Purpurogemutantin and Purpurogemutantidin, New Drimenyl Cyclohexenone Derivatives Produced by a Mutant Obtained by Diethyl Sulfate Mutagenesis of a Marine-Derived Penicillium purpurogenum G59

 ,
, 
Abstract
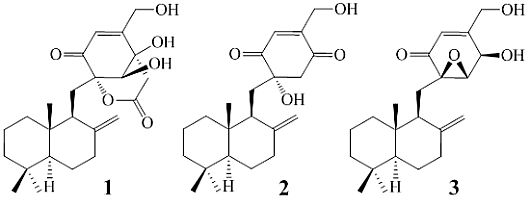
:
1. Introduction

2. Results and Discussion
 +25.7 (c 1.0, MeOH), afforded a molecular weight of 360 Dalton by positive and negative ESI-MSs and was identified as macrophorin A [24] according to the physicochemical and spectroscopic data. Full 1H NMR data of 3are reported for the first time.
+25.7 (c 1.0, MeOH), afforded a molecular weight of 360 Dalton by positive and negative ESI-MSs and was identified as macrophorin A [24] according to the physicochemical and spectroscopic data. Full 1H NMR data of 3are reported for the first time.2.1. Structure Determination of 1 and 2
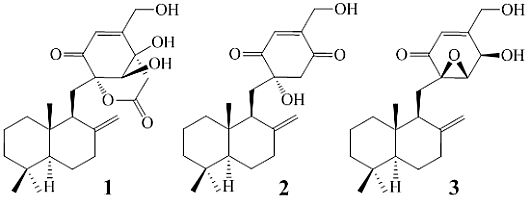
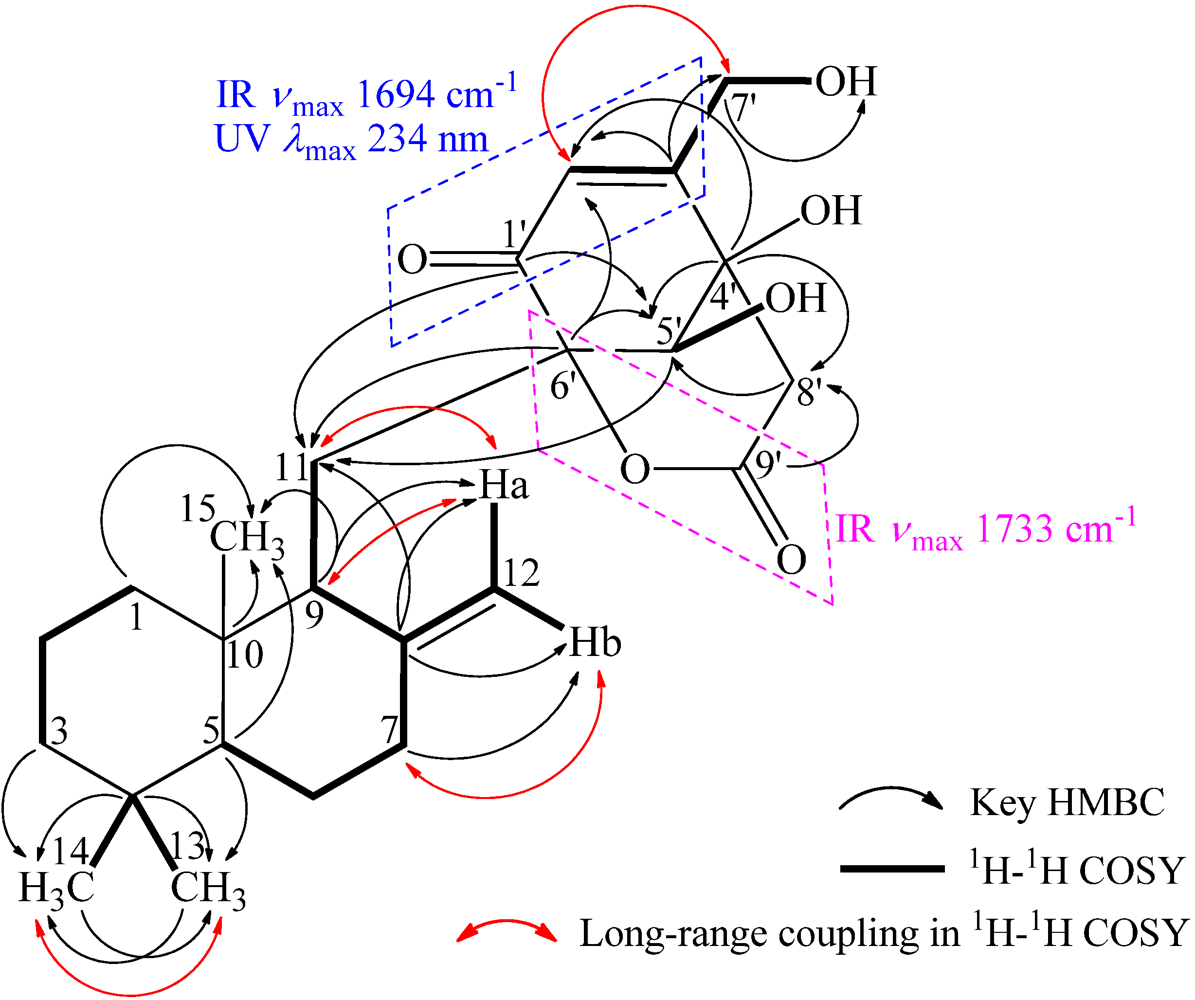
+21.0 (c 1.0, MeOH), and its molecular formula C24H34O6 was determined by HRESIMS (m/z 419.2431 [M + H]+; Δ = +0.3 mmu). Its UV (λmax 234 nm, log ε 3.87) and IR (νmax 1694, 889 cm−1) absorptions revealed an α,β-unsaturated ketone chromophore [24] in 1. The IR spectrum of 1 further indicated the presence of hydroxyl (3405 cm−1) and ester carbonyl (1733 cm−1) groups. The 1H NMR spectrum of 1 in acetone-d6 showed signals due to three tert-methyl groups, three olefinic and three exchangeable protons together with several methine and methylene proton signals (Table 1). The 13C NMR spectrum in acetone-d6, analyzed by DEPT, revealed the presence of a conjugated ketone carbonyl, an ester carbonyl, two sp2 and four sp3 quaternary carbons, one sp2 and three sp3 methine, one sp2 and eight sp3 methylene, and three methyl groups in 1 (Table 1). The ESIMS, HRESIMS, UV, IR, 1H and 13C NMR, DEPT, 1H–1H COSY, HMQC, HMBC, and NOESY spectra of 1 are given as supporting information in Supplementary Data S1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δCb,c | δHb (J in Hz) | COSY d | NOE e | HMBC f |
|---|---|---|---|---|---|
| 1 | 39.3 t | Ha
1.12 td (13.7, 3.4) H e 1.723 br d (13.7) | He-1, H2-2 Ha-1, Ha-2 | He-1, He-2, Ha-2 g, Ha-7, H-9 Ha-1, H2-2,Ha-11, H3-15 | C-3,5 |
