Isolation, Structure Elucidation and Total Synthesis of Lajollamide A from the Marine Fungus Asteromyces cruciatus


 and
and
Abstract
:
1. Introduction
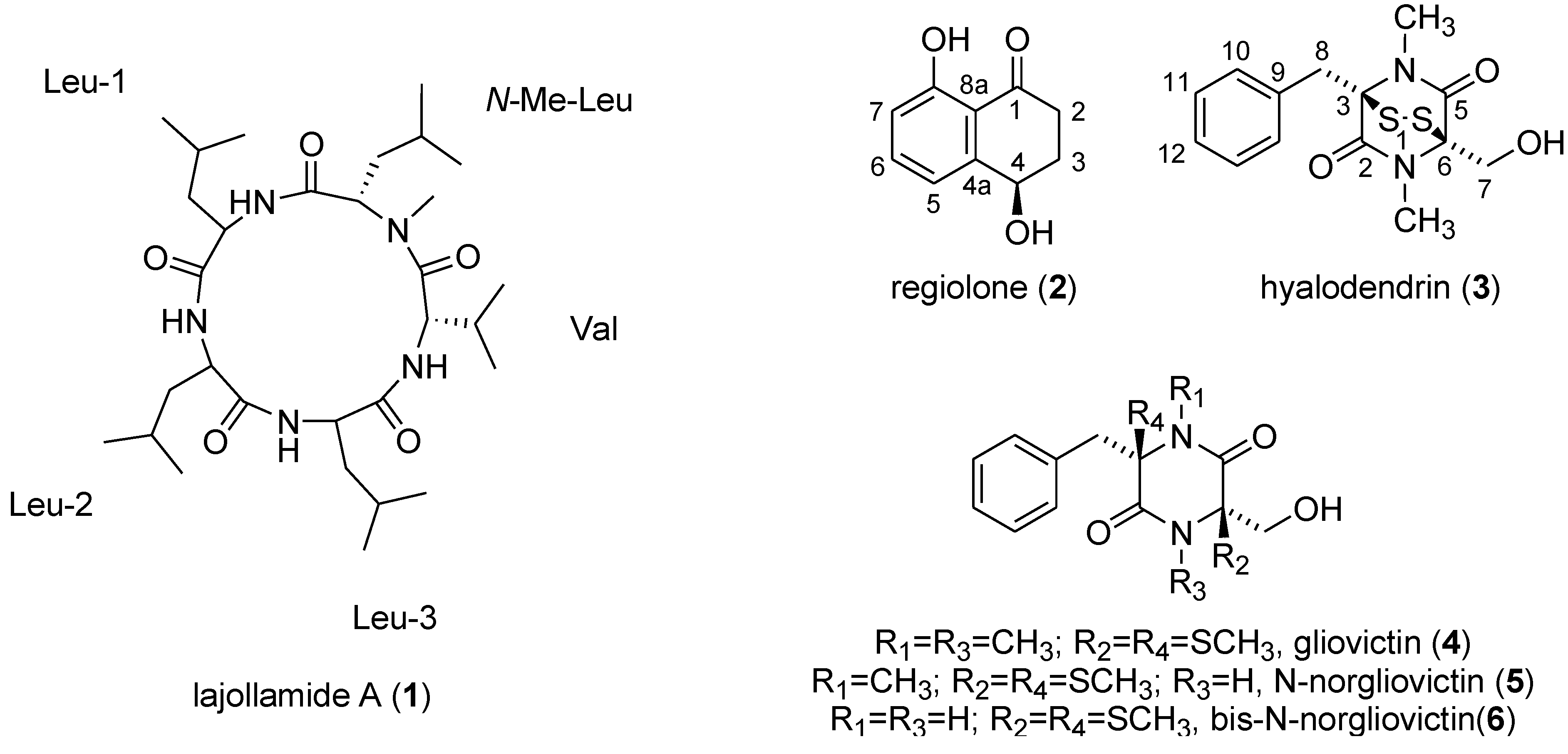
2. Results and Discussion
