Conotoxins that Confer Therapeutic Possibilities


Abstract
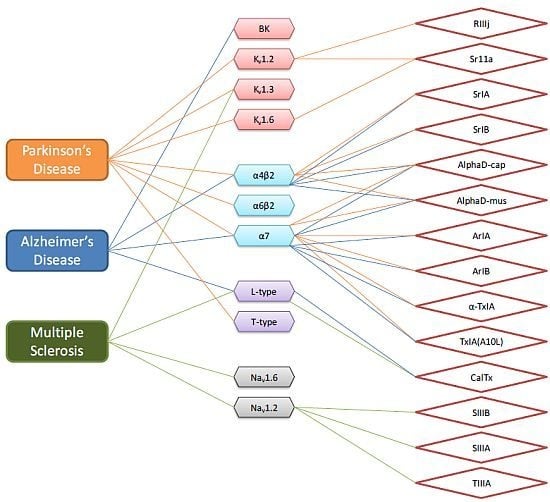
:
1. Introduction

2. Conus Peptides That Exhibit Therapeutic Potential
2.1. Voltage-Gated Ion Channels Targeted by Conotoxins
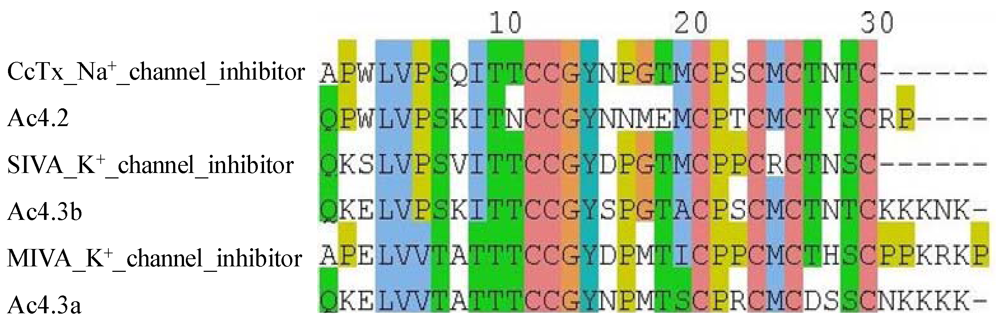
2.1.1. Na+ Channel Inhibitors

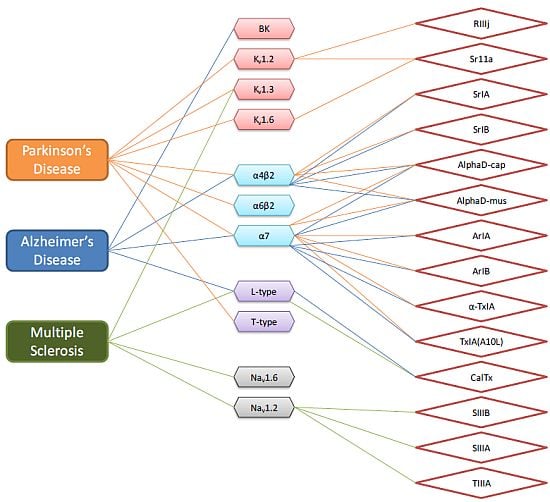{kind=link}
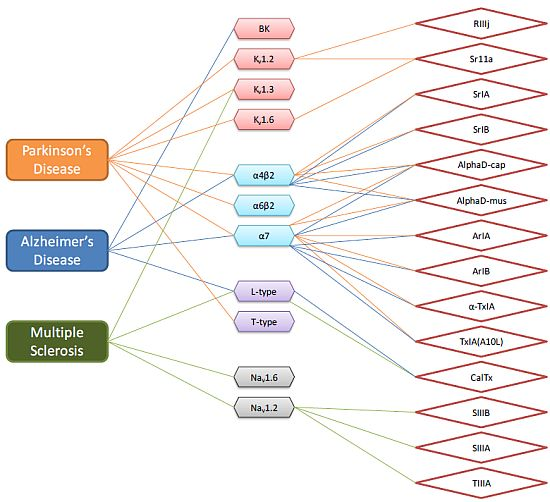{kind=link}
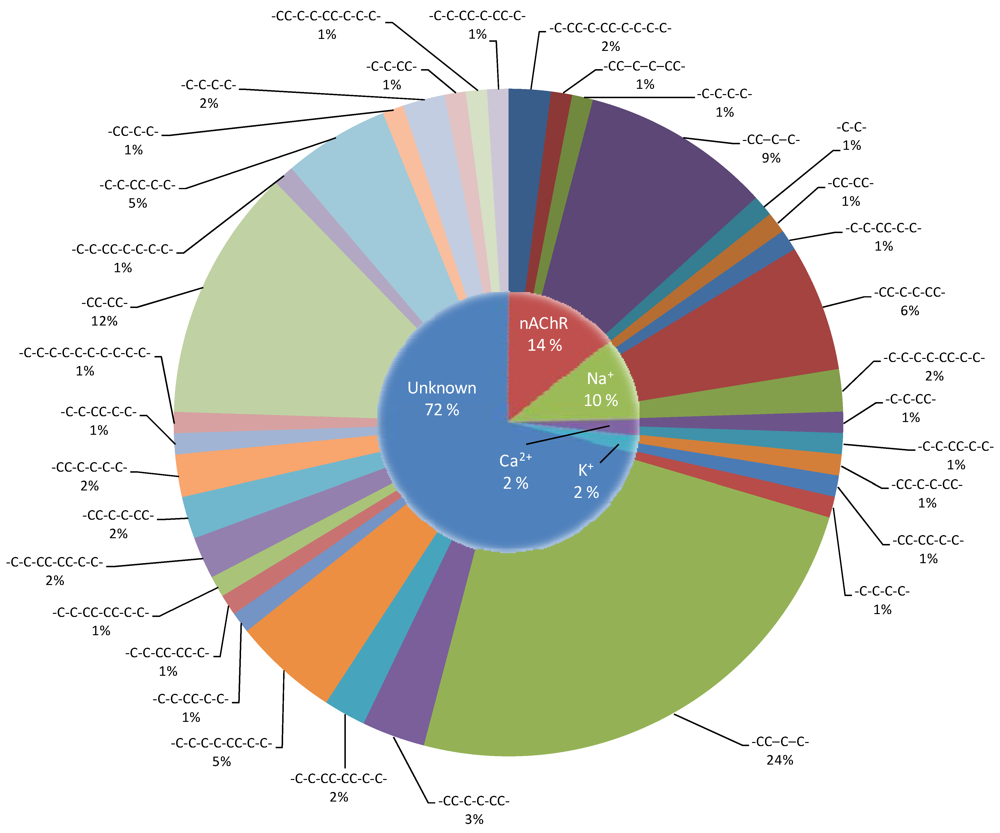{kind=link}
{kind=link}
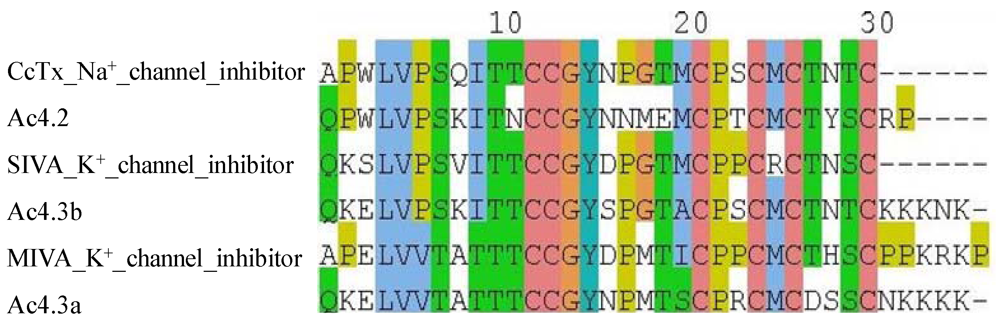{kind=link}
{kind=link}
| Peptide | AA Sequence | Gene Family with Cysteine Framework and Residues | Targets | Has no Effect on | Reference |
|---|---|---|---|---|---|
| Lt5d | DCCPAKLLCCNP | T superfamily V [connectivity I–III, II–IV] -CC-CC- | Na+ channel | ND | [42] |
| Lt6c | WPCKVAGSPCGLVSECCGTCNVLRNRCV | O1 superfamily VI/VII [connectivity I–IV, II–V, III–VI] -C-C-CC-C-C- | Na+ channel | ND | [43] |
| TIIIA | RHGCCKGOKGCSSRECRPQHCC | M superfamily III -CC-C-C-CC- | rNav1.2 rNav1.4 | rNav1.3 rNav1.5 rNav1.7 rNav1.8 | [44] |
| Cal12a | DVCDSLVGGHCIHNGCWCDQEAPHGNCCDTDGCTAAWWCPGTKWD | O2 superfamily XII -C-C-C-C-CC-C-C- | Na+ channel | ND | [45] |
| Cal12b | DVCDSLVGGHCIHNGCWCDQDAPHGNCCDTDGCTAAWWCPGTKWD | O2 superfamily XII -C-C-C-C-CC-C-C- | Na+ channel | ND | [45] |
| BuIIIA | VTDRCCKGKRECGRWCRDHSRCC | M superfamily III -CC-C-C-CC- | Nav1.4 | ND | [46] |
| BuIIIB | VGERCCKNGKRGCGRWCRDHSRCC | M superfamily III -CC-C-C-CC- | Nav1.4 | ND | [46] |
| BuIIIC | IVDRCCNKGNGKRGCSRWCRDHSRCC | M superfamily III -CC-C-C-CC- | Nav1.4 | ND | [46] |
| SIIIA | ZNCCNGGCSSKWCRDHARCC | M superfamily III -CC-C-C-CC- | rNav1.2 rNav1.4 | rNav1.3 rNav1.5 rNav1.7 rNav1.8 | [47] |
| SIIIB | ZNCCNGGCSSKWCKGHARCC | M superfamily III -CC-C-C-CC- | rNav1.2 rNav1.4 | rNav1.3 rNav1.5 rNav1.7 rNav1.8 | [47] |
2.1.2. Ca2+ Channel Inhibitors
| Peptide | AA Sequence | Gene Family with Cysteine Framework and Residues | Targets | Has no Effect on | Reference |
|---|---|---|---|---|---|
| CalTx | NCPAGCRSQGCCM | XVI -C-C-CC- | N-type L-type P/Q-type R-type | T-type | [49] |
| FVIA | CKGTGKSCSRIAYNCCTGSCRSGKC | O1 superfamily VI/VII [connectivity I–IV, II–V, III–VI] -C-C-CC-C-C- | N-type | T-type P/Q-type TTX-S Na+ channel | [48] |
2.1.3. K+ Channel Inhibitors
| Peptide | AA Sequence | Gene Family with Cysteine Framework and Residues | Targets | Has no Effect on | Reference |
|---|---|---|---|---|---|
| Sr11a | NQQCCWRSCCRGECEAPCRFGP | I2 superfamily XI [connectivity I–IV, II–VI, III–VII, V–VIII] -CC-CC-C-C- | Kv1.2 Kv1.6 | Kv1.3 | [50,51] |
| RIIIj | LPPCCTPPKKHCPAPACKYKPCCKS | M superfamily III -CC-C-C-CC- | Kv1.2 | Kv1.1 Kv1.3 Kv1.4 Kv1.5 Kv1.6 KCNQ2/KCNQ3 BK | [20] |
2.2. Ligand-Gated Ion Channels Targeted by Conotoxins
nAChR Inhibitors
| Peptide | AA Sequence | Gene Family with Cysteine Framework and Residues | nAChR Targets | Has no Effect on | Reference |
|---|---|---|---|---|---|
| AlphaD-cap | EVQECQVDTPGSSWGKCCMTRMCGTMCCSRSVCTCVYHWRRGHGCSCPG | D superfamily XX -C-CC-C-CC-C-C-C-C- | α7 α3β2 α4β2 | α3β4 α4β4 | [57] |
