Chondrosterins A–E, Triquinane-Type Sesquiterpenoids from Soft Coral-Associated Fungus Chondrostereum sp.


Abstract
:1. Introduction
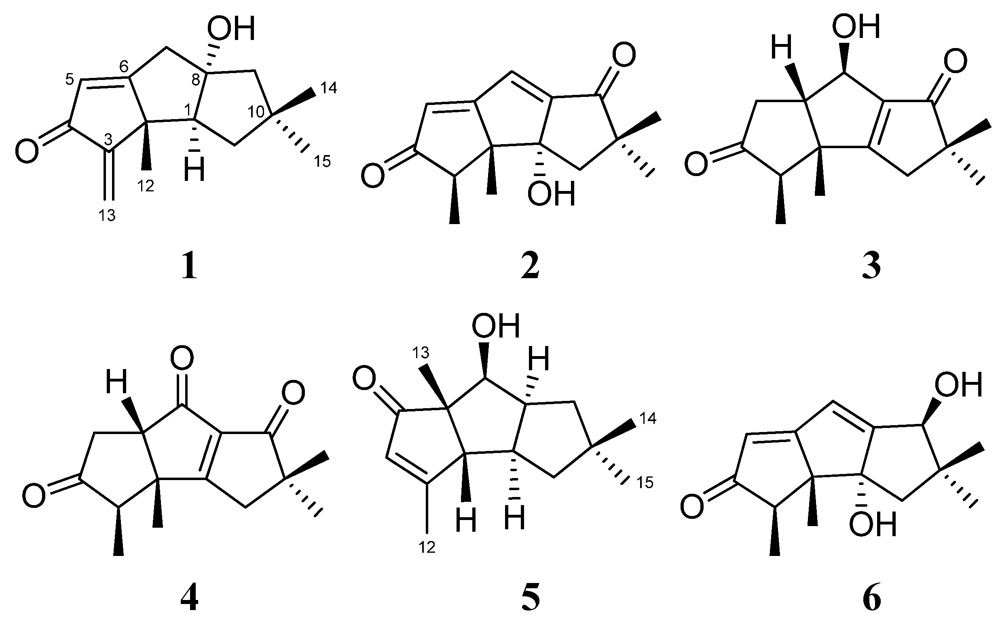
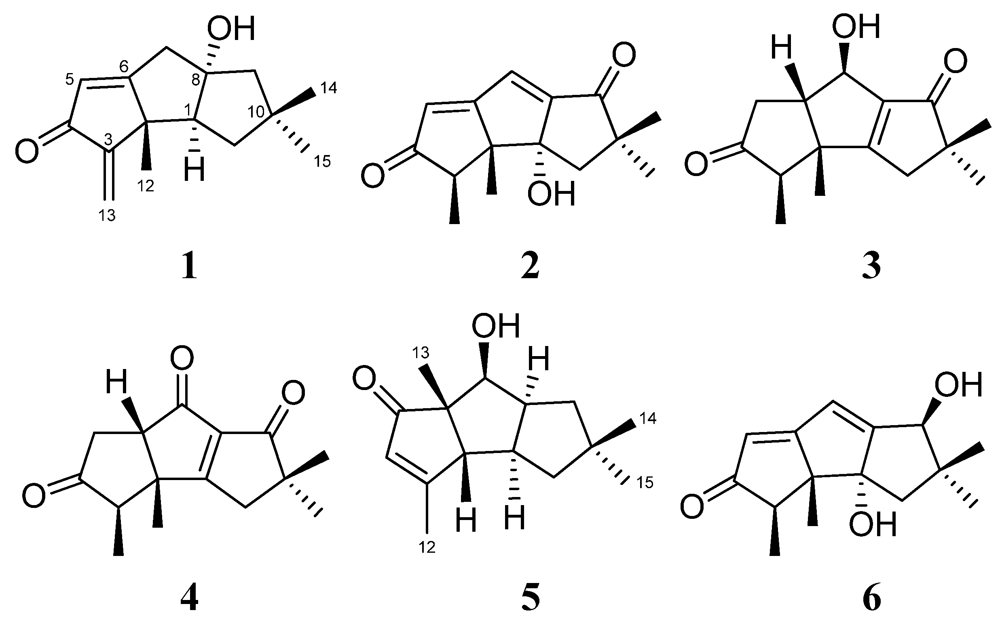
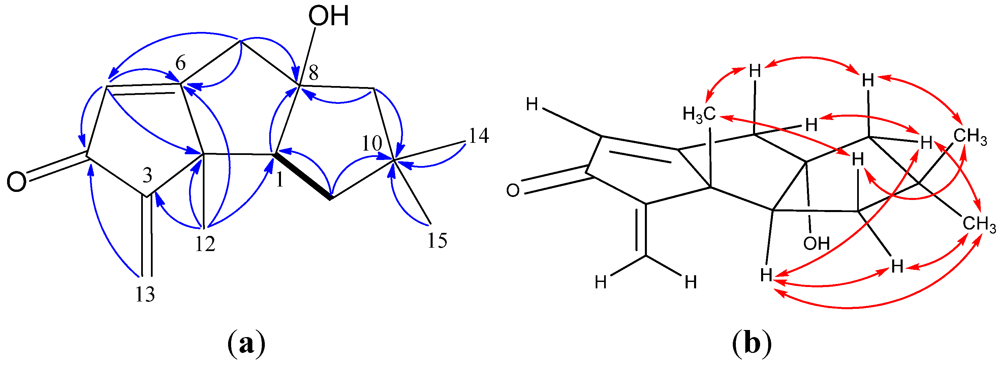
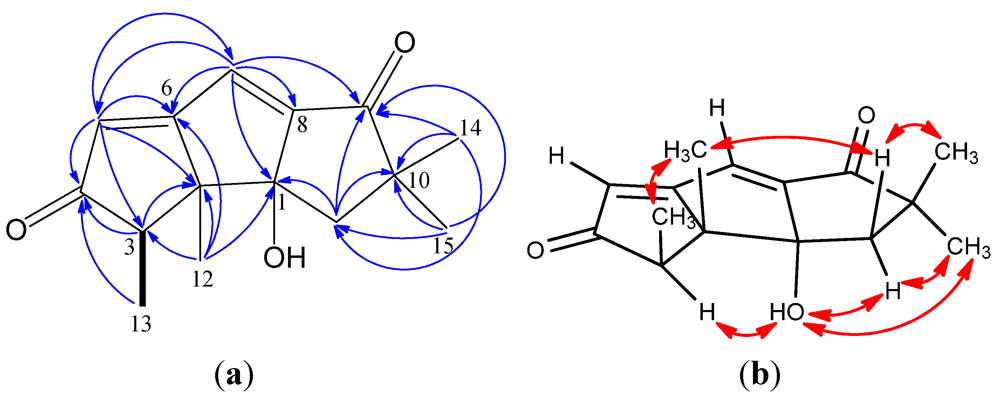
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
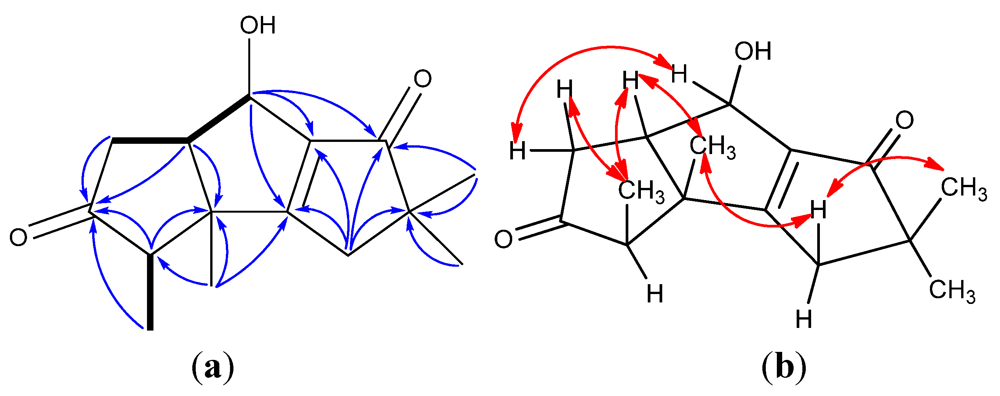{kind=link}
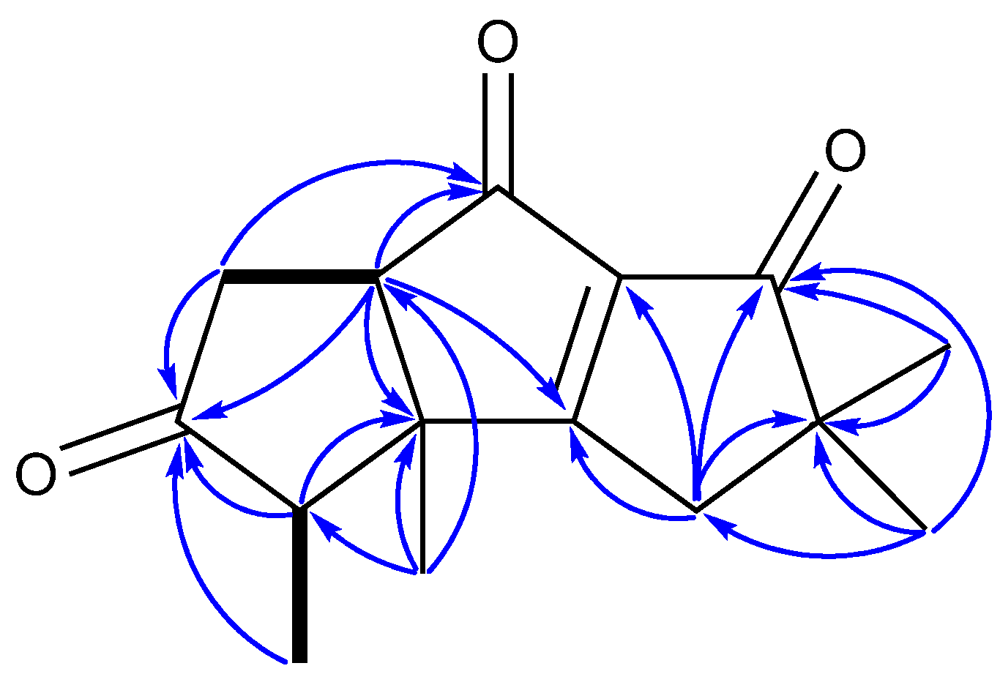{kind=link}
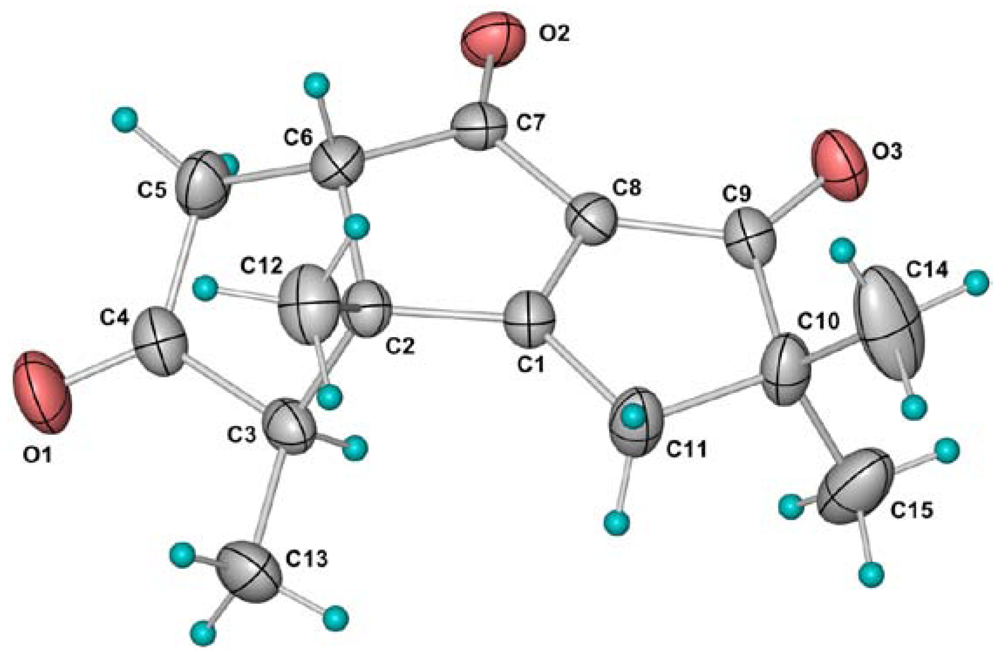{kind=link}
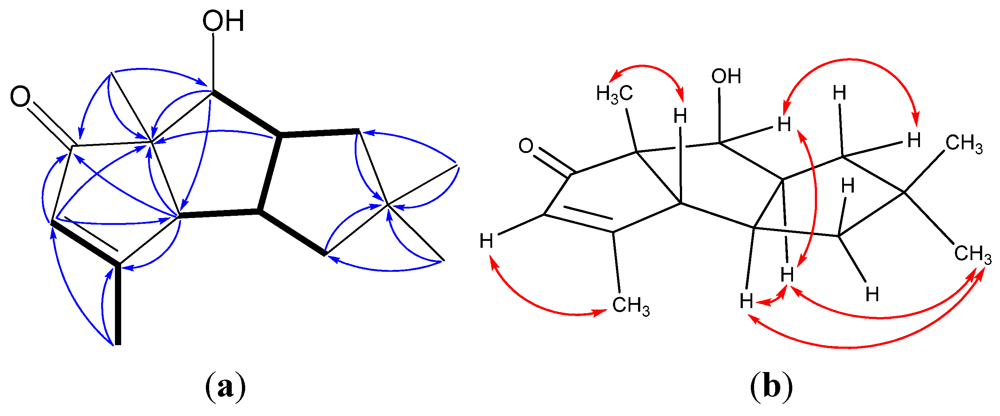{kind=link}
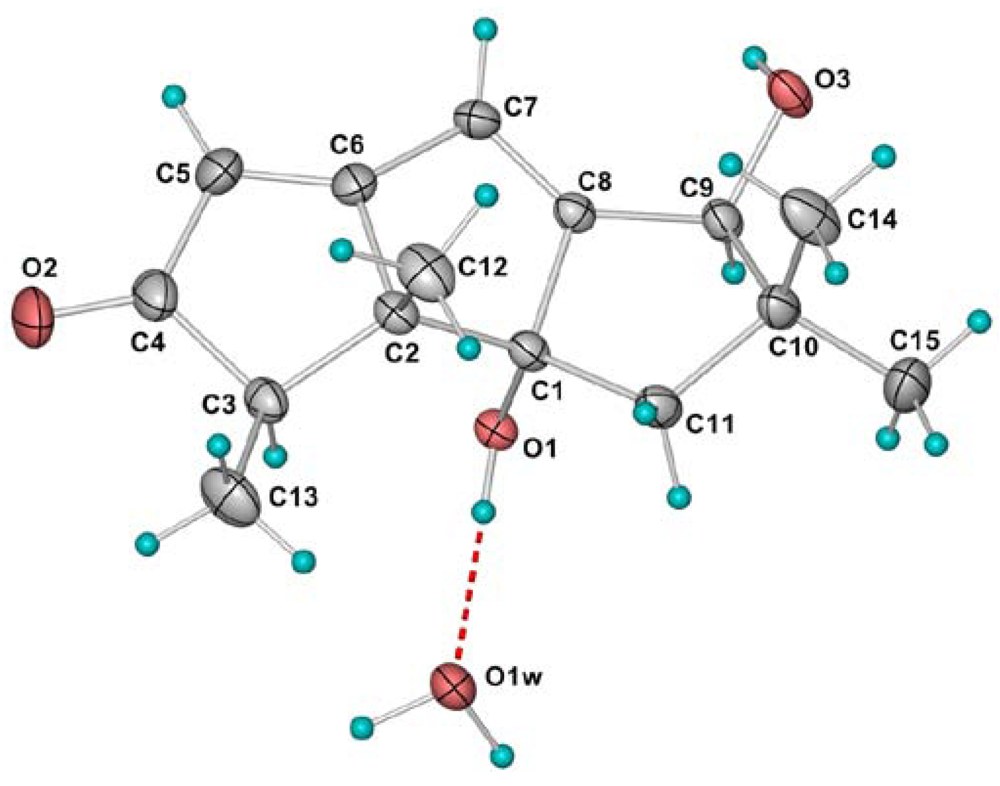{kind=link}
| Position | 1 a | 2 a | 3 a | 4 a | 5 a | 6 b |
|---|---|---|---|---|---|---|
| 1 | 60.6, CH | 84.0, C | 191.8, C | 197.6, C | 46.2, CH | 82.1, C |
| 2 | 52.6, C | 62.9, C | 53.1, C | 49.5, C | 63.8, CH | 61.6, C |
| 3 | 154.0, C | 48.5, CH | 53.1, CH | 52.4, CH | 182.6, C | 47.0, CH |
| 4 | 197.3, C | 210.5, C | 218.5, C | 214.3, C | 128.4, CH | 210.4, C |