| 2 | 19.9 t | Ha 1.58 qt (13.7, 3.4) He 1.45 dquint (13.7, 3.4) | H2-1, He-2, H2-3 Ha-1, Ha-2, H2-3 | He-1, Ha-1 g, H3-14, H3-15 | |
| 3 | 42.8 t | Ha 1.14 td (13.7, 3.4) He 1.36 dt (13.7, 3.4) | H2-2, He-3 H2-2, Ha-3 | He-3, H3-13 H3-13, H3-14 | C-14 C-1,5 |
| 4 | 34.2 s | — | — | — | — |
| 5 | 56.2 d | 1.17 dd (12.9, 2.5) | H2-6 | He-6, Ha-7, H-9, H3-13 | C-4,6,9,10,14,15 |
| 6 | 25.2 t | Ha 1.31 qd (12.9, 3.9) He 1.728 br d (12.9) | H-5, He-6, H2-7 H-5, Ha-6, Ha-7 | He-6, He-7, H3-14, H3-15 H-5, Ha-6, H2-7, H3-13 | |
| 7 | 38.8 t | Ha 2.11 td (12.9, 4.8) He 2.36 ddd (12.9, 3.9, 2.5) | H2-6, He-7, Ha-12 H2-6, Ha-7 | H-5, He-6, He-7 H2-6, Ha-7, Hb-12 | C-8,12 C-5,9,12 |
| 8 | 150.1 s | — | — | — | — |
| 9 | 50.2 d | 2.03–1.95 AB type | H2-11, H2-12 | Ha-1, H-5, H-5′ | C-8,10,11,6′ |
| 10 | 41.0 s | — | — | — | — |
| 11 | 22.2 t | Ha 2.20 dd (14.8, 4.6) Hb 2.03–1.95 AB type | H-9, Hb-11, Ha-12 H-9, Ha-11, Hb-12 | He-1, Hb-11, H3-15, H-5′ Ha-11, Ha-12, H3-15, H-5′ | C-8,9,10,1′,5′,6′ C-8,9,10 |
| 12 | 108.0 t | Ha 4.90 br s Hb 4.79 br s | Ha-7, H-9, Ha-11, Hb-12 H-9, Hb-11, Ha-12 | Hb-11, Hb-12, H3-15, H-5′ He-7, Ha-12 | C-7,8,9 C-7,9 |
| 13 | 33.8 q | 0.84 3H, s | H3-14 | H2-3, H-5, He-6 | C-3,4,5,14 |
| 14 | 22.0 q | 0.78 3H, s | H3-13 | Ha-2, He-3, Ha-6, H3-15 | C-3,4,5,13 |
| 15 | 15.1 q | 0.70 3H, s | He-1, Ha-2, Ha-6, H2-11, Ha-12, H3-14 | C-1,5,9,10 | |
| 1′ | 192.3 s | — | — | — | — |
| 2′ | 120.4 d | 6.12 br s | H2-7′ | H2-7′ | C-3′,4′,6′,7′ |
| 3′ | 164.5 s | — | — | — | — |
| 4′ | 71.8 s | — | — | — | — |
| 5′ | 74.7 d | 3.96 s | HO-5′ | H-9, Hb-11, Ha-12, Ha-8′ | C-11,1′,3′,4′,6′,8′,7′ |
| 6′ | 85.2 s | — | — | — | — |
| 7′ | 60.7 t | 4.41 2H, br s | H-2′, HO-7′ | H-2′, He-8′ | C-2′,3′ |
| 8′ | 43.3 t | Ha 2.91 d (17.2)He 3.05 d (17.2) | He-8′ Ha-8′ | H-5′H2-7′ | C-3′,4′,5′,9′ C-3′,4′,5′,9′ |
| 9′ | 167.8 s | — | — | — | — |
| 4′-OH | — | 4.90 br s | |||
| 5′-OH | — | 4.73 br s | H-5′ | ||
| 7′-OH | — | 4.34 br s | H2-7′ | C-7′ |

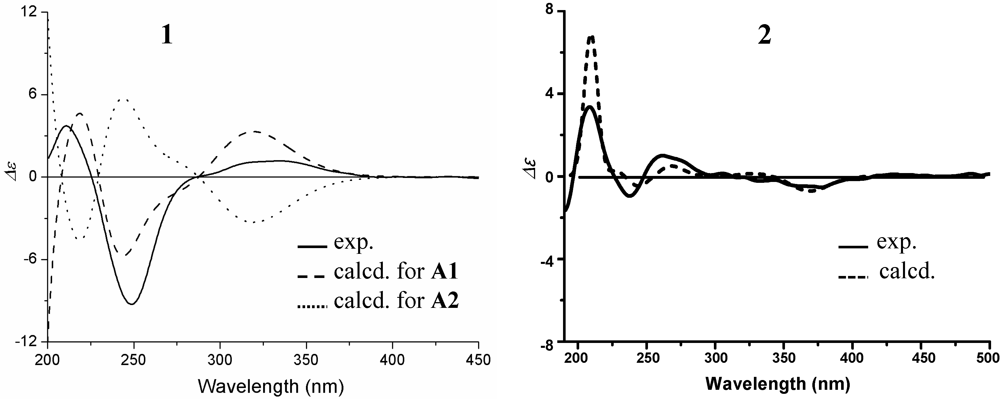
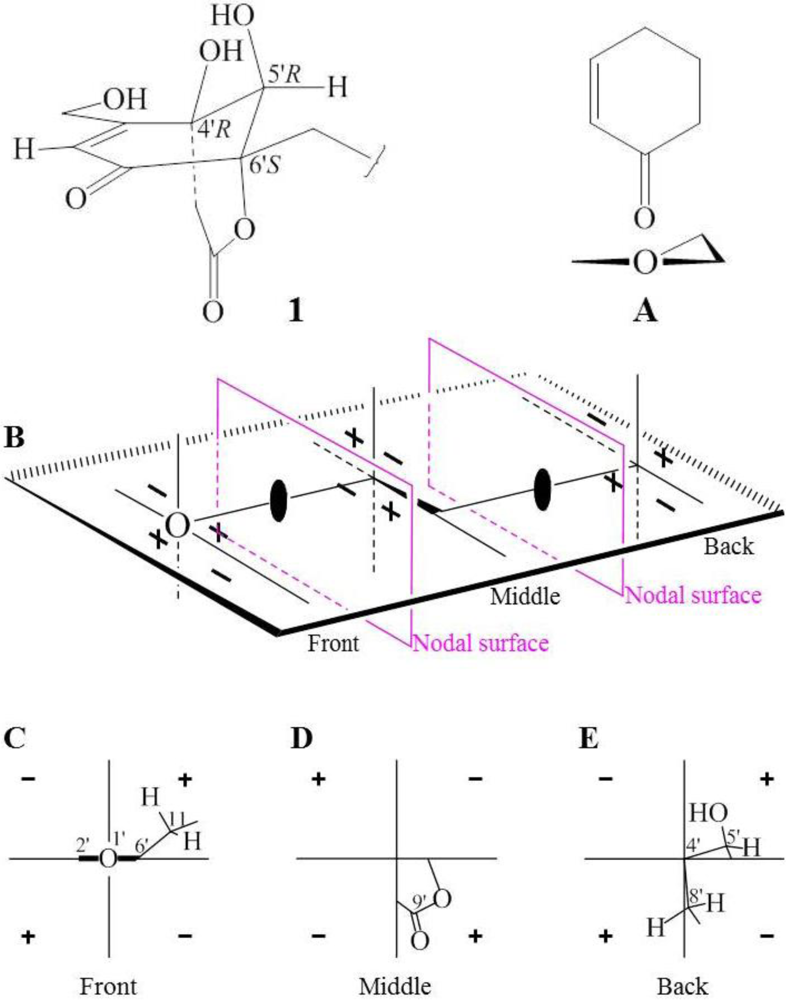
 +15.1 for A1 and +42.8 for B2, respectively. The calculated value of A1 much better matched the measured +21.0 (1.0, MeOH) of 1. Thus, the absolute configuration of 1 was assigned as 5S9S10S4′R5′R6′S.