2.1. OSMAC Studies
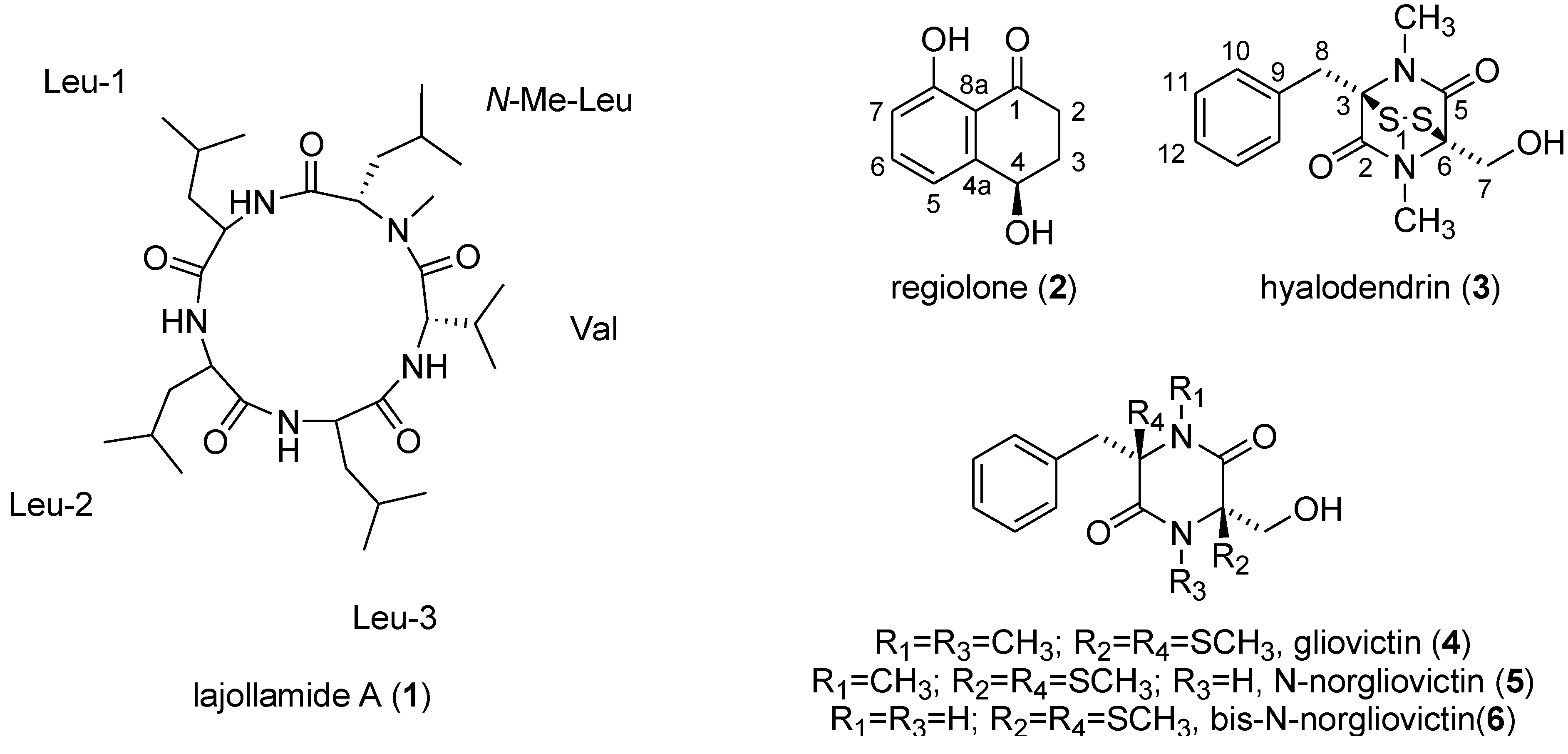
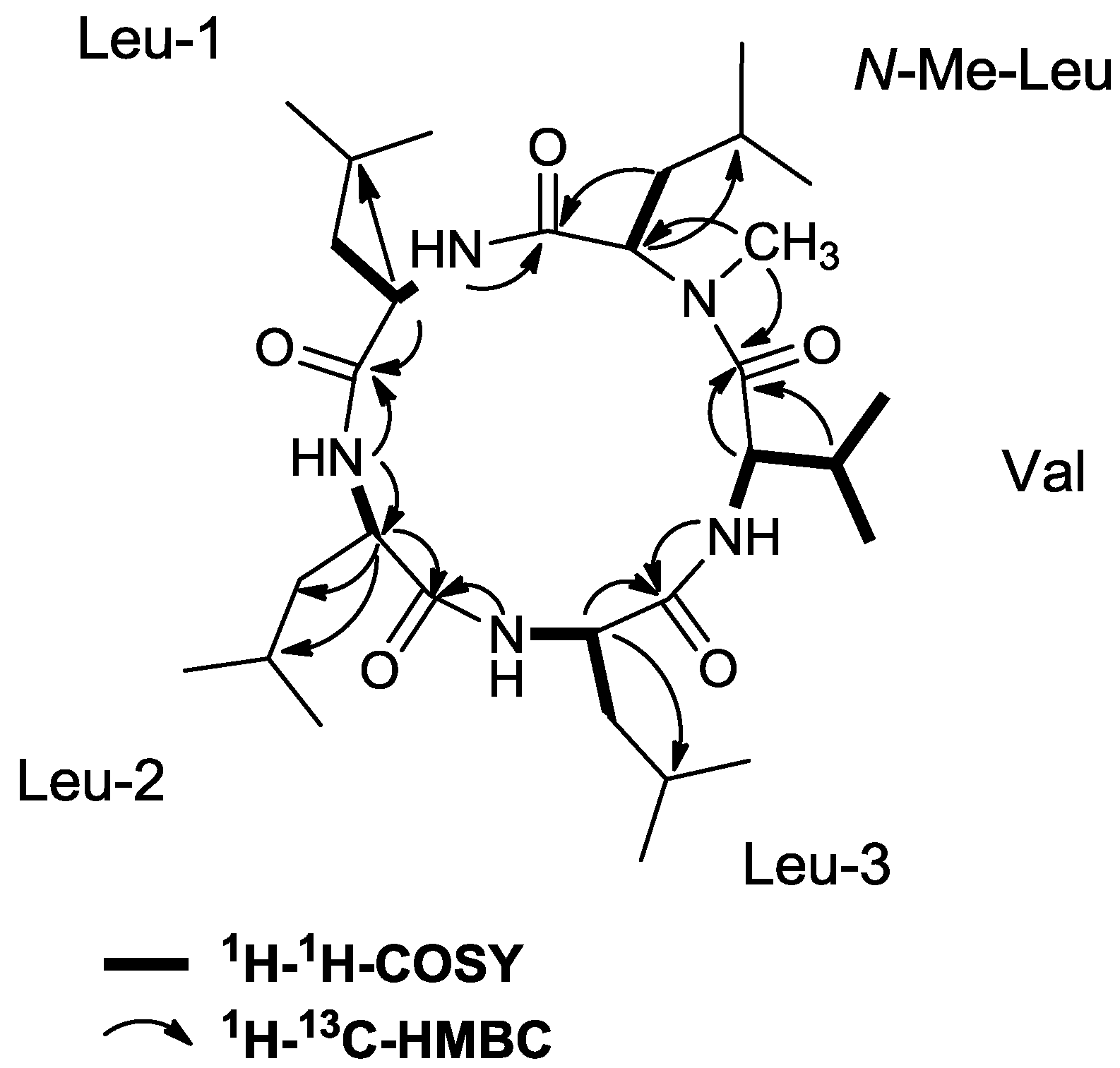
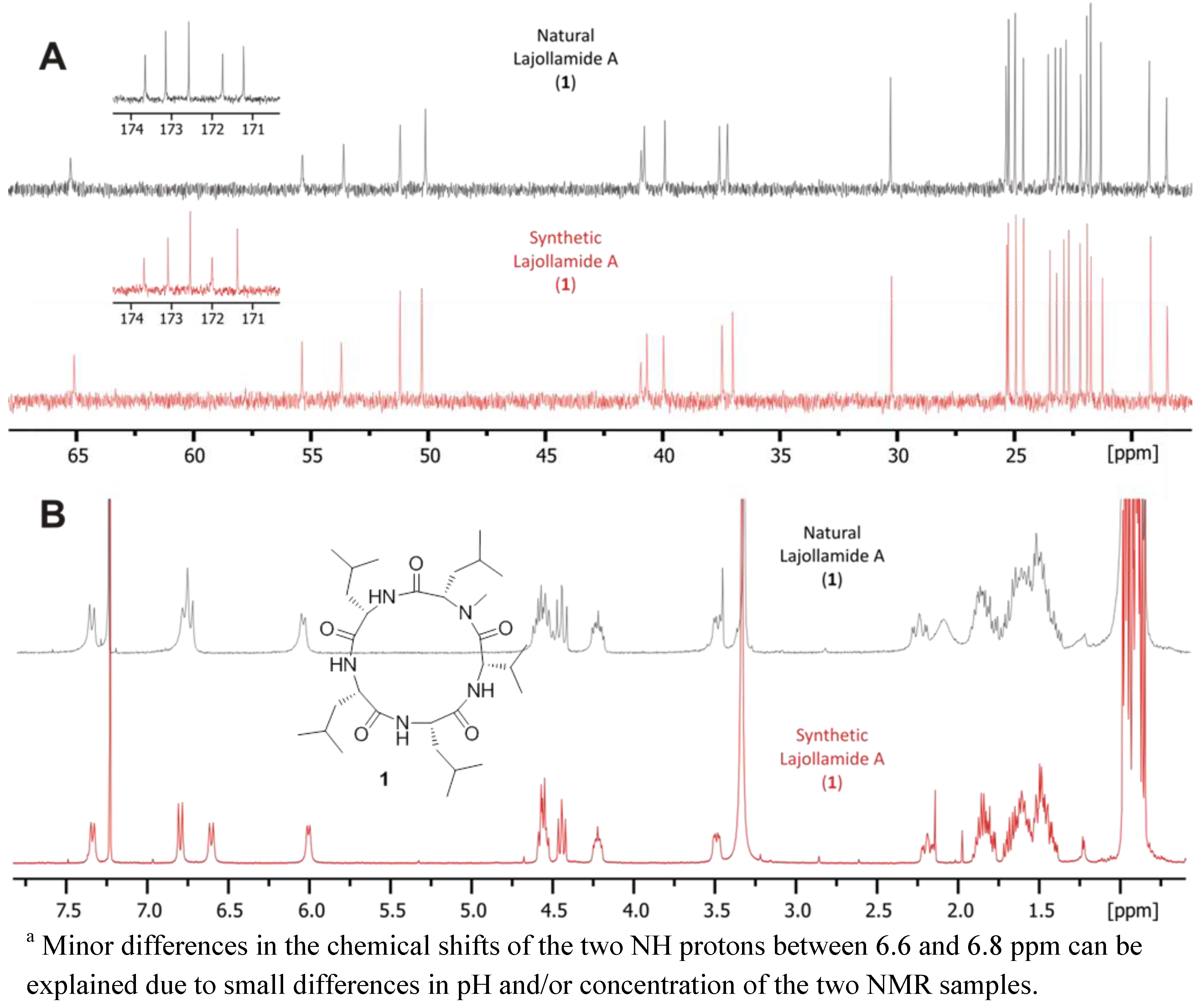
2.2. Isolation and Structure Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | Position | δCb | δH (mult, J in Hz) |
|---|---|---|---|
| Leu-1 | C=O | 171.7, C | |
| α | 53.6, CH | 4.23, m | |
| β | 40.7, CH2 | 1.56, m; 1.77, m | |
| γ | 24.9, CH | 1.54 m | |
| δ | 23.0, CH3c | 0.94, d c | |
| 21.3, CH3c | 0.90, d c | ||
| NH | 6.21, brs | ||
| Leu-2 | C=O | 172.5, C | |
| α | 51.2, CH | 4.48, m | |
| β | 39.9, CH2 | 1.49, m; 1.86, m | |
| γ | 25.2, CH | 1.59, m | |
| δ | 22.1, CH3c | 0.86, d c | |
| 22.8, CH3c | 0.90, d c | ||
| NH | 7.42, d (8.3) | ||
| Leu-3 | C=O | 173.1, C | |
| α | 50.1, CH | 4.58, m | |
| β | 37.2, CH2 | 1.48, m; 1.69, m | |
| γ | 24.6, CH | 1.51, m | |
| δ | 23.2, CH3c | 0.92, d c | |
| 21.9, CH3c | 0.88, d c | ||
| NH | 7.27, m | ||
| Val | C=O | 173.6, C | |
| α | 55.3, C | 4.48, m | |
| β | 30.3, CH | 1.89, m | |
| γ | 18.5, CH3 | 0.94 | |
| 19.2, CH3 | 0.92 | ||
| NH | 6.92, d (8.9) | ||