| AlphaD-mus | DVRECQVNTPGSKWGKCCMTRMCGTMCCARSGCTCVYHWRRGHGCSCPG | D superfamily XX -C-CC-C-CC-C-C-C-C- | α7 α3β2 α4β2 | α3β4 α4β4 | [57] |
| α-PIB | ZSOGCCWNPACVKNRC | A superfamily I [connectivity I–III, II–IV] -CC-C-C- | α1β1εδ α1β1γδ | α7 α3β4 α3β2 α2β4 α9α10 | [63] |
| SrIA | RTCCSROTCRMγYPγLCG | A superfamily I [connectivity I–III, II–IV] -CC-C-C- | α4β2 α1β1γδ | α3β4 | [64] |
| SrIB | RTCCSROTCRMEYPγLCG | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α4β2 α1β1γδ | α3β4 | [64] |
| Pu14a | DCPPHPVPGMHKCVCLKTC | A superfamily XIV [connectivity I–III, II–IV] -C-C-C-C- | α3β2 α6α3β2 α1β1γδ | K+ channels | [62] |
| PrIIIE | AARCCTYHGSCLKEKCRRKYCCGR | M superfamily III -CC-C-C-CC- | α1β1εδ α1β1γδ | Nav1.4 | [60] |
| ArIA | IRDECCSNPACRVNNOHVCRRR | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α7 α3β2 | ND | [68] |
| ArIB | DECCSNPACRVNNPHVCRRR | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α7 α3β2 | ND | [68] |
| Ac1.1a | NGRCCHPACGKHFNCGR | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α1β1γδ α1β1εδ α2β2 α3β4 α1γβ1 | ND | [66,67] |
| Ac1.1b | NGRCCHPACGKHFNCGR | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α1β1εδ α1β1γδ α2β2 α3β4 α1γβ1 | ND | [66,67] |
| PrXA | TYGIYDAKPOFSCAGLRGGCVLPONLROKFKE | -C-C- | α1β1εδ α1β1γδ | α7 α3β2 α3β4 α2β4 α4β2 α9 α10 Nav1.4 Nav1.6 NR2A NR2B | [59] |
| α-TxIA | GCCSRPPCIANNPDLC | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α7 α3β2 | α4β2 α1β1εδ α1β1γδ | [69] |
| TxIA(A10L) | GCCSRPPCILNNPDLC | A superfamily I [connectivity I-III, II-IV] -CC-C-C- | α7 α3β2 | α4β2 α1β1εδ α1β1γδ | [69] |
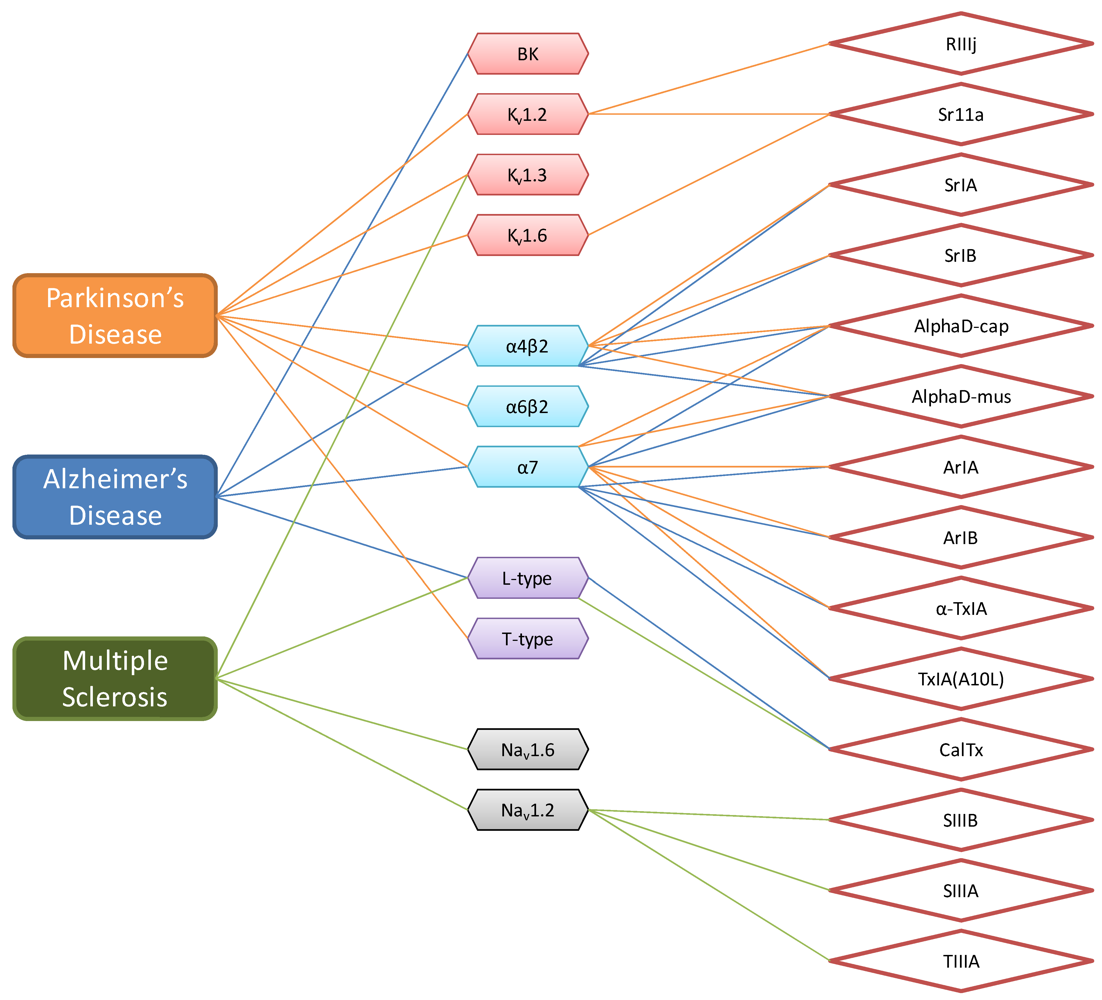
3. Prediction of Conotoxin Targets

4. Literature Analysis of Conotoxins Suggest Specific Therapeutic Potential

5. Concluding Remarks
References
- Olivera, B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef]
- Olivera, B.M.; Cruz, L.J. Conotoxins, in retrospect. Toxicon 2001, 39, 7–14. [Google Scholar] [CrossRef]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R.; et al. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar]
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among neuronal subtypes of voltage-activated sodium channels by mu-conotoxin piiia. J. Neurosci. 2000, 20, 76–80. [Google Scholar]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Ichida, S.; Abe, J.; Zhang, Y.A.; Sugihara, K.; Imoto, K.; Wada, T.; Fujita, N.; Sohma, H. Characteristics of the inhibitory effect of calmodulin on specific [125i]omega-conotoxin GVIA binding to crude membranes from chick brain. Neurochem. Res. 2000, 25, 1629–1635. [Google Scholar] [CrossRef]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef]
- Sher, E.; Gotti, C.; Canal, N.; Scoppetta, C.; Piccolo, G.; Evoli, A.; Clementi, F. Specificity of calcium channel autoantibodies in lambert-eaton myasthenic syndrome. Lancet 1989, 2, 640–643. [Google Scholar]
- Bowersox, S.S.; Luther, R. Pharmacotherapeutic potential of omega-conotoxin MVIIA (SNX-111), an N-type neuronal calcium channel blocker found in the venom of Conus magus. Toxicon 1998, 36, 1651–1658. [Google Scholar] [CrossRef]
