional intercellular communication by green tea in the liver of mice fed pentachlorophenol. Carcinogenesis 2000, 21, 1671–1676. [Google Scholar] [CrossRef]
- Sai, K.; Kang, K.-S.; Hirose, A.; Hasegawa, R.; Trosko, J.E.; Inoue, T. Inhibition of apoptosis by pentachlorophenol in v-myc-transfected rat liver epithelial cells: Relation to down-regulation of gap junctional intercellular communication. Cancer Lett. 2001, 173, 163–174. [Google Scholar] [CrossRef]
- Kang, K.S.; Lee, Y.S.; Kim, H.S. Di-(2-ethylhexyl) phthalate-induced cell proliferation is involved in the inhibition of gap junctional intercellular communication and blockage of apoptosis in mouse Sertoli cells. J. Toxicol. Environ. Health A 2002, 65, 447–459. [Google Scholar] [CrossRef]
- Lee, D.E.; Kang, N.J.; Lee, K.M.; Lee, B.K.; Kim, J.H.; Lee, K.W.; Lee, H.J. Cocoa polyphenols attenuate hydrogen peroxideinduced inhibition of gap-junction intercellular communication by blocking phosphorylation of connexin 43 via the MEK/ERK signaling pathway. J. Nutr. Biochem. 2010, 21, 680–686. [Google Scholar] [CrossRef]
- Lee, D.E.; Shin, B.J.; Hur, H.J.; Kim, J.H.; Kim, J.; Kang, N.J.; Kim, D.O.; Lee, C.Y.; Lee, K.W.; Lee, H.J. Quercetin, the active phenolic component in kiwifruit, prevents hydrogen peroxide-induced inhibition of gap-junction intercellularcommunication. Br. J. Nutr. 2010, 104, 164–170. [Google Scholar] [CrossRef]
- Rangel, M.; Ionta, M.; Pfister, S.C.; Sant’Anna Ferreira, R.A.; Machado-Santelli, G.M. Marine sponge depsipeptide increases gap junction length in HTC cells transfected with Cx43-GFP. Cell Biol. Int. Rep. 2010, 17, e00003. [Google Scholar]
- Daubrawa, F.; Sies, H.; Stahl, W. Astaxanthin diminishes gap junctional intercellular communication in primary human fibroblasts. J. Nutr. 2005, 135, 2507–2511. [Google Scholar]
- Stahl, W.; Sies, H. Bioactivity and protective effects of natural carotenoids. Biochim. Biophys. Acta 2005, 1740, 101–107. [Google Scholar] [CrossRef]
- Nováková, K.; Babica, P.; Adamovský, O.; Bláha, L.; Nováková, K. Modulation of gap-junctional intercellular communication by a series of cyanobacterial samples from nature and laboratory cultures. Toxicon 2011, 58, 76–84. [Google Scholar] [CrossRef]
- Patel, S.K.; Prasanth Kumar, S.; Jasrai, Y.T.; Pandya, H.A.; Rawal, R.M. Computational analysis of naturally occurring marine compounds (NOMC) targeting gap junctions and cell adhesive molecules for the identification of anticancer drug targets. JCIB 2011, 4, 161–171. [Google Scholar]
- Ebada, S.S.; Lin, W.; Proksch, P. Bioactive sesterterpenes and triterpenes from marine sponges: Occurrence and pharmacological significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef]
- Folmer, F.; Jaspars, M.; Schumacher, M.; Dicato, M.; Diederich, M. Marine natural products targeting phospholipases A2. Biochem. Pharmacol. 2010, 80, 1793–1800. [Google Scholar] [CrossRef]
- Soriente, A.; de Rosa, M.M.; Scettri, A.; Sodano, G.; Terencio, M.C.; Payá, M.; Alcaraz, M.J. Manoalide. Curr. Med. Chem. 1999, 6, 415–431. [Google Scholar]
- Gunstone, F.D. Novel Long-Chain Compounds Produced through Neighbouring Group Participation. In Recent Developments in the Synthesis of Fatty Acid Derivatives; Knothe, G., Derksen, J.T.P., Eds.; AOCS Press: Champaign, IL, USA, 1999; pp. 1–19. [Google Scholar]
- Osterhage, C.; Kaminsky, R.; König, G.M.; Wright, A.D. Ascosalipyrrolidinone A, an antimicrobial alkaloid, from the obligate marine fungus Ascochyta salicornia. J. Org. Chem. 2000, 65, 6412–6417. [Google Scholar] [CrossRef]