+15.1 for A1 and +42.8 for B2, respectively. The calculated value of A1 much better matched the measured +21.0 (1.0, MeOH) of 1. Thus, the absolute configuration of 1 was assigned as 5S9S10S4′R5′R6′S.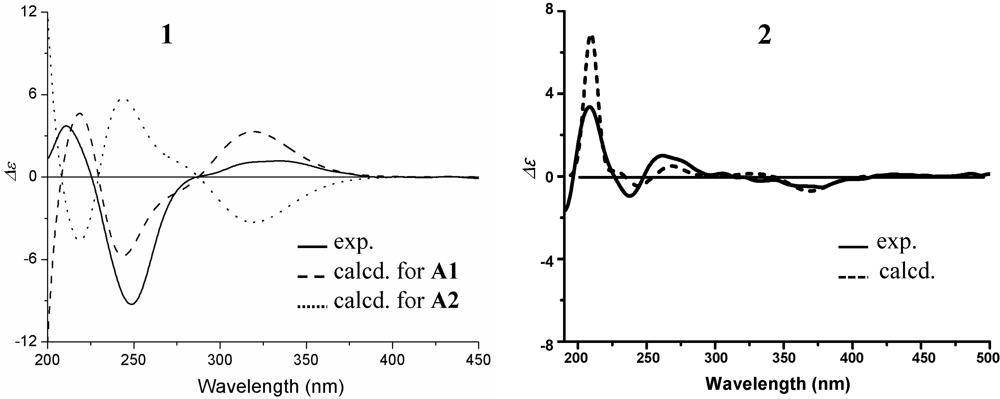
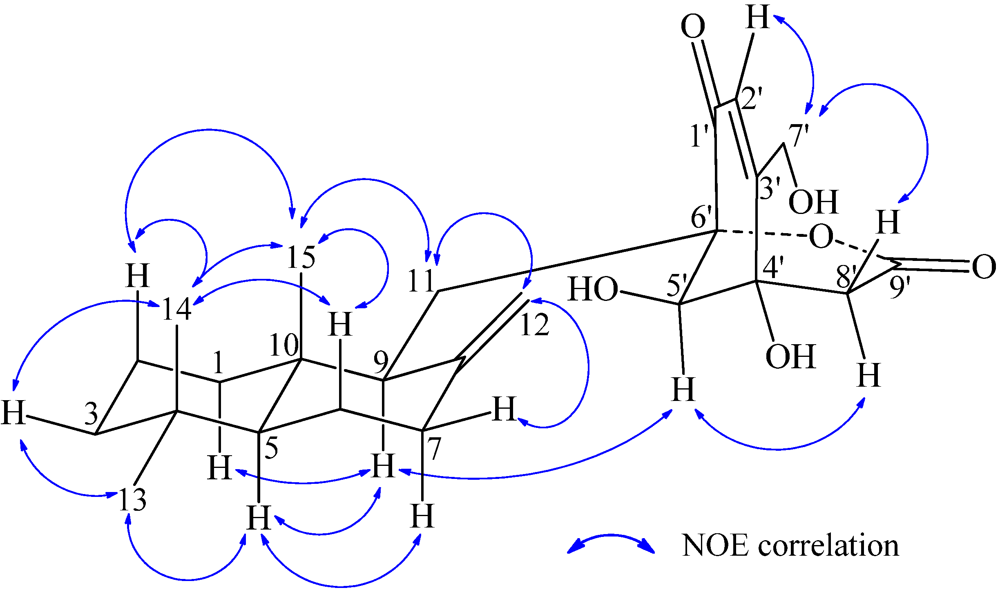
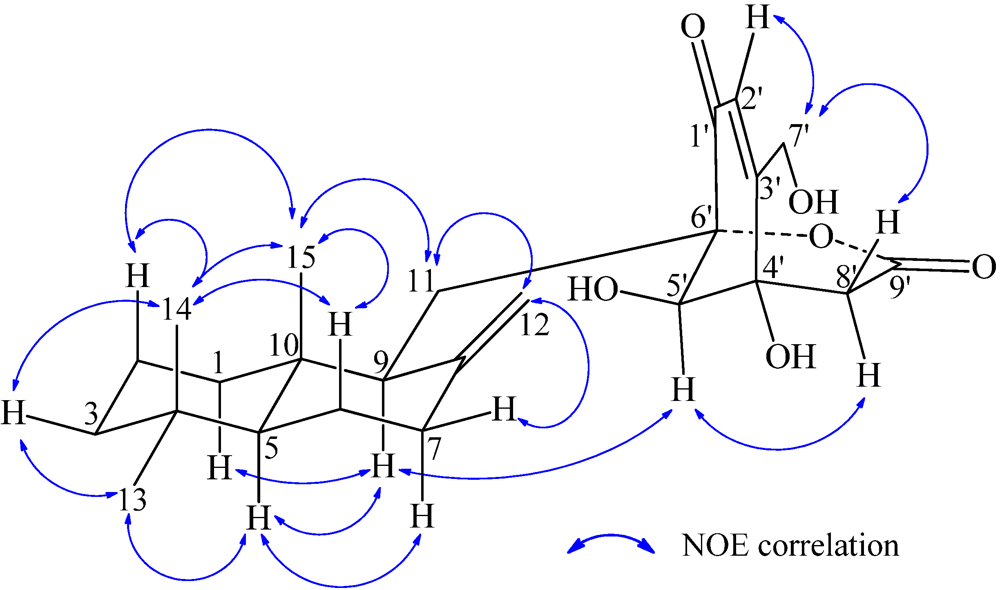
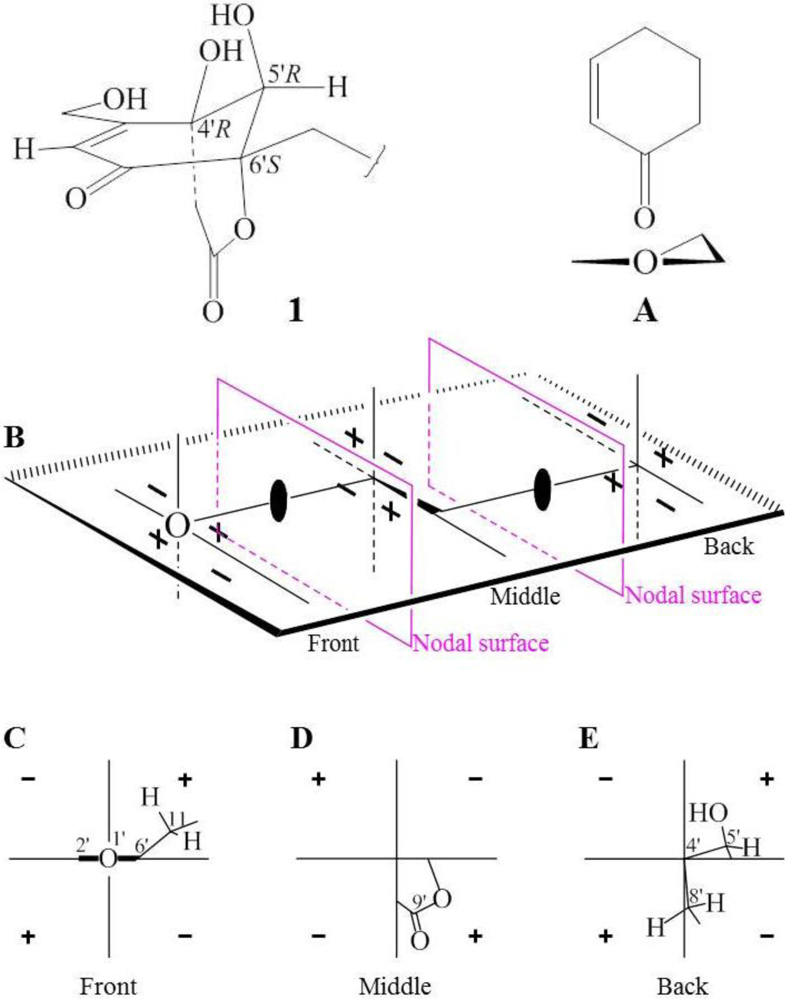 −13.7 (c 0.1, CHCl3). The elemental composition of 2, C22H32O4 (7 double bond equivalents), was established by HRESIMS (m/z361.2377 [M + H]+; Δ = +0.2 mmu). The UV and IR absorptions suggested the presence of hydroxyl (IR νmax 3392 cm−1) and ene(di)one (UV λmax 236 nm, log ε 4.00; IR νmax 1682 cm−1) [28] groups in 2. Its 1H and 13C NMR data in CDCl3 (Table 2), analyzed with the aid of DEPT, 1H–1H COSY, HMQC and HMBC, indicated the characteristic signals of drimene residue seen both in macrophorins [27,28] and purpurogemutantin (1). The same chair-chair conformation of the drimenyl residue in 2 as that in 1 (Figure 3) was established by NOESY and difference NOE experiments: Key NOEs were detected between protons, Ha-1/H-9, He-1/Ha-2, He-1/H3-15, Ha-2/H3-14, Ha-2/H3-15, He-3/H3-13, He-3/H3-14, Ha-3/H3-13, H-5/He-6, H-5/Ha-7, H-5/H-9, H-5/H3-13, Ha-6/He-7, Ha-6/H3-14, Ha-6/H3-15, He-7/Ha-12, H2-11/Hb-12, H2-11/H3-15, and H3-14/H3-15, respectively. The other NOEs and the splitting patterns and J values of relevant protons (Table 2) also supported the same conformation. Then, further detailed analysis of the 1H–1H COSY, HMQC and HMBC data (Table 2) demonstrated the presence of a cyclo-2-hexene-1,4-dione derived moiety attached to C-11 in 2: HMBC correlations were observed from H2-11 to C-8, C-9, C-10, C-1′ and C-6′; from H-2′ to C-3′, C-4′, C-6′ and C-7′; from H2-7′ to C-2′ and C-3′; from Ha-5′ to C-11, C-1′, C-3′, C-4′ and C-6′; from Hb-5′ to C-11, C-1′, C-4′ and C-6′; and from the 6′-OH proton to C-1′, C-5′ and C-6′. A remaining hydroxyl group was thus reasonably located at C-7′ of 2. The ESIMS, HRESIMS, UV, IR, 1H and 13C NMR, DEPT, 1H–1H COSY, HMQC, HMBC, and NOESY spectra of 2 are provided as supporting information in Supplementary Data S3.