- Barton, M.E.; White, H.S.; Wilcox, K.S. The effect of CGX-1007 and CI-1041, novel nmda receptor antagonists, on NMDA receptor-mediated EPSCs. Epilepsy Res. 2004, 59, 13–24. [Google Scholar] [CrossRef]
- Craig, A.G.; Norberg, T.; Griffin, D.; Hoeger, C.; Akhtar, M.; Schmidt, K.; Low, W.; Dykert, J.; Richelson, E.; Navarro, V.; et al. Contulakin-G, an O-glycosylated invertebrate neurotensin. J. Biol. Chem. 1999, 274, 13752–13759. [Google Scholar]
- Kern, S.E.; Allen, J.; Wagstaff, J.; Shafer, S.L.; Yaksh, T. The pharmacokinetics of the conopeptide contulakin-G (CGX-1160) after intrathecal administration: An analysis of data from studies in beagles. Anesth. Analg. 2007, 104, 1514–1520. [Google Scholar] [CrossRef]
- Lubbers, N.L.; Campbell, T.J.; Polakowski, J.S.; Bulaj, G.; Layer, R.T.; Moore, J.; Gross, G.J.; Cox, B.F. Postischemic administration of CGX-1051, a peptide from cone snail venom, reduces infarct size in both rat and dog models of myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 2005, 46, 141–146. [Google Scholar] [CrossRef]
- Livett, B.G.; Sandall, D.W.; Keays, D.; Down, J.; Gayler, K.R.; Satkunanathan, N.; Khalil, Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [Google Scholar] [CrossRef]
- Nielsen, C.K.; Lewis, R.J.; Alewood, D.; Drinkwater, R.; Palant, E.; Patterson, M.; Yaksh, T.L.; McCumber, D.; Smith, M.T. Anti-allodynic efficacy of the chi-conopeptide, Xen2174, in rats with neuropathic pain. Pain 2005, 118, 112–124. [Google Scholar] [CrossRef]
- Obata, H.; Conklin, D.; Eisenach, J.C. Spinal noradrenaline transporter inhibition by reboxetine and Xen2174 reduces tactile hypersensitivity after surgery in rats. Pain 2005, 113, 271–276. [Google Scholar] [CrossRef]
- Yan, L.D.; Liu, Y.L.; Zhang, L.; Dong, H.J.; Zhou, P.L.; Su, R.B.; Gong, Z.H.; Huang, P.T. Spinal antinociception of synthetic omega-conotoxin SO-3, a selective N-type neuronal voltage-sensitive calcium channel blocker, and its effects on morphine analgesia in chemical stimulus tests in rodent. Eur. J. Pharmacol. 2010, 636, 73–81. [Google Scholar] [CrossRef]
- Gasior, M.; White, N.A.; Rogawski, M.A. Prolonged attenuation of amygdala-kindled seizure measures in rats by convection-enhanced delivery of the N-type calcium channel antagonists omega-conotoxin GVIA and omega-conotoxin MVIIA. J. Pharmacol. Exp. Ther. 2007, 323, 458–468. [Google Scholar] [CrossRef]
- Shahlaie, K.; Lyeth, B.G.; Gurkoff, G.G.; Muizelaar, J.P.; Berman, R.F. Neuroprotective effects of selective N-type VGCC blockade on stretch-injury-induced calcium dynamics in cortical neurons. J. Neurotrauma 2010, 27, 175–187. [Google Scholar] [CrossRef]
- Chen, P.; Dendorfer, A.; Finol-Urdaneta, R.K.; Terlau, H.; Olivera, B.M. Biochemical characterization of kappam-RIIIJ, a Kv1.2 channel blocker: Evaluation of cardioprotective effects of kappam-conotoxins. J. Biol. Chem. 2010, 285, 14882–14889. [Google Scholar]
- Lahiry, A.; Dave, K. Conotoxins: Review and docking studies to determine potentials of conotoxin as an anticancer drug molecule. Curr. Top. Med. Chem. 2012, in press.. [Google Scholar]