- Kwak, J.H.; Schmitz, F.J.; Williams, G.C. Milolides, new briarane diterpenoids from the western Pacific octocoral Briareum stechei. J. Nat. Prod. 2001, 64, 754–760. [Google Scholar] [CrossRef]
- Groweiss, A.; Look, S.A.; Fenical, W. Solenolides new antiinflammatory and antiviral diterpenoids from a marine octocoral of the genus Solenopodium. J. Org. Chem. 1988, 53, 2401–2406. [Google Scholar] [CrossRef]
- Wakefield, T.S.; Kempf, S.C. Development of host- and symbiont-specific monoclonal antibodies and confirmation of the origin of the symbiosome membrane in a Cnidarian-Dinoflagellate symbiosis. Biol. Bull. 2001, 200, 127–143. [Google Scholar] [CrossRef]
- Starcevic, A.; Dunlap, W.C.; Cullum, J.; Shick, J.M.; Hranueli, D.; Long, P.F. Gene expression in the scleractinian Acropora microphthalma exposed to high solar irradiance reveals elements of photoprotection and coral bleaching. PLoS One 2010, 5, e13975. [Google Scholar]
- Bay, L.K.; Cumbo, V.R.; Abrego, D.; Kool, J.T.; Ainsworth, T.D.; Willis, B.L. Infection dynamics vary between Symbiodinium types and cell surface treatments during establishment of endosymbiosis with coral larvae. Diversity 2011, 3, 356–374. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Dobbs, F.C. Green autofluorescence in dinoflagellates, diatoms, and other microalgae and its implications for vital staining and morphological studies. Appl. Environ. Microbiol. 2007, 73, 2306–2313. [Google Scholar] [CrossRef]
- Hense, B.A.; Gais, P.; Jütting, U.; Scherb, H.; Rodenacker, K. Use of fluorescence information for automated phytoplankton investigation by image analysis. J. Plankton Res. 2008, 30, 587–606. [Google Scholar] [CrossRef]
- Alpert, A.K.; Shukla, A.J. Precipitation of Large, High-Abundance Proteins from Serum with Organic Solvents. ABRF, 2003. Available online: http://abrf.org/Other/ABRFMeetings/ABRF2003/Alpert.pdf (accessed on 13 May 2013).
- Romitelli, F.; Santini, S.A.; Chierici, E.; Pitocco, D.; Tavazzi, B.; Amorini, A.M.; Lazzarino, G.; di Stasio, E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC-MS: The importance of a correct removal of proteins in the Griess assay. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 851, 257–267. [Google Scholar] [CrossRef]
- Ghasemi, A.; Hedayati, M.; Biabani, H. Protein precipitation methods evaluated for determination of serum nitric oxide end products by the Griess assay. J. Med. Sci. Res. 2007, 1, 43–46. [Google Scholar]
- Mills, J.B.; Mant, C.T.; Hodges, R.S. One-step purification of a recombinant protein from a whole cell extract by reversed phase high performance liquid chromatography. J. Chromatogr. A 2006, 1133, 248–253. [Google Scholar] [CrossRef]
- Dhamole, P.B.; Mahajan, P.; Feng, H. Sugaring out: A new method for removal of acetonitrile from preparative RP-HPLC eluent for protein purification. Proc. Biochem. 2010, 45, 1672–1676. [Google Scholar]
- Mathies, J.C.; Austin, M.A. Modified acetonitrile protein-precipitation method of sample preparation for drug assay by liquid chromatography. Clin. Chem. 1980, 26, 1760. [Google Scholar]
- Gu, T.; Gu, Y.; Zheng, Y.; Wiehl, P.E.; Kopchick, J.J. Phase separation of acetonitrile-water mixture in protein purification. Sep. Technol. 1994, 4, 258–261. [Google Scholar] [CrossRef]