−13.7 (c 0.1, CHCl3). The elemental composition of 2, C22H32O4 (7 double bond equivalents), was established by HRESIMS (m/z361.2377 [M + H]+; Δ = +0.2 mmu). The UV and IR absorptions suggested the presence of hydroxyl (IR νmax 3392 cm−1) and ene(di)one (UV λmax 236 nm, log ε 4.00; IR νmax 1682 cm−1) [28] groups in 2. Its 1H and 13C NMR data in CDCl3 (Table 2), analyzed with the aid of DEPT, 1H–1H COSY, HMQC and HMBC, indicated the characteristic signals of drimene residue seen both in macrophorins [27,28] and purpurogemutantin (1). The same chair-chair conformation of the drimenyl residue in 2 as that in 1 (Figure 3) was established by NOESY and difference NOE experiments: Key NOEs were detected between protons, Ha-1/H-9, He-1/Ha-2, He-1/H3-15, Ha-2/H3-14, Ha-2/H3-15, He-3/H3-13, He-3/H3-14, Ha-3/H3-13, H-5/He-6, H-5/Ha-7, H-5/H-9, H-5/H3-13, Ha-6/He-7, Ha-6/H3-14, Ha-6/H3-15, He-7/Ha-12, H2-11/Hb-12, H2-11/H3-15, and H3-14/H3-15, respectively. The other NOEs and the splitting patterns and J values of relevant protons (Table 2) also supported the same conformation. Then, further detailed analysis of the 1H–1H COSY, HMQC and HMBC data (Table 2) demonstrated the presence of a cyclo-2-hexene-1,4-dione derived moiety attached to C-11 in 2: HMBC correlations were observed from H2-11 to C-8, C-9, C-10, C-1′ and C-6′; from H-2′ to C-3′, C-4′, C-6′ and C-7′; from H2-7′ to C-2′ and C-3′; from Ha-5′ to C-11, C-1′, C-3′, C-4′ and C-6′; from Hb-5′ to C-11, C-1′, C-4′ and C-6′; and from the 6′-OH proton to C-1′, C-5′ and C-6′. A remaining hydroxyl group was thus reasonably located at C-7′ of 2. The ESIMS, HRESIMS, UV, IR, 1H and 13C NMR, DEPT, 1H–1H COSY, HMQC, HMBC, and NOESY spectra of 2 are provided as supporting information in Supplementary Data S3.| Position | δCb,c | δHb (J in Hz) | COSY d | NOE e | HMBC f |
|---|---|---|---|---|---|
| 1 | 38.7 t | Ha 1.07 td (12.6, 4.8) He 1.63 br d (12.6) | He-1, H2-2 Ha-1, Ha-2 | He-1, H-9 Ha-1, Ha-2, H3-15 | C-3,5 |
| 2 | 19.3 t | 1.56–1.45 2H, AB type m Ha at lower field He at higher field | H2-1, H2-3 H2-1, H2-3 | H3-14, H3-15 Ha-1 | C-4,10 |
| 3 | 42.0 t | Ha 1.18 td (12.4, 4.8) He 1.37 br d (12.4) | H2-2, He-3 H2-2, Ha-3 | He-3, H3-13 Ha-3, H3-13, H3-14 | C-14 C-1,5,14 |
| 4 | 33.7 s | — | — | — | — |
| 5 | 55.6 d | 1.13 dd (12.8, 2.4) | H2-6 | He-6, Ha-7, H-9, H3-13 | C-6,9,10,14,15 |
| 6 | 24.6 t | Ha 1.25 qd (12.8, 3.8 )He 1.74 dm (12.8) | H-5, He-6, H2-7 H-5, Ha-6, H2-7 | He-6, He-7, H3-14, H3-15 H-5, Ha-6, H3-13 | C-5,7 |
| 7 | 38.1 t | Ha 1.89 td (12.8, 5.2) He 2.28 ddd (12.8, 3.8, 2.4) | H2-6, He-7 H2-6, Ha-7 | H-5, He-6, He-7 Ha-6, Ha-7, Ha-12 | C-8,12 C-5,6,8,9,12 |
| 8 | 148.9 s | — | — | — | — |
| 9 | 50.5 d | 1.77 t (4.7) | H2-11 | Ha-1, H-5 | C-8,10,6′ |
| 10 | 39.9 s | — | — | — | — |
| 11 | 34.8 t | 1.88 2H, d (4.7) | H-9 | He-1, Hb-12, H3-15 | C-8,9,10,1′,6′ |
| 12 | 107.1 t | Ha 4.75 br s Hb 4.25 br s | Ha-7, Hb-12 Ha-12 | He-7, Hb-12 H2-11, Ha-12, H3-15 | C-7,9 C-7,9 |
| 13 | 33.5 q | 0.85 3H, s | H3-14 | Ha-3, H-5, He-6 | C-3,4,5,14 |
| 14 | 21.6 q | 0.75 3H, s | H3-13 | Ha-2, He-3, Ha-6, H3-15 | C-3,4,5,13 |
| 15 | 15.0 q | 0.57 3H, s | He-1, Ha-2, Ha-6, H2-11, Hb-12, H3-14 | C-1,5,9,10 | |
| 1′ | 201.2 s | — | — | — | — |
| 2′ | 134.4 d | 6.82 br s | H2-7′ | — | C-3′,4′,6′,7′ |
| 3′ | 150.9 s | — | — | — | — |
| 4′ | 196.6 s | — | — | — | — |
| 5′ | 53.1 t | Ha 3.12 d (16.0) Hb 2.97 d (16.0) | Hb-5′ Ha-5′ | Hb-5′, H2-11 Ha-5′ | C-11,1′,3′,4′,6′ C-11,1′,4′,6′ |
| 6′ | 77.4 s | — | — | — | — |
| 7′ | 59.6 t | Ha 4.54 br d (17.2) Hb 4.44 br d (17.2) | H-2′, Hb-7′ H-2′, Ha-7′ | Hb-7′ Ha-7′ | C-2′,3′ C-2′,3′ |
| 6′-OH | — | 3.87 br s | — | C-1′,5′,6′ | |
| 7′-OH | — | 3.48 br s | — |
−13.7 (c 0.1, CHCl3), −9.3 (c 0.5, MeOH); penicilliumin A, white crystalline solid, −0.008 (c 0.85, CHCl3). In view of the presence of four possible stereoisomers together with the incompletely defined structure of penicilliumin A, we designated purpurogemutantidin (2) as a new compound.