- Waxman, S.G. Axonal conduction and injury in multiple sclerosis: The role of sodium channels. Nat. Rev. Neurosci. 2006, 7, 932–941. [Google Scholar] [CrossRef]
- Haydar, S.N.; Dunlop, J. Neuronal nicotinic acetylcholine receptors—Targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top. Med. Chem. 2010, 10, 144–152. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2010, 38, D5–D16. [Google Scholar]
- French, R.J.; Terlau, H. Sodium channel toxins—Receptor targeting and therapeutic potential. Curr. Med. Chem. 2004, 11, 3053–3064. [Google Scholar]
- Wood, J.N.; Boorman, J.P.; Okuse, K.; Baker, M.D. Voltage-gated sodium channels and pain pathways. J. Neurobiol. 2004, 61, 55–71. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar]
- Xu, R.; Thomas, E.A.; Jenkins, M.; Gazina, E.V.; Chiu, C.; Heron, S.E.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol. Cell. Neurosci. 2007, 35, 292–301. [Google Scholar] [CrossRef]
- Catterall, W.A.; Kalume, F.; Oakley, J.C. Nav1.1 channels and epilepsy. J. Physiol. 2010, 588, 1849–1859. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef]
- Mani, B.K.; Brueggemann, L.I.; Cribbs, L.L.; Byron, K.L. Activation of vascular KCNQ (Kv7) potassium channels reverses spasmogen-induced constrictor responses in rat basilar artery. Br. J. Pharmacol. 2011, 164, 237–249. [Google Scholar] [CrossRef]
- Takeda, M.; Tanimoto, T.; Nasu, M.; Matsumoto, S. Temporomandibular joint inflammation decreases the voltage-gated K+ channel subtype 1.4-immunoreactivity of trigeminal ganglion neurons in rats. Eur. J. Pain 2008, 12, 189–195. [Google Scholar] [CrossRef]
- Martel, P.; Leo, D.; Fulton, S.; Berard, M.; Trudeau, L.E. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS One 2011, 6. [Google Scholar]
- Cahalan, M.D.; Chandy, K.G. Ion channels in the immune system as targets for immunosuppression. Curr. Opin. Biotechnol. 1997, 8, 749–756. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, W.P.; Feng, N.; Wang, L.; Wang, X.L. Donepezil attenuated oxygen-glucose deprivation insult by blocking Kv2.1 potassium channels. Eur. J. Pharmacol. 2011, 657, 76–83. [Google Scholar] [CrossRef]
- MacDonald, P.E.; Sewing, S.; Wang, J.; Joseph, J.W.; Smukler, S.R.; Sakellaropoulos, G.; Wang, J.; Saleh, M.C.; Chan, C.B.; Tsushima, R.G.; et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cells enhances glucose-dependent insulin secretion. J. Biol. Chem. 2002, 277, 44938–44945. [Google Scholar]
- Ellison, D.H. The voltage-gated K+ channel subunit Kv1.1 links kidney and brain. J. Clin. Invest. 2009, 119, 763–766. [Google Scholar] [CrossRef]
- Su, X.; Leon, L.A.; Laping, N.J. Role of spinal Cav2.2 and Cav2.1 ion channels in bladder nociception. J. Urol. 2008, 179, 2464–2469. [Google Scholar] [CrossRef]
- Ilijic, E.; Guzman, J.N.; Surmeier, D.J. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 43, 364–371. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Lin-Ye, A.; Baird, M.A.; Lei, D.; Bao, J. Neuroprotective effects of blockers for T-type calcium channels. Mol. Neurodegener. 2009, 4. [Google Scholar]