- Hoepffner, N.; Sathyendranath, S. Effect of pigment composition on absorption properties of phytoplankton. Mar. Ecol. Prog. Ser. 1991, 73, 11–23. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Richa, S.R.P.; Singh, S.P.; Häder, D.P. Photoprotective compounds from marine organisms. J. Ind. Microbiol. Biotechnol. 2010, 37, 537–558. [Google Scholar] [CrossRef]
- Korbee, N.; Teresa Mata, M.; Figueroa, F.L. Photoprotection mechanisms against ultraviolet radiation in Heterocapsa sp. (Dinophyceae) are influenced by nitrogen availability: Mycosporine-like amino acids vs. xanthophyll cycle. Limnol. Oceanogr. 2010, 55, 899–908. [Google Scholar]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef]
- Findlay, J.A.; Patil, A.D. Antibacterial constituents of the diatom Navicula delognei. J. Nat. Prod. 1984, 47, 815–818. [Google Scholar] [CrossRef]
- Ohta, S.; Chang, T.; Kawashima, A.; Nagate, T.; Murase, M.; Nakanishi, H.; Miyata, H.; Kondo, M. Anti methicillin-resistant Staphylococcus aureus (MRSA) activity by linolenic acid isolated from the marine microalga Chlorococcum HS-101. Bull. Environ. Contam. Toxicol. 1994, 52, 673–680. [Google Scholar]
- Desbois, A.P.; Mearns-Spragg, A.; Smith, V.J. A fatty acid from the diatom Phaeodactylum tricornutum is antibacterial against diverse bacteria including multi-resistant Staphylococcus aureus (MRSA). Mar. Biotechnol. (NY) 2009, 11, 45–52. [Google Scholar] [CrossRef]
- Nappo, M.; Berkov, S.; Massucco, C.; Di Maria, V.; Bastida, J.; Codina, C.; Avila, C.; Messina, P.; Zupo, V.; Zupo, S. Apoptotic activity of the marine diatom Cocconeis scutellum and eicosapentaenoic acid in BT20 cells. Pharm. Biol. 2012, 50, 529–535. [Google Scholar] [CrossRef]
- Zheng, L.H.; Wang, Y.J.; Sheng, J.; Wang, F.; Zheng, Y.; Lin, X.K.; Sun, M. Antitumor peptides from marine organisms. Mar. Drugs 2011, 9, 1840–1859. [Google Scholar] [CrossRef]
- Ngo, D.H.; Vo, T.S.; Ngo, D.N.; Wijesekara, I.; Kim, S.K. Biological activities and potential health benefits of bioactive peptides derived from marine organisms. Int. J. Biol. Macromol. 2012, 51, 378–383. [Google Scholar] [CrossRef]
- Lazcano-Pérez, F.; Román-González, S.A.; Sánchez-Puig, N.; Arreguin-Espinosa, R. Bioactive peptides from marine organisms: A short overview. Protein Pept. Lett. 2012, 19, 700–707. [Google Scholar] [CrossRef]
- Suarez-Jimenez, G.M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef]
- Kim, S.K.; Kang, K.H. Chapter 25—Medicinal effects of peptides from marine microalgae. Adv. Food. Nutr. Res. 2011, 64, 313–323. [Google Scholar]
- Kang, K.H.; Qian, Z.J.; Ryu, B.M.; Kim, S.K. Characterization of growth and protein contents from microalgae Navicula incerta with the investigation of antioxidant activity of enzymatic hydrolysates. Food Sci. Biotechnol. 2011, 20, 183–191. [Google Scholar] [CrossRef]
- Brown, M.R. The amino acid and sugar composition of sixteen species of microalgae used in mariculture. J. Exp. Mar. Biol. Ecol. 1991, 145, 79–99. [Google Scholar]
- Giri, A.; Ohshima, T. Chapter 5—Bioactive marine peptides: Nutraceutical value and novel approaches. Adv. Food Nutr. Res. 2012, 65, 73–105. [Google Scholar] [CrossRef]
- Sheih, I.C.; Wu, T.K.; Fang, T.J. Antioxidant properties of a new antioxidative peptide from algae protein waste hydrolysate in different oxidation systems. Bioresour. Technol. 2009, 100, 3419–3425. [Google Scholar] [CrossRef]
- Spolaore, P.; Joannis-Cassana, C.; Duranb, E.; Isamberta, A. Commercial applications of microalgae. J. Biosci. Bioeng. 2006, 101, 87–96. [Google Scholar] [CrossRef]