| 5 | 125.0, CH | 126.9, CH | 34.4, CH2 | 36.4, CH2 | 211.1, C | 116.8, CH |
| 6 | 186.9, C | 187.5, C | 54.3, CH | 59.5, CH | 61.8, C | 192.3, C |
| 7 | 43.6, CH2 | 126.2, CH | 67.4, CH | 212.8, C | 76.9, CH | 116.0, CH |
| 8 | 92.5, C | 158.0, C | 145.2, C | 139.8, C | 49.8, CH | 173.9, C |
| 9 | 55.9, CH2 | 207.6, C | 209.1, C | 202.6, C | 39.9, CH2 | 75.8, CH |
| 10 | 43.4, C | 51.0, C | 50.6, C | 51.7, C | 41.3, C | 42.3, C |
| 11 | 41.2, CH2 | 41.7, CH2 | 40.1, CH2 | 41.0, CH2 | 49.1, CH2 | 44.5, CH2 |
| 12 | 22.8, CH3 | 22.9, CH3 | 17.6, CH3 | 17.5, CH3 | 15.0, CH3 | 23.9, CH3 |
| 13 | 113.4, CH2 | 9.5, CH3 | 9.3, CH3 | 9.4, CH3 | 19.7, CH3 | 9.4, CH3 |
| 14 | 30.2, CH3 | 27.7, CH3 | 25.4, CH3 | 25.2, CH3 | 29.3, CH3 | 29.6, CH3 |
| 15 | 28.1, CH3 | 26.2, CH3 | 25.0, CH3 | 25.0, CH3 | 29. 2, CH3 | 23.0, CH3 |
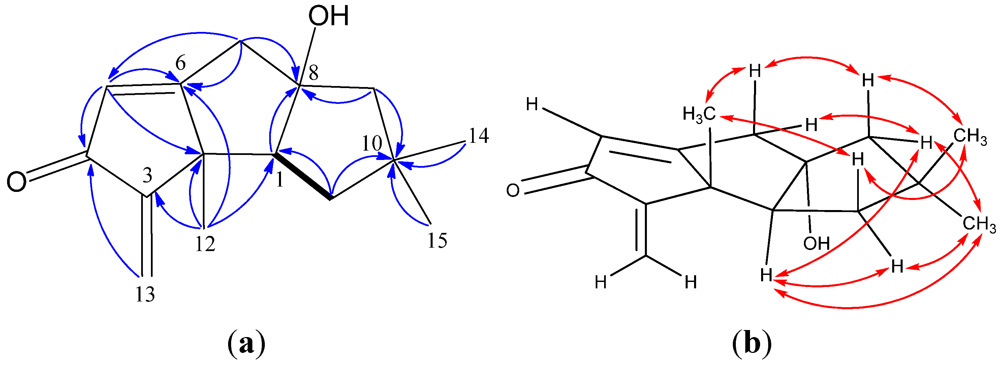

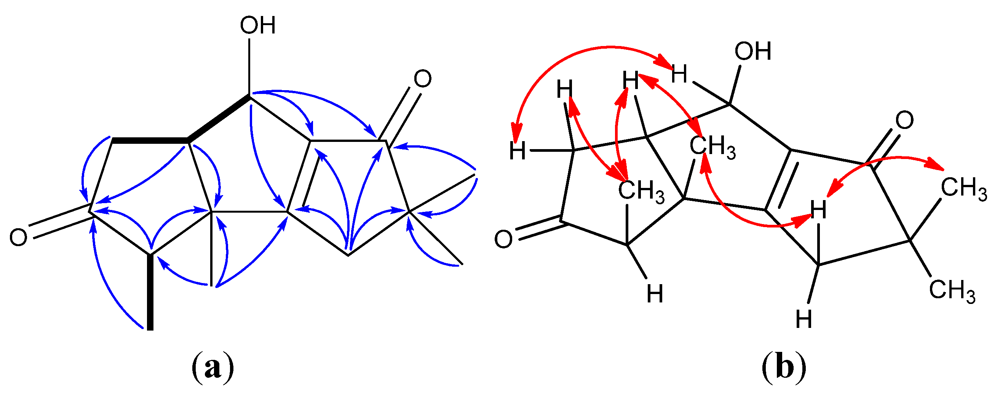
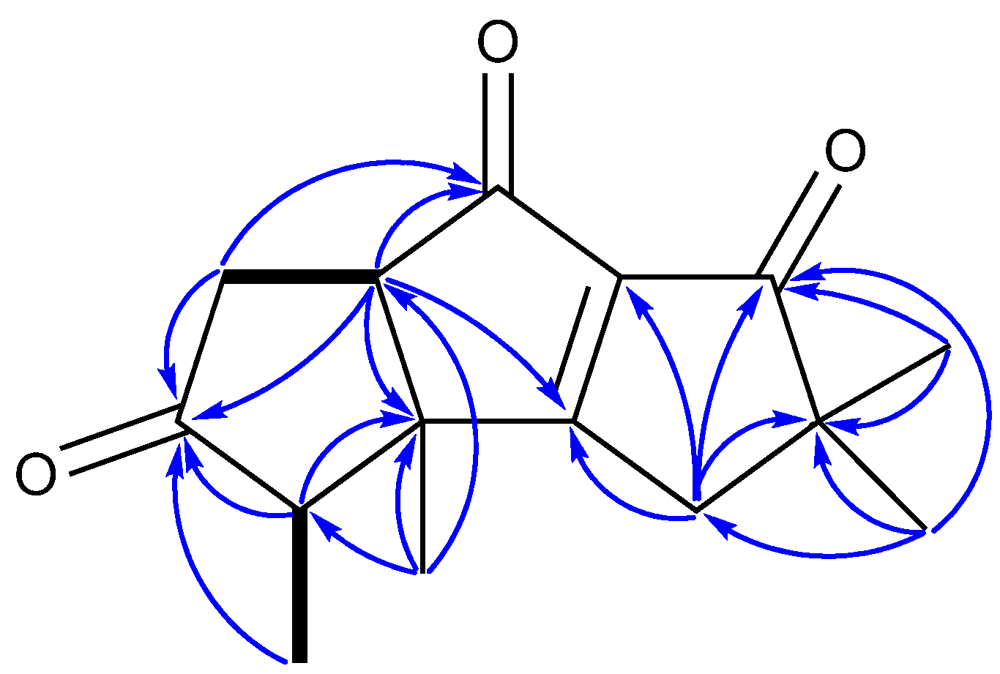