- Jing, X.; Li, D.Q.; Olofsson, C.S.; Salehi, A.; Surve, V.V.; Caballero, J.; Ivarsson, R.; Lundquist, I.; Pereverzev, A.; Schneider, T.; et al. Cav2.3 calcium channels control second-phase insulin release. J. Clin. Invest. 2005, 115, 146–154. [Google Scholar]
- Liu, J.; Wu, Q.; Pi, C.; Zhao, Y.; Zhou, M.; Wang, L.; Chen, S.; Xu, A. Isolation and characterization of a T-superfamily conotoxin from Conus litteratus with targeting tetrodotoxin-sensitive sodium channels. Peptides 2007, 28, 2313–2319. [Google Scholar] [CrossRef]
- Wang, L.; Pi, C.; Liu, J.; Chen, S.; Peng, C.; Sun, D.; Zhou, M.; Xiang, H.; Ren, Z.; Xu, A. Identification and characterization of a novel O-superfamily conotoxin from Conus litteratus. J. Pept. Sci. 2008, 14, 1077–1083. [Google Scholar] [CrossRef]
- Lewis, R.J.; Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Loughnan, M.; Thomas, L.; Adams, D.A.; Drinkwater, R.; Adams, D.J.; Alewood, P.F. Isolation and structure-activity of mu-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol. Pharmacol. 2007, 71, 676–685. [Google Scholar]
- Gilly, W.F.; Richmond, T.A.; Duda, T.F., Jr.; Elliger, C.; Lebaric, Z.; Schulz, J.; Bingham, J.P.; Sweedler, J.V. A diverse family of novel peptide toxins from an unusual cone snail, Conus californicus. J. Exp. Biol. 2011, 214, 147–161. [Google Scholar]
- Holford, M.; Zhang, M.M.; Gowd, K.H.; Azam, L.; Green, B.R.; Watkins, M.; Ownby, J.P.; Yoshikami, D.; Bulaj, G.; Olivera, B.M. Pruning nature: Biodiversity-derived discovery of novel sodium channel blocking conotoxins from Conus bullatus. Toxicon 2009, 53, 90–98. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Adams, D.; Loughnan, M.L.; Thomas, L.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Neuronally micro-conotoxins from Conus striatus utilize an alpha-helical motif to target mammalian sodium channels. J. Biol. Chem. 2008, 283, 21621–21628. [Google Scholar]
- Lee, S.; Kim, Y.; Back, S.K.; Choi, H.W.; Lee, J.Y.; Jung, H.H.; Ryu, J.H.; Suh, H.W.; Na, H.S.; Kim, H.J.; et al. Analgesic effect of highly reversible omega-conotoxin fvia on N type Ca2+ channels. Mol. Pain 2010, 6. [Google Scholar]
- Bernaldez, J.; Lopez, O.; Licea, A.; Salceda, E.; Arellano, R.O.; Vega, R.; Soto, E. Electrophysiological characterization of a novel small peptide from the venom of Conus californicus that targets voltage-gated neuronal Ca2+ channels. Toxicon 2011, 57, 60–67. [Google Scholar] [CrossRef]
- Aguilar, M.B.; Lopez-Vera, E.; de la Cotera, E.P.H.; Falcon, A.; Olivera, B.M.; Maillo, M. I-conotoxins in vermivorous species of the west atlantic: Peptide sr11a from Conus spurius. Peptides 2007, 28, 18–23. [Google Scholar] [CrossRef]
- Aguilar, M.B.; Perez-Reyes, L.I.; Lopez, Z.; de la Cotera, E.P.H.; Falcon, A.; Ayala, C.; Galvan, M.; Salvador, C.; Escobar, L.I. Peptide sr11a from Conus spurius is a novel peptide blocker for Kv1 potassium channels. Peptides 2010, 31, 1287–1291. [Google Scholar] [CrossRef]
- Quik, M.; Wonnacott, S. α6β2* and α4β2* nicotinic acetylcholine receptors as drug targets for Parkinson’s disease. Pharmacol. Rev. 2011, 63, 938–966. [Google Scholar] [CrossRef]