- Suput, D. In vivo effects of cnidarian toxins and venoms. Toxicon 2009, 54, 1190–1200. [Google Scholar] [CrossRef]
- Brinkman, D.L.; Burnell, J.N. Biochemical and molecular characterisation of cubozoan protein toxins. Toxicon 2009, 54, 1162–1173. [Google Scholar] [CrossRef]
- Lee, H.; Jung, E.S.; Kang, C.; Yoon, W.D.; Kim, J.S.; Kim, E. Scyphozoan jellyfish venom metalloproteinases and their role in the cytotoxicity. Toxicon 2011, 58, 277–284. [Google Scholar] [CrossRef]
- Lassen, S.; Helmholz, H.; Ruhnau, C.; Prange, A. A novel proteinaceous cytotoxin from the northern Scyphozoa Cyanea capillata (L.) with structural homology to cubozoan haemolysins. Toxicon 2011, 57, 721–729. [Google Scholar] [CrossRef]
- Ayed, Y.; Chayma, B.; Hayla, A.; Abid, S.; Bacha, H. Is cell death induced by nematocysts extract of medusa Pelagia noctiluca related to oxidative stress? Environ. Toxicol. 2011. [Google Scholar] [CrossRef]
- Ayed, Y.; Bouaziz, C.; Brahmi, D.; Zaid, C.; Abid, S.; Bacha, H. Cell death in relation to DNA damage after exposure to the jellyfish Pelagia noctiluca nematocysts. Environ. Toxicol. 2012. [Google Scholar] [CrossRef]
- Suput, D. Interactions of cnidarian toxins with the immune system. Inflamm. Allergy Drug Targets 2011, 10, 429–437. [Google Scholar]
- Simstein, R.; Burow, M.; Parker, A.; Weldon, C.; Beckman, B. Apoptosis, chemoresistance, and breast cancer: Insights from the MCF-7 cell model system. Exp. Biol. Med. (Maywood) 2003, 228, 995–1003. [Google Scholar]
- Fornelli, F.; Leone, A.; Verdesca, I.; Minervini, F.; Zacheo, G. The influence of lycopene on the proliferation of human breast cell line (MCF-7). Toxicol. In Vitro 2007, 21, 217–223. [Google Scholar] [CrossRef]
- Zefferino, R.; Leone, A.; Piccaluga, S.; Cincione, R. Mercury modulates interplay between IL-1β, TNF-α, and gap junctional intercellular communication in keratinocytes: Mitigation by lycopene. J. Immunotoxicol. 2008, 4, 353–360. [Google Scholar]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3242. [Google Scholar] [CrossRef]
- Sarrazin, J.F.; Comeau, G.; Daleau, P.; Kingma, J.; Plante, I.; Fournier, D.; Molin, F. Reduced incidence of vagally induced atrial fibrillation and expression levels of connexins by n-3 polyunsaturated fatty acids in dogs. J. Am. Coll. Cardiol. 2007, 50, 1505–1512. [Google Scholar]
- Champeil-Potokar, G.; Chaumontet, C.; Guesnet, P.; Lavialle, M.; Denis, I. Docosahexaenoic acid (22:6 n-3) enrichment of membrane phospholipids increases gap junction coupling capacity in cultured astrocytes. Eur. J. Neurosci. 2006, 24, 3084–3090. [Google Scholar] [CrossRef]
- Mitasíková, M.; Smidová, S.; Macsaliová, A.; Knezl, V.; Dlugosová, K.; Okruhlicová, L.; Weismann, P.; Tribulová, N. Aged male and female spontaneously hypertensive rats benefit from n-3 polyunsaturated fatty acids supplementation. Physiol. Res. 2008, 57, 39–48. [Google Scholar]
- Stoffel, W.; Holz, B.; Jenke, B.; Binczek, E.; Günter, R.H.; Kiss, C.; Karakesisoglou, I.; Thevis, M.; Weber, A.A.; Arnhold, S.; et al. ∆6-Desaturase (FADS2) deficiency unveils the role of ω3- and ω6-polyunsaturated fatty acids. EMBO J. 2008, 27, 2281–2292. [Google Scholar] [CrossRef]
- Harris, W.S. Omega-3 fatty acids and cardiovascular disease: A case for omega-3 index as a new risk factor. Pharmacol. Res. 2007, 55, 217–223. [Google Scholar] [CrossRef]