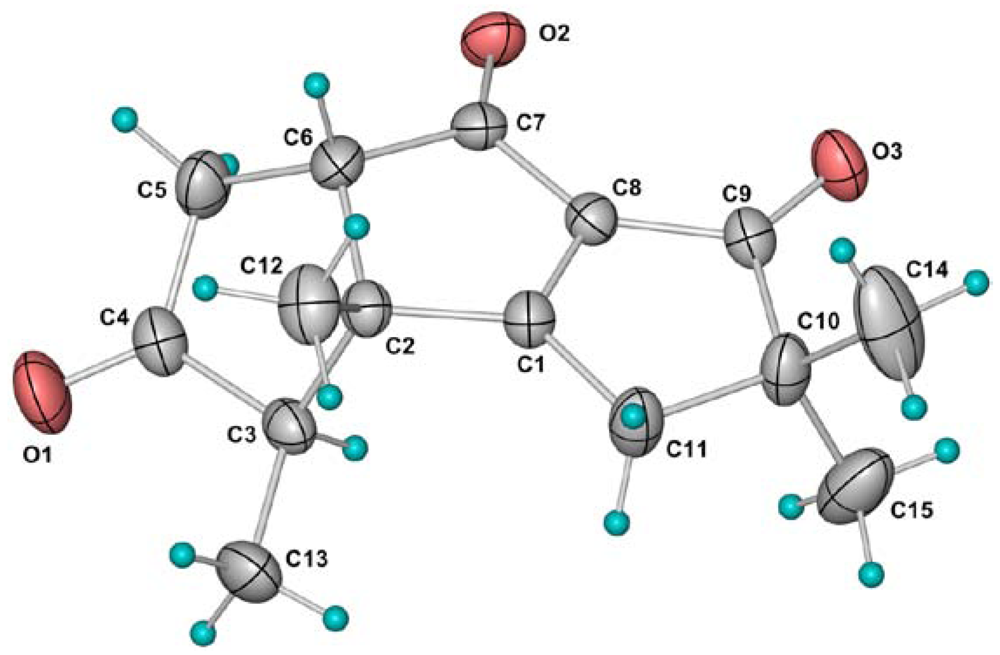
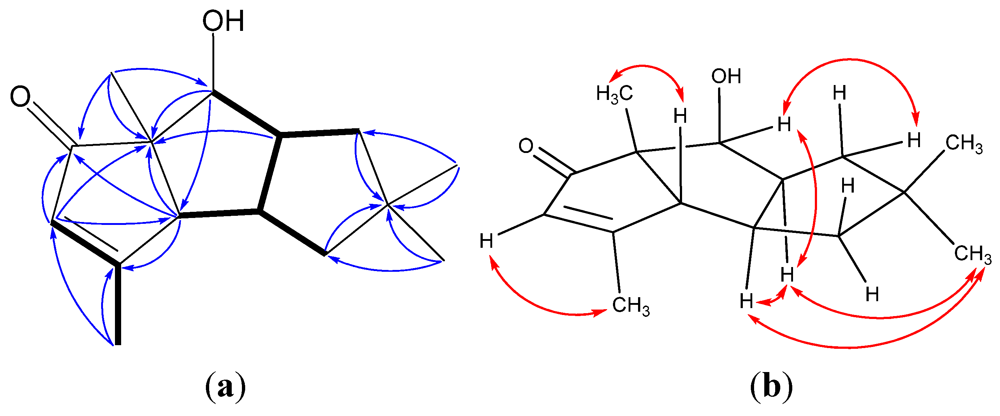
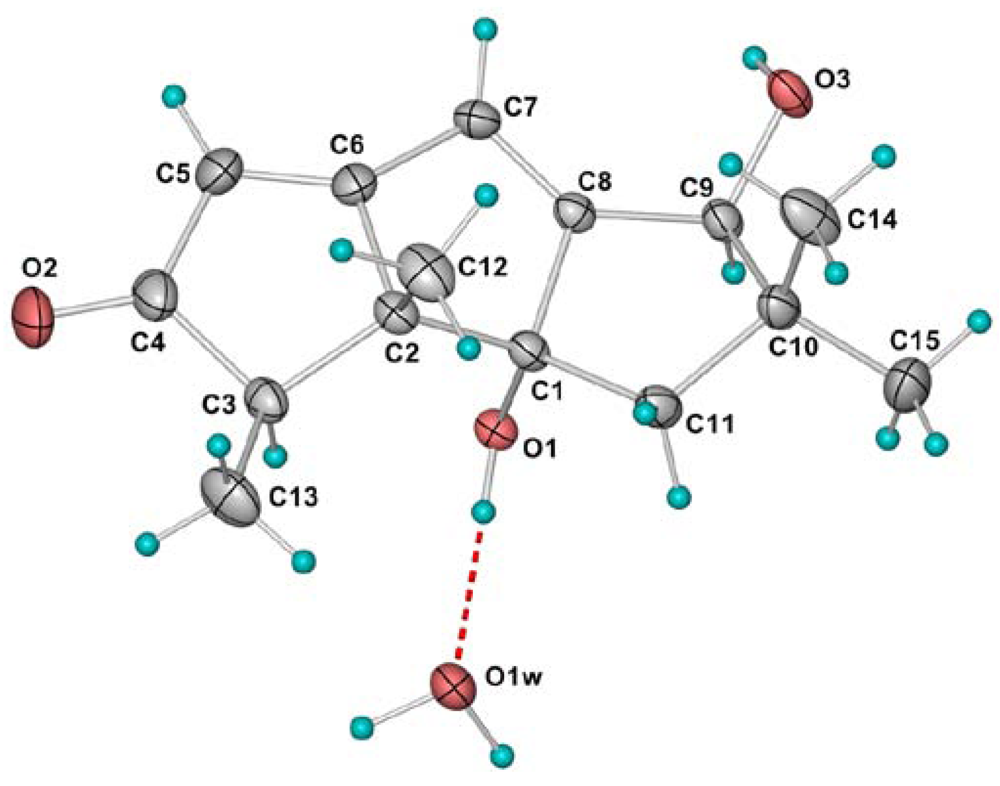
| Position | 1 a | 2 a | 3 a | 4 a | 5 a | 6 b |
|---|---|---|---|---|---|---|
| 1 | 2.33, dd (10.0, 9.0) | 2.58, dddd (10.5, 8.5, 7.5, 2.5) | ||||
| 2 | 2.34, d (2.5) | |||||
| 3 | 2.99, q (7.0) | 2.41, qd (7.0, 1.5) | 2.13, qd (7.0, 1.5) | 2.77, q (7.0) | ||
| 4 | 5.73, q (1.0) | |||||
| 5 | 6.02, d (1.5) | 6.20, s | α: 2.91, ddd (19.0, 3.5, 1.5); β: 2.32, dd (19.0, 10.0) | α: 2.79, ddd (19.5, 4.5, 1.5); β: 2.62, dd (19.5, 12.0) | 5.69, s | |
| 6 | 2.83, ddd (10.0, 7.0, 3.5) | 3.12, dd (12.0, 4.5) | ||||
| 7 | α: 2.76, d (15.5); β: 2.71, dd (15.5, 1.5) | 7.12, s | 4.80, dd (7.0, 1.0) | 3.92, d (6.5) | 6.32, d (2.5) | |
| 8 | 2.64, dddd (8.5, 8.5, 6.5, 6.5) | |||||
| 9 | α: 1.90, d (14.0); β: 1.65, d (14.0) | α: 1.66, dd (13.5, 6.5); β: 1.45, dd (13.5, 8.5) | 4.55 dd (6.0, 2.5) | |||
| 10 | ||||||