2.2. Inhibitory Effects of 1–3 on Several Human Cancer Cell Lines
| Sample | IR% Value at the 100 µg/mL Test Sample | ||||
|---|---|---|---|---|---|
| K562 | HL-60 | HeLa | BGC-823 | MCF-7 | |
| 1 | 62.8% | 74.5% | 88.0% | 87.3% | 86.5% |
| 2 | 71.3% | 70.9% | 83.1% | 81.3% | 77.7% |
| docetaxol | 71.2% | 74.9% | 84.4% | 70.2% | 69.7% |
| 5-fluorouracil | 57.6% | 66.3% | 78.4% | 69.1% | 66.6% |
| Compound | K562 | HL-60 | HeLa | BGC-823 | MCF-7 |
|---|---|---|---|---|---|
| 1 | 13.4 | 18.1 | 18.9 | 33.0 | 29.3 |
| 2 | 0.93 | 2.48 | 16.6 | 31.0 | 26.3 |
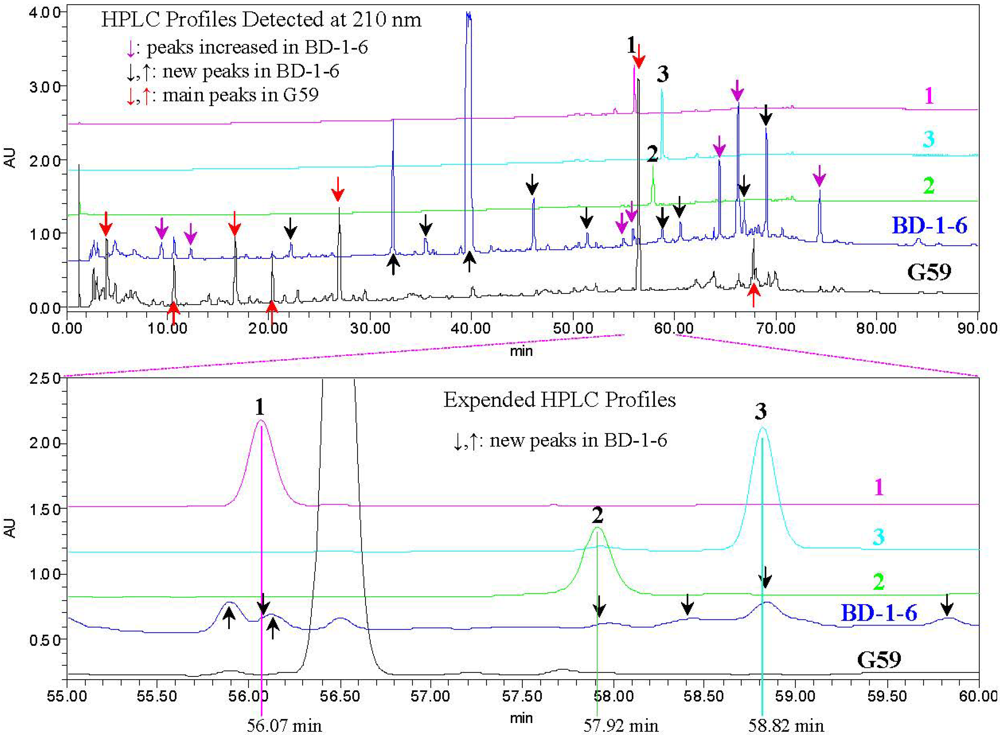
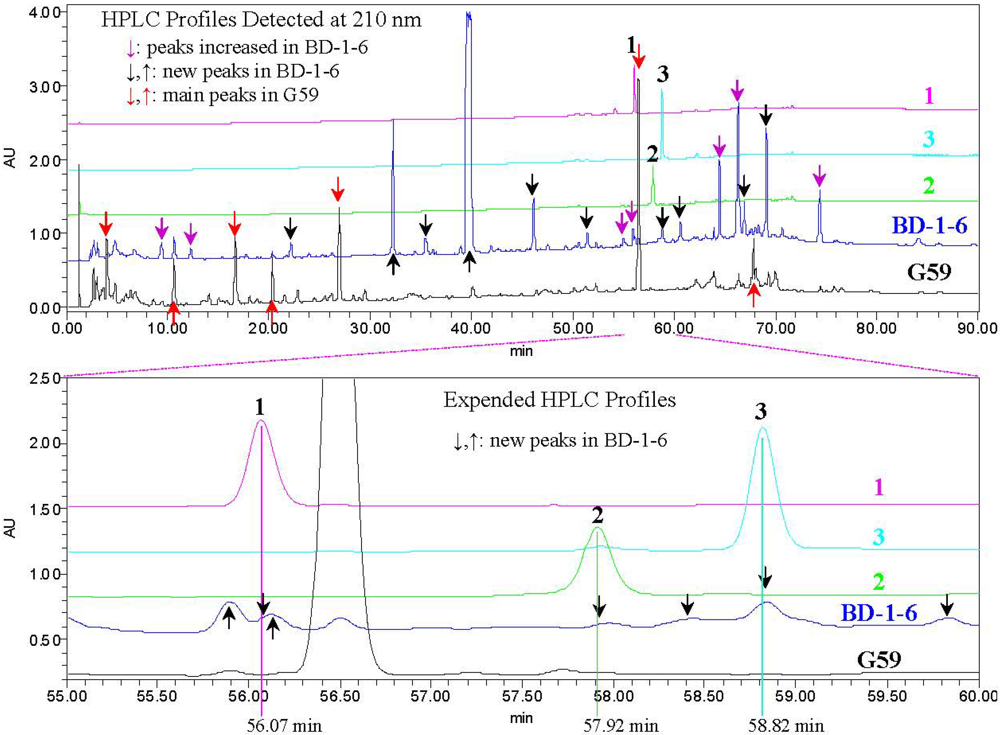
2.3. Experimental Verification of the Absence of 1–3 in the G59 Products by HPLC and LC-ESIMS
3. Experimental Section
3.1. General Experimental
3.2. Fungal Strain and Human Cancer Cell Line
3.3. Fermentation and EtOAc Extract Preparation
3.4. Isolation of Compounds 1–3
3.5. MTT Assay
3.6. Physicochemical and Spectral Data for Compounds 1–3
+21.0 (c 1.0, MeOH). UV λmax nm (log ε) in MeOH: 234 (3.87). IR νmax cm−1: 3405, 2934, 1733, 1694, 1459, 1442, 1388, 1365, 1277, 1246, 1229, 1114, 1067, 1044, 1021, 889. CD (0.48 mM, MeOH) Δε (nm): 0 (388.5), +1.16 (335.5), 0 (286.5), −9.27 (249), 0 (225.5), +3.72 (210). 1H and 13C NMR data in acetone-d6: see Table 1. Positive ESIMS m/z: 419 [M + H]+, 441 [M + Na]+, 837 [2M + H]+, 859 [2M + Na]+; negative ESI-MS m/z: 531 [M + CF3CO2]−. Positive HRESIMS m/z: measured 419.2431 [M + H]+, calculated for C24H35O6 [M + H] 419.2434; measured 436.2692 [M + NH4]+, calculated for C24H38NO6 [M + NH4] 436.2699; measured 441.2234 [M + Na]+, calculated for C24H34O6Na [M + Na] 441.2253. −13.7 (c 0.1, CHCl3), −9.3 (c 0.5, MeOH); (penicilliumin A, white crystalline solid, −0.008 (c 0.85, CHCl3) in literature [31]). UV λmax nm (log ε) in MeOH: 236 (4.00). IR νmax cm−1: 3392, 2941, 1682, 1457, 1443, 1388, 1368, 1243, 1208, 1092, 1036, 898, 829. CD (0.27 mM, MeOH) Δε (nm): 0 (415.5), −0.57 (360.5), 0 (320), +1.14 (260.5), 0 (247.6), −1.25 (237), 0 (226), +3.74 (209.5); CD λmax nm (mdeg) in MeOH at 100 μg/mL: 415.5 (0), 360.5 (−0.52177), 320 (0), 260.5 (+1.04783), 247.6 (0), 237 (−1.14715), 226 (0), 209.5 (+3.42439). 1H and 13C NMR data in CDCl3: see Table 2. Positive ESIMS m/z: 383 [M + Na]+; negative ESIMS m/z: 341 [M − H2O − H]−, 359 [M − H]−, 395 [M + Cl]−. Positive HRESIMS m/z: measured 743.4486 [2M + Na]+, calculated for C44H64O8Na [2M + Na] 743.4499; measured 399.1939 [M + K]+, calculated for C22H32O4K [M + K] 399.1938; measured 383.2193 [M + Na]+, calculated for C22H32O4Na [M + Na] 383.2198; measured 361.2377 [M + H]+, calculated for C22H33O4 [M + H] 361.2379; measured 343.2269 [M − H2O + H]+, calculated for C22H33O4 [M − H2O + H] 343.2273. Negative HRESIMS m/z: measured 395.1995 [M + Cl]−, calculated for C22H32O4Cl [M + Cl] 395.1989; measured 359.2223 [M − H]−, calculated for C22H31O4 [M − H] 359.2222; measured 341.2118 [M − H2O − H]−, calculated for C22H29O3 [M − H2O − H] 341.2117. +25.7 (c 1.0, MeOH), ([α]D +29 (MeOH) in literature [24]). Positive ESIMS m/z: 361 [M + H]+, 378 [M + NH4]+, 383 [M + Na]+; negative ESIMS m/z: 359 [M − H]−, 395 [M + Cl]−, 405 [M + HCOO]−. IR νmax cm−1: 3400, 2938.6, 2867.8, 2844.7, 1677.4, 1458.9, 1439.9, 1387.8, 1365.7, 1278.9, 1204.3, 1103.6, 1030.4, 882.1, 669.1. CD (2.78 mM, MeOH) Δε (nm): 0 (389), +2.76 (334), 0 (280), −4.41 (243), 0 (218), +0.31 (215), 0 (212), −1.92 (204). CD λmax nm (mdeg) in MeOH at 1.0 mg/mL: 389 (0), 334 (+25.33), 280 (0), 243 (−40.40), 218 (0), 215 (+2.80), 212 (0), 204 (−17.56). 1H NMR (400 MHz, CD3OD) δ: 5.91 (1H, d, J = 1.6, H-2′), 4.77 (1H, br s, Ha-12), 4.52 (2H, br s, Hb-12 and H-4′), 4.29 (1H, d, J = 17.7 Hz, Ha-7′), 4.21 (1H, d, J = 17.7 Hz, Hb-7′) , 3.67 (1H, d, J = 2.9 Hz, H-5′), 2.33 (1H, ddd, J = 12.6, 3.9, 2.5 Hz, He-7), 2.31 (1H, d, J = 14.0 Hz, Ha-11), 1.92 (1H, td, J = 12.6, 3.9 Hz, Ha-7), 1.81 (1H, dd, J = 14.0, 11.1 Hz, Hb-11), 1.76 (1H, dt, calcd J = 12.6, 3.9 Hz, He-1; overlapped with H-9 and He-6), 1.75 (1H, d, J = 11.1 Hz, H-9; overlapped with He-1 and He-6), 1.72 (1H, m, He-6; overlapped with He-1 and H-9), 1.60 (1H, qt, J = 13.6, 3.4 Hz, Ha-2), 1.49 (1H, dquint, J = 13.6, 3.4 Hz, He-2), 1.38 (1H, dm, J = 13.6 Hz, He-3), 1.30 (1H, qd, J = 12.6, 3.9 Hz, Ha-6), 1.21 (1H, td, J = 13.6, 4.6 Hz, Ha-3), 1.18 (1H, td, J = 13.6, 3.4 Hz, Ha-1), 1.12 (1H, dd, J = 12.6, 2.5 Hz, H-5), 0.70 (3H, s, H3-13), 0.79 (3H, s, H3-14), 0.85 (3H, s, H3-15). 13C NMR (100 MHz, CD3OD)δ: 195.4 (C-1′), 161.2 (C-3′), 150.5 (C-8), 120.2 (C-2′), 107.4 (C-12), 66.2 (C-4′), 62.3 (C-5′), 62.2 (C-7′), 61.2 (C-6′), 56.9 (C-5), 52.9 (C-9), 43.3 (C-3), 40.7 (C-10), 40.0 (C-1), 39.3 (C-7), 34.5 (C-4), 34.1 (C-13), 25.6 (C-6), 22.2 (C-14), 22.0 (C-11), 20.4 (C-2), 15.0 (C-15). The above 1H and 13C NMR data were assigned on the basis of DEPT, 1H–1H COSY, HMQC, HMBC and NOESY experiments.3.7. HPLC Analysis for 1–3 and the G59 and BD-1-6 Extracts