- Tong, M.; Arora, K.; White, M.M.; Nichols, R.A. Role of key aromatic residues in the ligand-binding domain of α7 nicotinic receptors in the agonist action of β-amyloid. J. Biol. Chem. 2011, 286, 34373–34381. [Google Scholar]
- Marquis, K.L.; Comery, T.A.; Jow, F.; Navarra, R.L.; Grauer, S.M.; Pulicicchio, C.; Kelley, C.; Brennan, J.A.; Roncarati, R.; Scali, C.; et al. Preclinical assessment of an adjunctive treatment approach for cognitive impairment associated with schizophrenia using the alpha7 nicotinic acetylcholine receptor agonist WYE-103914/SEN34625. Psychopharmacology (Berl.) 2011, 218, 635–647. [Google Scholar] [CrossRef]
- Raffa, R.B. Cancer “survivor-care”: I. The alpha7 nachr as potential target for chemotherapy-related cognitive impairment. J. Clin. Pharm. Ther. 2010, 36, 437–445. [Google Scholar] [CrossRef]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880–17884. [Google Scholar]
- Kauferstein, S.; Kendel, Y.; Nicke, A.; Coronas, F.I.; Possani, L.D.; Favreau, P.; Krizaj, I.; Wunder, C.; Kauert, G.; Mebs, D. New conopeptides of the D-superfamily selectively inhibiting neuronal nicotinic acetylcholine receptors. Toxicon 2009, 54, 295–301. [Google Scholar] [CrossRef]
- Loughnan, M.; Nicke, A.; Jones, A.; Schroeder, C.I.; Nevin, S.T.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Identification of a novel class of nicotinic receptor antagonists: Dimeric conotoxins VxXIIA, VxXIIB, and VxXIIC from Conus vexillu. J. Biol. Chem. 2006, 281, 24745–24755. [Google Scholar]
- Jimenez, E.C.; Olivera, B.M.; Teichert, R.W. Alphac-conotoxin PrXA: A new family of nicotinic acetylcholine receptor antagonists. Biochemistry 2007, 46, 8717–8724. [Google Scholar] [CrossRef]
- Lluisma, A.O.; Lopez-Vera, E.; Bulaj, G.; Watkins, M.; Olivera, B.M. Characterization of a novel psi-conotoxin from Conus parius reeve. Toxicon 2008, 51, 174–180. [Google Scholar] [CrossRef]
- Shon, K.J.; Grilley, M.; Jacobsen, R.; Cartier, G.E.; Hopkins, C.; Gray, W.R.; Watkins, M.; Hillyard, D.R.; Rivier, J.; Torres, J.; et al. A noncompetitive peptide inhibitor of the nicotinic acetylcholine receptor from Conus purpurascens venom. Biochemistry 1997, 36, 9581–9587. [Google Scholar]
- Peng, C.; Ye, M.; Wang, Y.; Shao, X.; Yuan, D.; Liu, J.; Hawrot, E.; Wang, C.; Chi, C. A new subfamily of conotoxins belonging to the A-superfamily. Peptides 2010, 31, 2009–2016. [Google Scholar]
- Lopez-Vera, E.; Jacobsen, R.B.; Ellison, M.; Olivera, B.M.; Teichert, R.W. A novel alpha conotoxin (alpha-PIB) isolated from C. purpurascens is selective for skeletal muscle nicotinic acetylcholine receptors. Toxicon 2007, 49, 1193–1199. [Google Scholar] [CrossRef]
- Lopez-Vera, E.; Aguilar, M.B.; Schiavon, E.; Marinzi, C.; Ortiz, E.; Restano Cassulini, R.; Batista, C.V.; Possani, L.D.; de la Cotera, E.P.H.; Peri, F.; et al. Novel alpha-conotoxins from Conus spurius and the alpha-conotoxin ei share high-affinity potentiation and low-affinity inhibition of nicotinic acetylcholine receptors. FEBS J. 2007, 274, 3972–3985. [Google Scholar]