- Von Schacky, C.; Harris, W.S. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc. Res. 2007, 73, 310–315. [Google Scholar] [CrossRef]
- Leaf, A.; Xiao, Y.F.; Kang, J.X.; Billman, G.E. Prevention of sudden cardiac death by n-3 polyunsaturated fatty acids. Pharmacol. Ther. 2003, 98, 355–377. [Google Scholar] [CrossRef]
- Messerli, S.M.; Greenberg, R.M. Cnidarian toxins acting on voltage-gated ion channels. Mar. Drugs 2006, 4, 70–81. [Google Scholar] [CrossRef]
- Torres, M.; Aguilar, M.B.; Falcón, A.; Sánchez, L.; Radwan, F.F.; Burnett, J.W.; Heimer-de la Cotera, E.P.; Arellano, R.O. Electrophysiological and hemolytic activity elicited by the venom of the jellysh Cassiopea xamachana. Toxicon 2001, 39, 1297–1307. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Szczęsna-Antczak, M.; Antczak, T.; Piotrowicz-wasiak, M.; Rzyska, M.; Binkowska, N.; Bielecki, S. Relationships between lipases and lipids in mycelia of two mucor strains. Enzyme Microb. Technol. 2006, 39, 1214–1222. [Google Scholar] [CrossRef]
- Guaratini, T.; Cardozo, K.H.M.; Pinto, E.; Colepicolo, P. Comparison of diode array and electrochemical detection in the C30 reverse phase HPLC analysis of algae carotenoids. J. Braz. Chem. Soc. 2009, 20, 1609–1616. [Google Scholar] [CrossRef]
- Fraser, P.D.; Pinto, M.E.S.; Holloway, D.E.; Bramley, P.M. Application of high-performance liquid chromatography with photodiode array detection to the metabolic profiling of plant isoprenoids. Plant J. 2000, 24, 551–558. [Google Scholar] [CrossRef]
- Talà, A.; Lenucci, M.S.; Gaballo, A.; Durante, M.; Tredici, S.M.; Debowles, D.A.; Pizzolante, G.; Marcuccio, C.; Carata, E.; Piro, G.; et al. Sphingomonas cynarae sp. nov., a proteobacterium that produces an unusual type of sphingan. Int. J. Syst. Evol. Microbiol. 2013, 63, 72–79. [Google Scholar] [CrossRef]
- Huang, K.T.; Chen, Y.H.; Walker, A.M. Inaccuracies in MTS assays: Major distorting effects of medium, serum albumin, and fatty acids. Biotechniques 2004, 37, 406–412. [Google Scholar]
- Wang, P.; Henning, S.M.; Heber, D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS One 2010, 5, e10202. [Google Scholar] [CrossRef]
- El-Fouly, M.H.; Trosko, J.E.; Chang, C.-C. Scrape-loading and dye transfer: A rapid and simple technique to study gap junctional intercellular communication. Exp. Cell Res. 1987, 168, 422–430. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leone, A.; Lecci, R.M.; Durante, M.; Piraino, S. Extract from the Zooxanthellate Jellyfish Cotylorhiza tuberculata Modulates Gap Junction Intercellular Communication in Human Cell Cultures. Mar. Drugs 2013, 11, 1728-1762. https://doi.org/10.3390/md11051728
Leone A, Lecci RM, Durante M, Piraino S. Extract from the Zooxanthellate Jellyfish Cotylorhiza tuberculata Modulates Gap Junction Intercellular Communication in Human Cell Cultures. Marine Drugs. 2013; 11(5):1728-1762. https://doi.org/10.3390/md11051728
Chicago/Turabian StyleLeone, Antonella, Raffaella Marina Lecci, Miriana Durante, and Stefano Piraino. 2013. "Extract from the Zooxanthellate Jellyfish Cotylorhiza tuberculata Modulates Gap Junction Intercellular Communication in Human Cell Cultures" Marine Drugs 11, no. 5: 1728-1762. https://doi.org/10.3390/md11051728
APA StyleLeone, A., Lecci, R. M., Durante, M., & Piraino, S. (2013). Extract from the Zooxanthellate Jellyfish Cotylorhiza tuberculata Modulates Gap Junction Intercellular Communication in Human Cell Cultures. Marine Drugs, 11(5), 1728-1762. https://doi.org/10.3390/md11051728