3.8. LC-ESIMS Analysis for 1–3 and the G59 and BD-1-6 Extracts
4. Conclusions
Acknowledgments
References
- Geris, R.; Simpson, T.J. Meroterpenoids produced by fungi. Nat. Prod. Rep. 2009, 26, 1063–1094. [Google Scholar]
- Swersey, J.C.; Barrows, L.R.; Ireland, C.M. Mamanuthaquinone: An antimicrobial and cytotoxic metabolite of Fasciospongia sp. Tetrahedron Lett. 1991, 32, 6687–6690. [Google Scholar] [CrossRef]
- Jankam, A.; Somerville, M.J.; Hooper, J.N.A.; Brecknell, D.J.; Suksamrarn, A.; Garson, M.J. Dactylospongiaquinone, a new meroterpenoid from the Australian marine sponge Dactylospongia n. sp. Tetrahedron 2007, 63, 1577–1582. [Google Scholar]
- Yong, K.W.L.; Jankam, A.; Hooper, J.N.A.; Suksamrarn, A.; Garson, M.J. Stereochemical evaluation of sesquiterpene quinones from two sponges of the genus Dactylospongia and the implication for enantioselective processes in marine terpene biosynthesis. Tetrahedron 2008, 64, 6341–6348. [Google Scholar]
- Capon, R.J.; Macleod, J.K. A revision of the absolute stereochemistry of ilimaquinone. J. Org. Chem. 1987, 52, 5059–5060. [Google Scholar]
- Urban, S.; Capon, R.J. 5-Epi-Isospongiaquinone, a new sesquiterpene/quinone antibiotic from an Australian marine sponge, Spongia hispid. J. Nat. Prod. 1992, 55, 1638–1642. [Google Scholar] [CrossRef]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Makarchenko, A.E. Determination of the absolute stereochemistry of cyclosmenospongine. J. Nat. Prod. 2003, 66, 1263–1265. [Google Scholar]
- Carté, B.; Rose, C.B.; Faulkner, D.J. 5-Epi-Ilimiquinone, a metabolite of the sponge Fenestraspongia sp. J. Org. Chem. 1985, 50, 2785–2787. [Google Scholar] [CrossRef]
- Ochi, M.; Kotsuki, H.; Muraoka, K.; Tokoroyama, T. The structure of yahazunol, a new sesquiterpene-substituted hydroquinone from the brown seaweed Dictyopteris undulata Okamura. Bull. Chem. Soc. Jpn. 1979, 52, 629–630. [Google Scholar] [CrossRef]
- Fenical, W.; Sims, J.J. Zonarol and isozonarol, fungitoxic hydroquinones from the brown seaweed Dictyopteris zonarioides. J. Org. Chem. 1973, 38, 2383–2386. [Google Scholar] [CrossRef]
- Talpir, R.; Rudi, A.; Kashman, Y.; Loya, Y.; Hizi, A. Three new sesquiterpene hydroquinones from marine origin. Tetrahedron 1994, 50, 4179–4184. [Google Scholar]
- Kawashima, K.; Nakanishi, K.; Nishikawa, H. Structure of tauranin and a note on the “C16-acids” obtained from di and triterpenoids. Chem. Pharm. Bull. 1964, 12, 796–803. [Google Scholar]
- Kawashima, K.; Nakanishi, K.; Tada, M.; Nishikawa, H. Structure of tauranin. Tetrahedron Lett. 1964, 5, 1227–1231. [Google Scholar]
- Kono, K.; Tanaka, M.; Ogita, T.; Hosoya, T.; Kohama, T. F-12509A, a new sphingosine kinase inhibitor, produced by a discomycet. J. Antibiot. 2000, 53, 459–466. [Google Scholar]
- Wijeratne, E.M.K.; Paranagama, P.A.; Marron, M.T.; Gunatilaka, M.K.; Arnold, A.E.; Gunatilaka, A.A.L. Sesquiterpene quinones and related metabolites from Phyllosticta spinarum, a fungal strain endophytic in Platycladus orientalis of the Sonoran desert. J. Nat. Prod. 2008, 71, 218–222. [Google Scholar] [CrossRef]
- Chen, L.; Li, D.H.; Cai, S.X.; Wang, F.P.; Xiao, X.; Gu, Q.Q. A new cytotoxic metabolite from a deep sea derived fungus, Phialocephala sp. Acta Pharm. Sin. 2010, 45, 1275–1278. [Google Scholar]
- Ishii, S.; Fujii, M.; Akita, H. First syntheses of (−)-tauranin and antibiotic (−)-BE-40644 based on lipase-catalyzed optical resolution of albicanol. Chem. Pharm. Bull. 2009, 57, 1103–1106. [Google Scholar]
- Maezawa, N.; Furuichi, N.; Tsuchikawa, H.; Katsumura, S. Synthesis of a novel sphingosine kinase inhibitor (−)-F-12509A and determination of its absolute configuration. Tetrahedron Lett. 2007, 48, 4865–4867. [Google Scholar]
- Poigny, S.; Huor, T.; Guyot, M.; Samadi, M. Synthesis of (−)-hyatellaquinone and revision of absolute configuration of naturally occurring (+)-hyatellaquinone. J. Org. Chem. 1999, 64, 9318–9320. [Google Scholar]
- Akita, H.; Nozawa, M.; Shimizu, H. Synthesis of decalin type chiral synthons based on enzymatic functionalisation and their application to the synthesis of (−)-ambrox and (+)-zonarol. Tetrahedron Asymmetry 1998, 9, 1789–1799. [Google Scholar]
- Schröder, J.; Magg, C.; Seifert, K. Total synthesis of the marine sesquiterpene hydroquinones zonarol and isozonarol and the sesquiterpene quinones zonarone and isozonarone. Tetrahedron Lett. 2000, 41, 5469–5473. [Google Scholar]
- Laube, T.; Schröder, J.; Stehle, R.; Seifert, K. Total synthesis of yahazunol, zonarone and isozonarone. Tetrahedron 2002, 58, 4299–4309. [Google Scholar]
- Toshima, H.; Oikawa, H.; Toyomasu, T.; Sassa, T. Total synthesis of (+)-albicanol and (+)-albicanyl acetate. Biosci. Biotechnol. Biochem. 2001, 65, 1244–1247. [Google Scholar]
- Sassa, T.; Yoshikoshi, H. New terpene-linked cyclohexenone epoxides, macrophorin A, B and C, produced by the fungus caused Macrophoma fruit rot of apple. Agric. Biol. Chem. 1983, 47, 187–189. [Google Scholar] [CrossRef]