- Park, K.H.; Suk, J.E.; Jacobsen, R.; Gray, W.R.; McIntosh, J.M.; Han, K.H. Solution conformation of alpha-conotoxin EI, a neuromuscular toxin specific for the alpha 1/delta subunit interface of torpedo nicotinic acetylcholine receptor. J. Biol. Chem. 2001, 276, 49028–49033. [Google Scholar]
- Yuan, D.D.; Han, Y.H.; Wang, C.G.; Chi, C.W. From the identification of gene organization of alpha conotoxins to the cloning of novel toxins. Toxicon 2007, 49, 1135–1149. [Google Scholar] [CrossRef]
- Liu, L.; Chew, G.; Hawrot, E.; Chi, C.; Wang, C. Two potent alpha3/5 conotoxins from piscivorous Conus achatinus. Acta Biochim. Biophys. Sin. (Shanghai) 2007, 39, 438–444. [Google Scholar] [CrossRef]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar]
- Dutertre, S.; Ulens, C.; Buttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. Achbp-targeted alpha-conotoxin correlates distinct binding orientations with nachr subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Ramoz, N.; Boni, C.; Downing, A.M.; Close, S.L.; Peters, S.L.; Prokop, A.M.; Allen, A.J.; Hamon, M.; Purper-Ouakil, D.; Gorwood, P. A haplotype of the norepinephrine transporter (Net) gene Slc6a2 is associated with clinical response to atomoxetine in attention-deficit hyperactivity disorder (ADHD). Neuropsychopharmacology 2009, 34, 2135–2142. [Google Scholar] [CrossRef]
- Brust, A.; Palant, E.; Croker, D.E.; Colless, B.; Drinkwater, R.; Patterson, B.; Schroeder, C.I.; Wilson, D.; Nielsen, C.K.; Smith, M.T.; et al. chi-Conopeptide pharmacophore development: Toward a novel class of norepinephrine transporter inhibitor (Xen2174) for pain. J. Med. Chem. 2009, 52, 6991–7002. [Google Scholar]
- Kits, K.S.; Lodder, J.C.; van der Schors, R.C.; Li, K.W.; Geraerts, W.P.; Fainzilber, M. Novel omega-conotoxins block dihydropyridine-insensitive high voltage-activated calcium channels in molluscan neurons. J. Neurochem. 1996, 67, 2155–2163. [Google Scholar]
- Kuo, I.Y.; Ellis, A.; Seymour, V.A.; Sandow, S.L.; Hill, C.E. Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J. Cereb. Blood Flow Metab. 2010, 30, 1226–1239. [Google Scholar] [CrossRef]
- Cummings, J.L.; Vinters, H.V.; Cole, G.M.; Khachaturian, Z.S. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51, S2–S17, discussion S65–S17.. [Google Scholar]
- Mousavi, M.; Hellstrom-Lindahl, E.; Guan, Z.Z.; Shan, K.R.; Ravid, R.; Nordberg, A. Protein and mrna levels of nicotinic receptors in brain of tobacco using controls and patients with Alzheimer’s disease. Neuroscience 2003, 122, 515–520. [Google Scholar] [CrossRef]
- Yuan, D.D.; Liu, L.; Shao, X.X.; Peng, C.; Chi, C.W.; Guo, Z.Y. New conotoxins define the novel I3-superfamily. Peptides 2009, 30, 861–865. [Google Scholar] [CrossRef]
- Srivareerat, M.; Tran, T.T.; Salim, S.; Aleisa, A.M.; Alkadhi, K.A. Chronic nicotine restores normal abeta levels and prevents short-term memory and e-ltp impairment in abeta rat model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 834–844. [Google Scholar] [CrossRef]
- Mousavi, M.; Hellstrom-Lindahl, E. Nicotinic receptor agonists and antagonists increase sappalpha secretion and decrease abeta levels in vitro. Neurochem. Int. 2009, 54, 237–244. [Google Scholar] [CrossRef]