| 11 | α: 1.76, dd (13.5, 9.0); β: 1.60, dd (13.5, 10.0) | α: 1.97, d(14.0); β: 2.10, d (14.0) | α: 2.38, d (19.0); β: 2.48, dd (19.0, 1.0) | 2.78, d (21.0); 2.58, d (21.0) | α: 1.76, dd (12.0, 7.5); β: 1.48, dd (12.0, 10.5) | 1.98, d (14.5); 1.52, d (14.5) |
| 12 | 1.14, s | 1.04, s | 1.08, s | 1.28, s | 2.03, d (1.0) | 0.89, s |
| 13 | 5.89, s; 5.16, s | 1.16,d (7.0) | 1.05, d (7.0) | 1.15, d (7.0) | 1.35, s | 0.93, d (7.0) |
| 14 | 1.12, s | 1.18, s | 1.17, s | 1.25, s | 1.09, s | 1.19, s |
| 15 | 1.19, s | 1.40, s | 1.16, s | 1.20, s | 0.93, s | 0.80, s |
| 1α-OH | 1.94, brs | 5.15, s | ||||
| 7β-OH | 3.42, brs | |||||
| 8α-OH | 2.05, brs | |||||
| 9β-OH | 5.250, d(6.0) |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Strain and Culture Method
3.3. Extraction and Isolation
3.4. Crystal Structure Determination of 4 and 6
3.5. Cytotoxicity Assay
4. Conclusions
Acknowledgments
References