- Ayer, W.A.; Altena, I.V.; Browne, L.M. Three piperazinediones and a drimane diterpenoid from Penicillium brevi-compactum. Phytochemistry 1990, 29, 1661–1665. [Google Scholar]
- Sassa, T.; Nukina, M. Macrophorin D, a new self-growth inhibitor of the causal fungus of Macrophoma fruit rot of apple. Agric. Biol. Chem. 1984, 48, 1923–1925. [Google Scholar] [CrossRef]
- Fujimoto, H.; Nakamura, E.; Kim, Y.P.; Okuyama, E.; Ishibashi, M.; Sassa, T. Immunomodulatory constituents from an ascomycete, Eupenicillium crustaceum, and revised absolute structure of macrophorin D. J. Nat. Prod. 2001, 64, 1234–1237. [Google Scholar] [CrossRef]
- Sassa, T.; Ishizaki, A.; Nukina, M.; Ikeda, M.; Sugiyama, T. Isolation and identification of new antifungal macrophorins E, F and G as malonyl meroterpenes from Botryosphaeria berengeriana. Biosci. Biotechnol. Biochem. 1998, 62, 2260–2262. [Google Scholar] [CrossRef]
- Schmidt, L.E.; Deyrup, S.T.; Baltrusaitis, J.; Swenson, D.C.; Wicklow, D.T.; Goler, J.B. Hymenopsins A and B and a macrophorin analogue from a fungicolous Hymenopsis sp. J. Nat. Prod. 2010, 73, 404–408. [Google Scholar] [CrossRef]
- Mohamed, I.E.; Gross, H.; Pontius, A.; Kehraus, S.; Krick, A.; Kelter, G.; Maier, A.; Fiebig, H.H.; König, G.M. Epoxyphomalin A and B, prenylated polyketides with potent cytotoxictiy from the marine-derived fungus Phoma sp. Org.Lett. 2009, 11, 5014–5017. [Google Scholar]
- Lin, X.; Zhou, X.; Wang, F.; Liu, K.; Yang, B.; Yang, X.; Peng, Y.; Liu, J.; Ren, Z.; Liu, Y. A new cytotoxic sesquiterpene quinone produced by Penicillium sp. F00120 isolated from a deep sea sediment sample. Mar. Drugs 2012, 10, 106–115. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, Y.; Wang, Y.; Pei, Y. Anti-tumor effects of rubratoxin B on cell toxicity, inhibition of cell proliferation, cytotoxic activity and matrix metalloproteinase-2,9. Toxicol In Vitro 2007, 21, 646–650. [Google Scholar] [CrossRef]
- King, T.J.; Roberts, J.C.; Thompson, D.J. Studies in mycological chemistry. Part XXX and Last. Isolation and structure of purpuride, a metabolite of Penicillium purpurogenum stoll. J. Chem. Soc. PerkinTrans. 1 1973, 78–80. [Google Scholar]
- Tomoda, H.; Nishida, H.; Masuma, R.; Cao, J.; Okuda, S.; Ōmura, S. Purpactins, new inhibitors of acyl-CoA: Cholesterol acyltransferase produced by Penicillium purpurogenum I. Production, isolation and physico-chemical and biological properties. J. Antibiot 1991, 44, 136–143. [Google Scholar] [CrossRef]
- Nishida, H.; Tomoda, H.; Cao, J.; Okuda, S.; Ōmura, S. Purpactins, new inhibitors of acyl-CoA: Cholesterol acyltransferase produced by Penicillium purpurogenum II. Structure elucidation of purpactins A, B and C. J. Antibiot 1991, 44, 144–151. [Google Scholar] [CrossRef]
- de Silva, E.D.; Williams, D.E.; Jayanetti, D.R.; Centko, R.M.; Patrick, B.O.; Wijesundera, R.L.; Andersen, R.J. Dhilirolides A–D, meroterpenoids produced in culture by the fruit-infecting fungus Penicillium purpurogenum collected in Sri Lanka. Org.Lett. 2011, 13, 1174–1177. [Google Scholar]
- Tian, C.K.; Cui, C.B.; Han, X.X. Isolation of fungal strains in unusual environment and screening for their antitumor activity. J. Int. Pharm. Res. 2008, 35, 401–405. [Google Scholar]
- Brakhage, A.A.; Schroeckh, V. Fungal secondary metabolites—Strategies to activate silent gene clusters. Fungal Genet. Biol. 2011, 48, 15–22. [Google Scholar]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. Chembiochem 2002, 3, 619–627. [Google Scholar]
- Hosaka, T.; Ohnishi-Kameyama, M.; Muramatsu, H.; Murakami, K.; Tsurumi, Y.; Kodani, S.; Yoshida, M.; Fujie, A.; Ochi, K. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat. Biotechnol. 2009, 27, 462–464. [Google Scholar]
- Ochi, K. From microbial differentiation to ribosome engineering. Biosci. Biothenol. Biochem. 2007, 71, 1373–1386. [Google Scholar] [CrossRef]
- Cichewicz, R.H. Epigenome manipulation as a pathway to new natural product scaffolds and their congeners. Nat. Prod. Rep. 2010, 27, 11–22. [Google Scholar]
- Wang, X.; Sena Filho, J.G.; Hoover, A.R.; King, J.B.; Ellis, T.K.; Powell, D.R.; Cichewicz, R.H. Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an atlantic-forest-soil-derived Penicillium citreonigrum. J. Nat. Prod. 2010, 73, 942–948. [Google Scholar]
- Chai, Y.J.; Cui, C.B.; Li, C.W.; Wu, C.J.; Tian, C.K.; Hua, W. Activation of the dormant secondary metabolite production by introducing gentamicin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 559–582. [Google Scholar] [CrossRef]
- Chai, Y.J.; Cui, C.B.; Li, C.W.; Hua, W. Antitumor metabolites newly produced by a gentamicin-resistant mutant of Penicillium purpurogenum G59. J. Int. Pharm. Res. 2011, 38, 216–222. [Google Scholar]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute sterostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 2009, 2717–2727. [Google Scholar]
- Gaussian 09, Revision A.02. Gaussian Inc.: Wallingford, CT, USA, 2009.