- Kim, S.; Rhim, H. Effects of amyloid-beta peptides on voltage-gated l-type Cav1.2 and Cav1.3 Ca2+ channels. Mol. Cells 2011, 32, 289–294. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590. [Google Scholar]
- Ye, H.; Jalini, S.; Mylvaganam, S.; Carlen, P. Activation of large-conductance Ca2+-activated K+ channels depresses basal synaptic transmission in the hippocampal CA1 area in APP (swe/ind) TgCRND8 mice. Neurobiol. Aging 2010, 31, 591–604. [Google Scholar] [CrossRef]
- Meyer, A.K.; Maisel, M.; Hermann, A.; Stirl, K.; Storch, A. Restorative approaches in Parkinson’s disease: Which cell type wins the race? J. Neurol. Sci. 2010, 289, 93–103. [Google Scholar] [CrossRef]
- Perez, X.A.; Bordia, T.; McIntosh, J.M.; Quik, M. Alpha6ss2* and alpha4ss2* nicotinic receptors both regulate dopamine signaling with increased nigrostriatal damage: Relevance to Parkinson’s disease. Mol. Pharmacol. 2010, 78, 971–980. [Google Scholar] [CrossRef]
- Kawamata, J.; Shimohama, S. Stimulating nicotinic receptors trigger multiple pathways attenuating cytotoxicity in models of Alzheimer’s and Parkinson’s diseases. J. Alzheimers Dis. 2011, 24, 95–109. [Google Scholar]
- Tai, C.H.; Yang, Y.C.; Pan, M.K.; Huang, C.S.; Kuo, C.C. Modulation of subthalamic T-type Ca2+ channels remedies locomotor deficits in a rat model of Parkinson disease. J. Clin. Invest. 2011, 121, 3289–3305. [Google Scholar] [CrossRef]
- Liu, X.K.; Wang, G.; Chen, S.D. Modulation of the activity of dopaminergic neurons by SK channels: A potential target for the treatment of Parkinson’s disease? Neurosci. Bull. 2010, 26, 265–271. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar]
- Craner, M.J.; Damarjian, T.G.; Liu, S.; Hains, B.C.; Lo, A.C.; Black, J.A.; Newcombe, J.; Cuzner, M.L.; Waxman, S.G. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 2005, 49, 220–229. [Google Scholar] [CrossRef]
- Brand-Schieber, E.; Werner, P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp. Neurol. 2004, 189, 5–9. [Google Scholar] [CrossRef]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J. Clin. Invest. 2003, 111, 1703–1713. [Google Scholar]
- Shimohama, S. Nicotinic receptor-mediated neuroprotection in neurodegenerative disease models. Biol. Pharm. Bull. 2009, 32, 332–336. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Essack, M.; Bajic, V.B.; Archer, J.A.C. Conotoxins that Confer Therapeutic Possibilities. Mar. Drugs 2012, 10, 1244-1265. https://doi.org/10.3390/md10061244
Essack M, Bajic VB, Archer JAC. Conotoxins that Confer Therapeutic Possibilities. Marine Drugs. 2012; 10(6):1244-1265. https://doi.org/10.3390/md10061244
Chicago/Turabian StyleEssack, Magbubah, Vladimir B. Bajic, and John A. C. Archer. 2012. "Conotoxins that Confer Therapeutic Possibilities" Marine Drugs 10, no. 6: 1244-1265. https://doi.org/10.3390/md10061244
APA StyleEssack, M., Bajic, V. B., & Archer, J. A. C. (2012). Conotoxins that Confer Therapeutic Possibilities. Marine Drugs, 10(6), 1244-1265. https://doi.org/10.3390/md10061244