- Su, J.Y.; Long, K.H.; Peng, T.S.; Zheng, Q.T.; Lin, X.Y. Chemical constituents of Chinese soft coral. 12. The structure of methyl sartortuoate, a unique new tetraterpenoid. Acta Chim. Sinica 1985, 43, 796–797. [Google Scholar]
- Su, J.Y.; Long, K.H.; Peng, T.S.; Zeng, L.M.; Zheng, Q.T.; Lin, X.Y. Determination of molecular and crystal structures of a unique new tetraterpenoid-methyl sartortuoate. Sci. China Ser. B 1988, 31, 1172–1184. [Google Scholar]
- Lan, W.J.; Su, J.Y.; Zeng, L.M. Isolation and 1H, 13C NMR assignments formethyl sartortuoate. Chinese J. Org. Chem. 2005, 25, 1465–1468. [Google Scholar]
- Su, J.Y.; Long, K.H.; Peng, T.S.; He, C.H.; Clardy, J. The structure ofmethylisosartortuoate, a novel tetracyclic tetraterpenoid from the soft coral Sarcophyton tortuosum. J. Am. Chem. Soc. 1986, 108, 177–178. [Google Scholar]
- Zeng, L.M.; Lan, W.J.; Su, J.Y.; Zhang, G.W.; Feng, X.L.; Liang, Y.J.; Yang, X.P. Two new cytotoxic tetracyclic tetraterpenoids from the soft coral Sarcophyton tortuosum. J. Nat. Prod. 2004, 67, 1915–1918. [Google Scholar] [CrossRef]