- Snatzk, G. Circular dichroism and absolute conformation: Application of qualitative MO theory to chiroptical phenomena. Angew. Chem. Int. Ed. Engl. 1979, 18, 363–377. [Google Scholar]
- Burnett, R.D.; Kirk, D.N. Chiroptical studies. Part 101. An empirical analysis of circular dichroism data for steroidal and related transoid α,β-unsaturated ketones. J. Chem. Soc. Perkin Trans. 1 1981, 1460-1468. [Google Scholar]
- Gawroński, J.K. Circular dichroism and stereochemistry of chiral conjugated cyclohexenones. Tetrahedron 1982, 38, 3–26. [Google Scholar]
- Mann, J.; Davidson, R.S.; Hobbs, J.B.; Banthorpe, D.V.; Harborne, J.B. Natural Products: Their Chemistry and Biological Significance, 1st ed; Addison Wesley Longman Ltd.: Essex, UK, 1996; pp. 289–331. [Google Scholar]
- Nabeta, K.; Ichihara, A.; Sakamura, S. Biosynthesis of epoxydon and related compounds by Phyllosticta sp. Agric. Biol. Chem. 1975, 39, 409–413. [Google Scholar] [CrossRef]
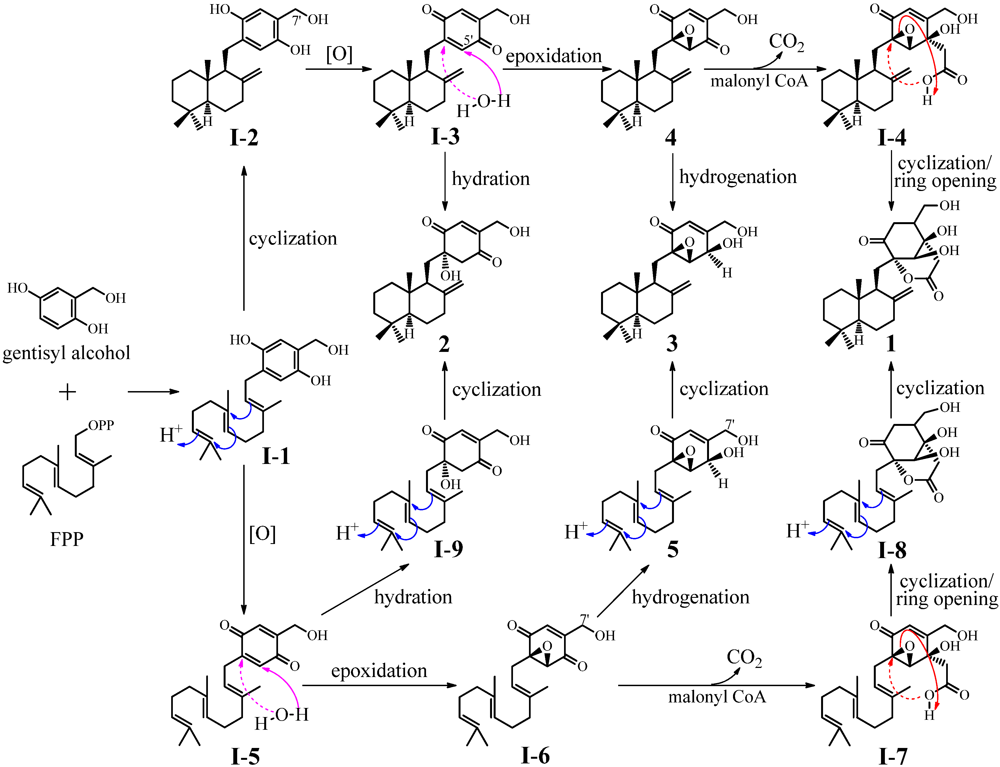
- Iijima, H.; Ebizuka, Y.; Sankawa, U. Biosynthesis of patulin; in vitro conversion of gentisyl alcohol into patulin by microbial enzyme(s) and retention of one of the carbinol protons in this reaction. Chem. Pharm. Bull. 1986, 34, 3534–3537. [Google Scholar] [CrossRef]
- Bugni, T.S.; Abbanat, D.; Bernan, V.S.; Maiese, W.M.; Greenstein, M.; van Wagoner, R.M.; Ireland, C.M. Yanuthones: Novel metabolites from a marine isolate of Aspergillus niger. J. Org. Chem. 2000, 65, 7195–7200. [Google Scholar]
- Samples Availability: Available from the authors.
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fang, S.-M.; Cui, C.-B.; Li, C.-W.; Wu, C.-J.; Zhang, Z.-J.; Li, L.; Huang, X.-J.; Ye, W.-C. Purpurogemutantin and Purpurogemutantidin, New Drimenyl Cyclohexenone Derivatives Produced by a Mutant Obtained by Diethyl Sulfate Mutagenesis of a Marine-Derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 1266-1287. https://doi.org/10.3390/md10061266
Fang S-M, Cui C-B, Li C-W, Wu C-J, Zhang Z-J, Li L, Huang X-J, Ye W-C. Purpurogemutantin and Purpurogemutantidin, New Drimenyl Cyclohexenone Derivatives Produced by a Mutant Obtained by Diethyl Sulfate Mutagenesis of a Marine-Derived Penicillium purpurogenum G59. Marine Drugs. 2012; 10(6):1266-1287. https://doi.org/10.3390/md10061266
Chicago/Turabian StyleFang, Shi-Ming, Cheng-Bin Cui, Chang-Wei Li, Chang-Jing Wu, Zhi-Jun Zhang, Li Li, Xiao-Jun Huang, and Wen-Cai Ye. 2012. "Purpurogemutantin and Purpurogemutantidin, New Drimenyl Cyclohexenone Derivatives Produced by a Mutant Obtained by Diethyl Sulfate Mutagenesis of a Marine-Derived Penicillium purpurogenum G59" Marine Drugs 10, no. 6: 1266-1287. https://doi.org/10.3390/md10061266
APA StyleFang, S.-M., Cui, C.-B., Li, C.-W., Wu, C.-J., Zhang, Z.-J., Li, L., Huang, X.-J., & Ye, W.-C. (2012). Purpurogemutantin and Purpurogemutantidin, New Drimenyl Cyclohexenone Derivatives Produced by a Mutant Obtained by Diethyl Sulfate Mutagenesis of a Marine-Derived Penicillium purpurogenum G59. Marine Drugs, 10(6), 1266-1287. https://doi.org/10.3390/md10061266