- Lan, W.J.; Li, H.J.; Yan, S.J.; Su, J.Y.; Zeng, L.M. New tetraterpenoidfrom the soft coral Sarcophyton tortuosum. J. Asian Nat. Prod. Res. 2007, 9, 267–271. [Google Scholar] [CrossRef]
- Lan, W.J.; Wang, S.L.; Li, H.J. Additional new tetracyclic tetraterpenoid:methyltortuoateD from soft coral Sarcophyton tortuosum. Nat. Prod. Commun. 2009, 4, 1193–1196. [Google Scholar]
- Li, H.J.; Lan, W.J.; Lam, C.K.; Yang, F.; Zhu, X.F. Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp. Chem. Biodivers. 2011, 8, 317–324. [Google Scholar] [CrossRef]
- Yang, F.; Gao, Y.H.; Wu, K.W.; Deng, R.; Li, D.D.; Wei, Z.X.; Jiang, S.; Wu, X.Q.; Feng, G.K.; Li, H.J.; Zhu, X.F. A novel sesquiterpene hirsutanolAinduces autophagical cell death in human hepatocellular carcinoma cells by increasing reactive oxygen species. Chin. J. Cancer 2010, 29, 655–660. [Google Scholar]
- Wang, G.Y.S.; Abrell, L.M.; Avelar, A.; Borgeson, B.M.; Crews, P. New hirsutane based sesquiterpenes from salt water cultures of a marine sponge-derived fungus and the terrestrial fungus Coriolus consors. Tetrahedron 1998, 54, 7335–7342. [Google Scholar]
- SADABS; University of Göttingen: Göttingen, Germany, 1996; Program for empirical absorption correction of area detector data.
- SHELXTL, 5.10 for Windows NT, Bruker Analytical X-Ray Systems, Inc.: Madison, WI, USA, 1998; Structure determination software programs.
- Available online: http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi (accessed on 12 March 2012). These data can be obtained free of charge.
- SPSS, student version 13.0 for Windows, Statistical Program for Social Sciences, IBM Company: New York, NY, USA, 2001.
- Sample Availability: Available from the authors.
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.-J.; Xie, Y.-L.; Xie, Z.-L.; Chen, Y.; Lam, C.-K.; Lan, W.-J. Chondrosterins A–E, Triquinane-Type Sesquiterpenoids from Soft Coral-Associated Fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627-638. https://doi.org/10.3390/md10030627
Li H-J, Xie Y-L, Xie Z-L, Chen Y, Lam C-K, Lan W-J. Chondrosterins A–E, Triquinane-Type Sesquiterpenoids from Soft Coral-Associated Fungus Chondrostereum sp. Marine Drugs. 2012; 10(3):627-638. https://doi.org/10.3390/md10030627
Chicago/Turabian StyleLi, Hou-Jin, Ying-Lu Xie, Zhong-Liang Xie, Ying Chen, Chi-Keung Lam, and Wen-Jian Lan. 2012. "Chondrosterins A–E, Triquinane-Type Sesquiterpenoids from Soft Coral-Associated Fungus Chondrostereum sp." Marine Drugs 10, no. 3: 627-638. https://doi.org/10.3390/md10030627
APA StyleLi, H.-J., Xie, Y.-L., Xie, Z.-L., Chen, Y., Lam, C.-K., & Lan, W.-J. (2012). Chondrosterins A–E, Triquinane-Type Sesquiterpenoids from Soft Coral-Associated Fungus Chondrostereum sp. Marine Drugs, 10(3), 627-638. https://doi.org/10.3390/md10030627