| N-Me-Leu | C=O | 171.2, C | |
| α | 65.2, CH | 3.49, dd (4.1, 3.6) | |
| β | 37.6, CH2 | 1.44, m; 2.24, m | |
| γ | 25.3, CH | 1.59, m | |
| δ | 23.5, CH3c | 0.96, d c | |
| 21.7, CH3c | 0.97, d c | ||
| N-Me | 40.9, CH3 | 3.29, s |
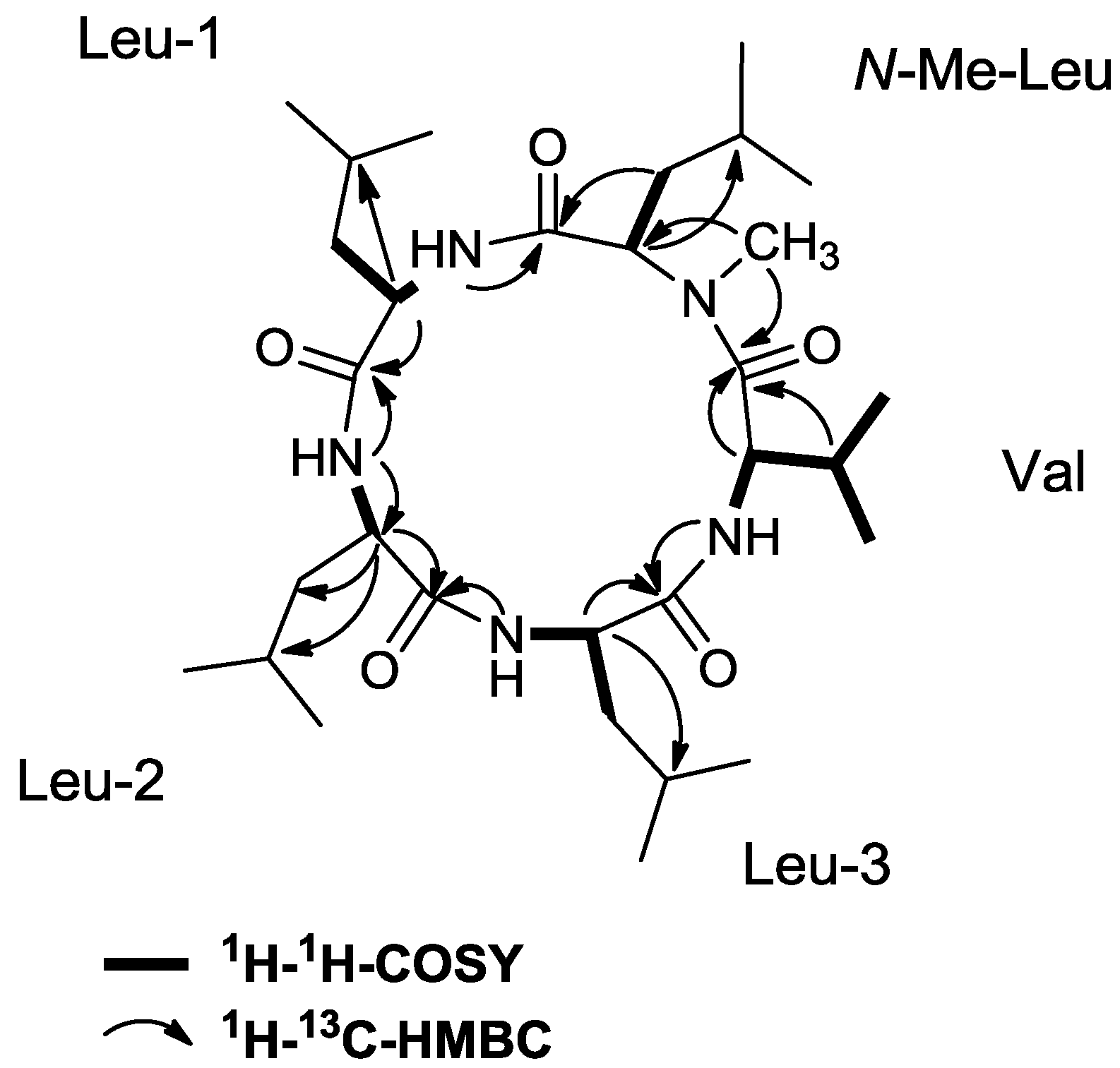
2.3. Initial Stereochemical Investigations

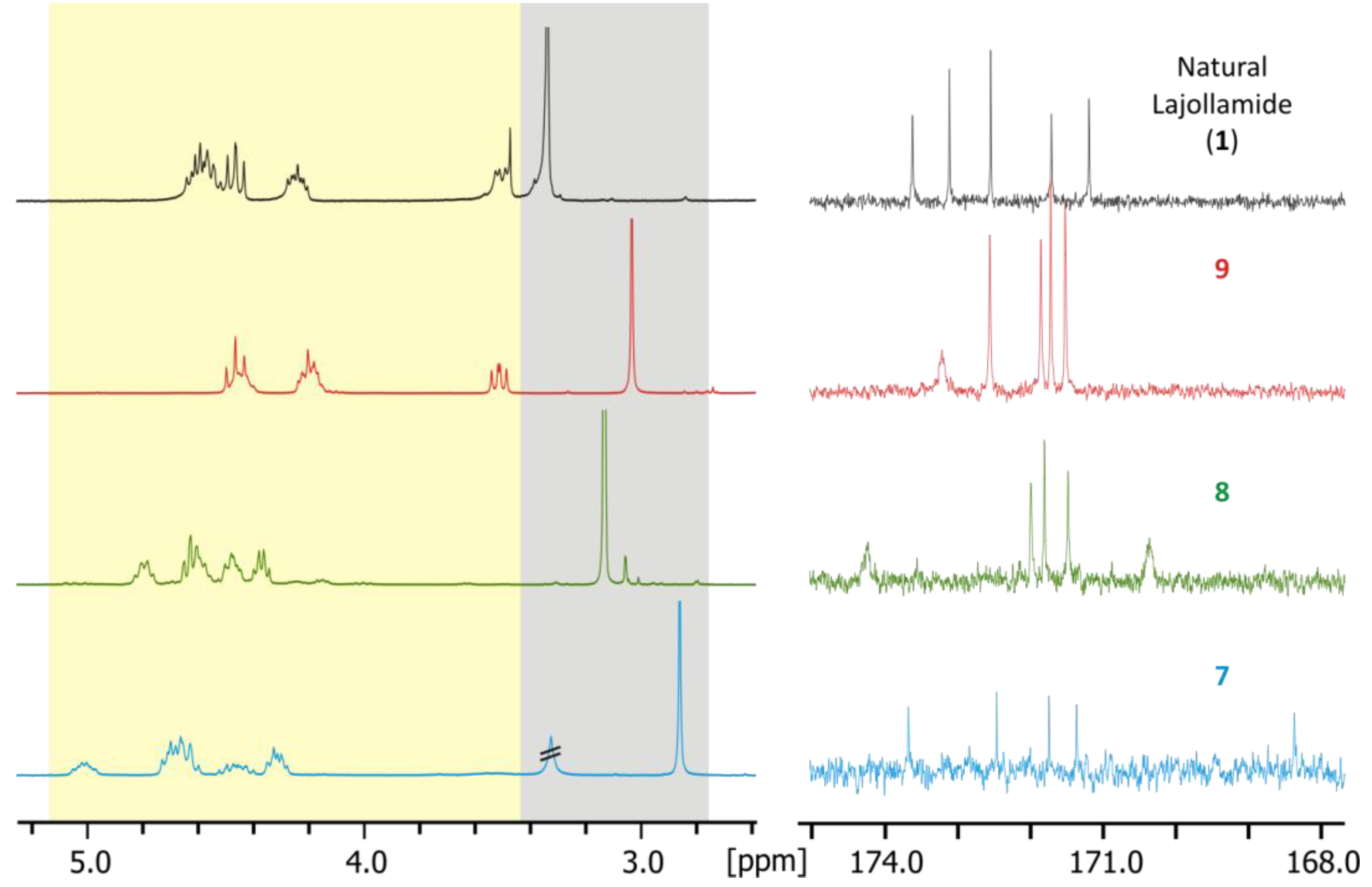
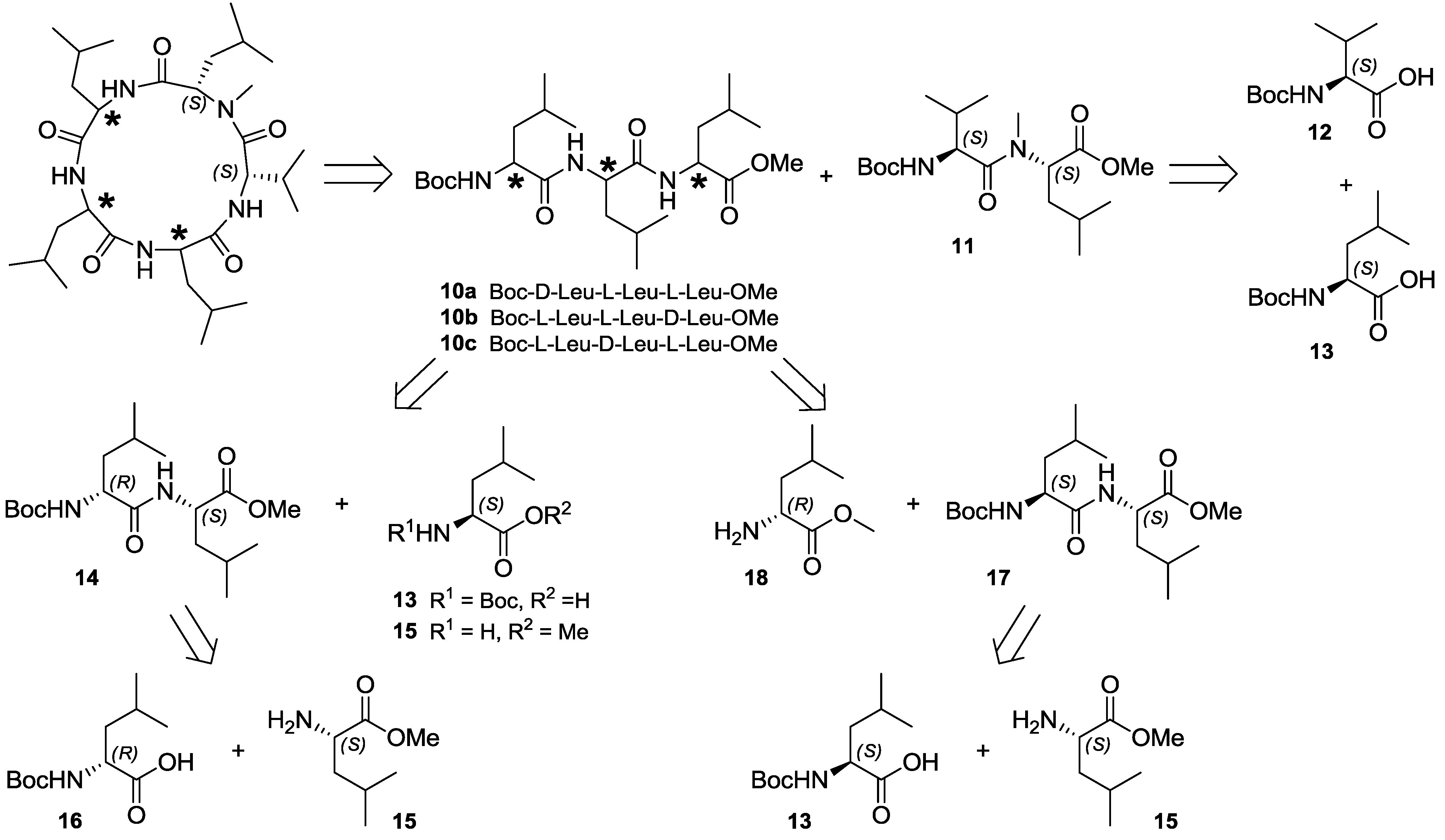
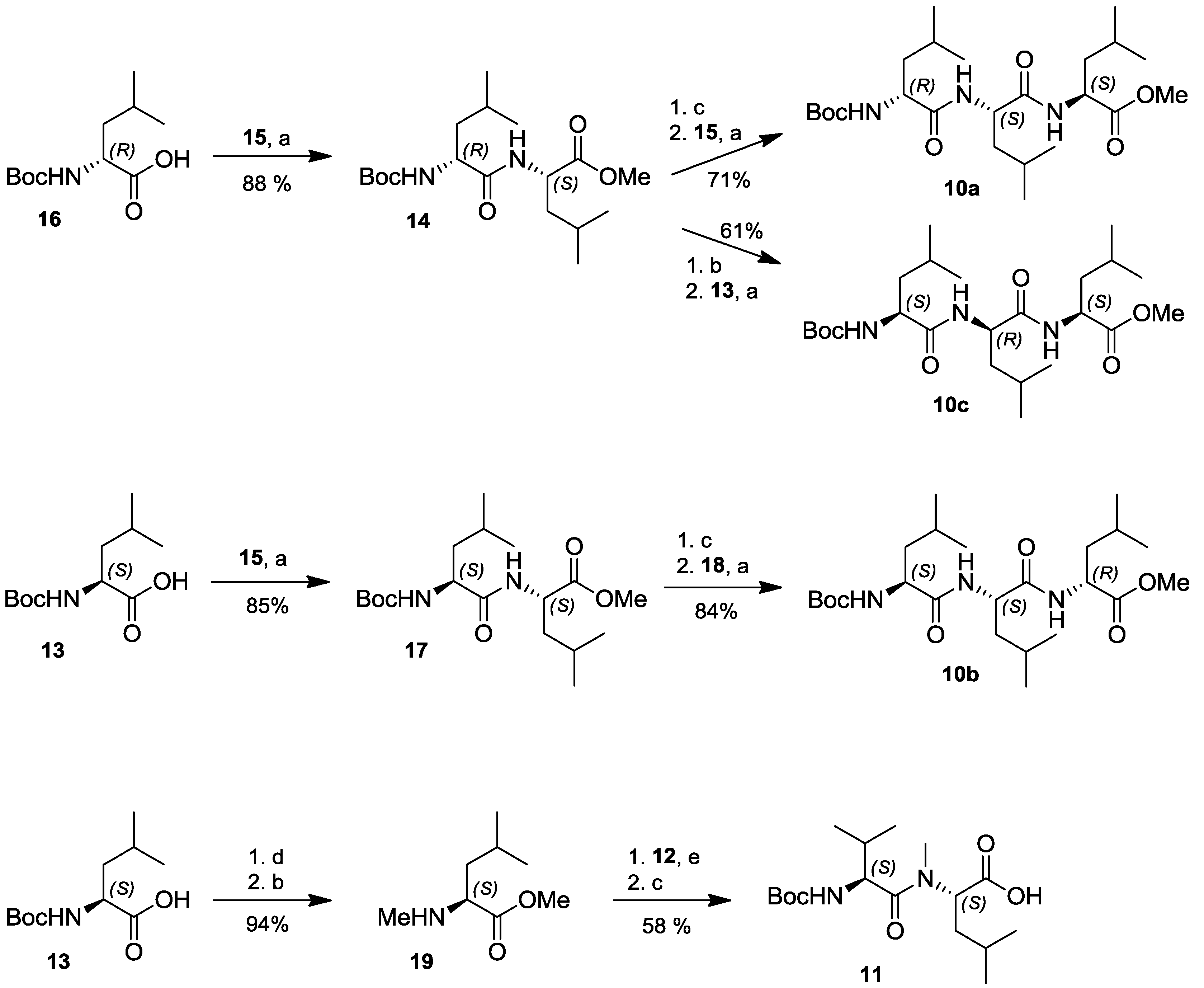
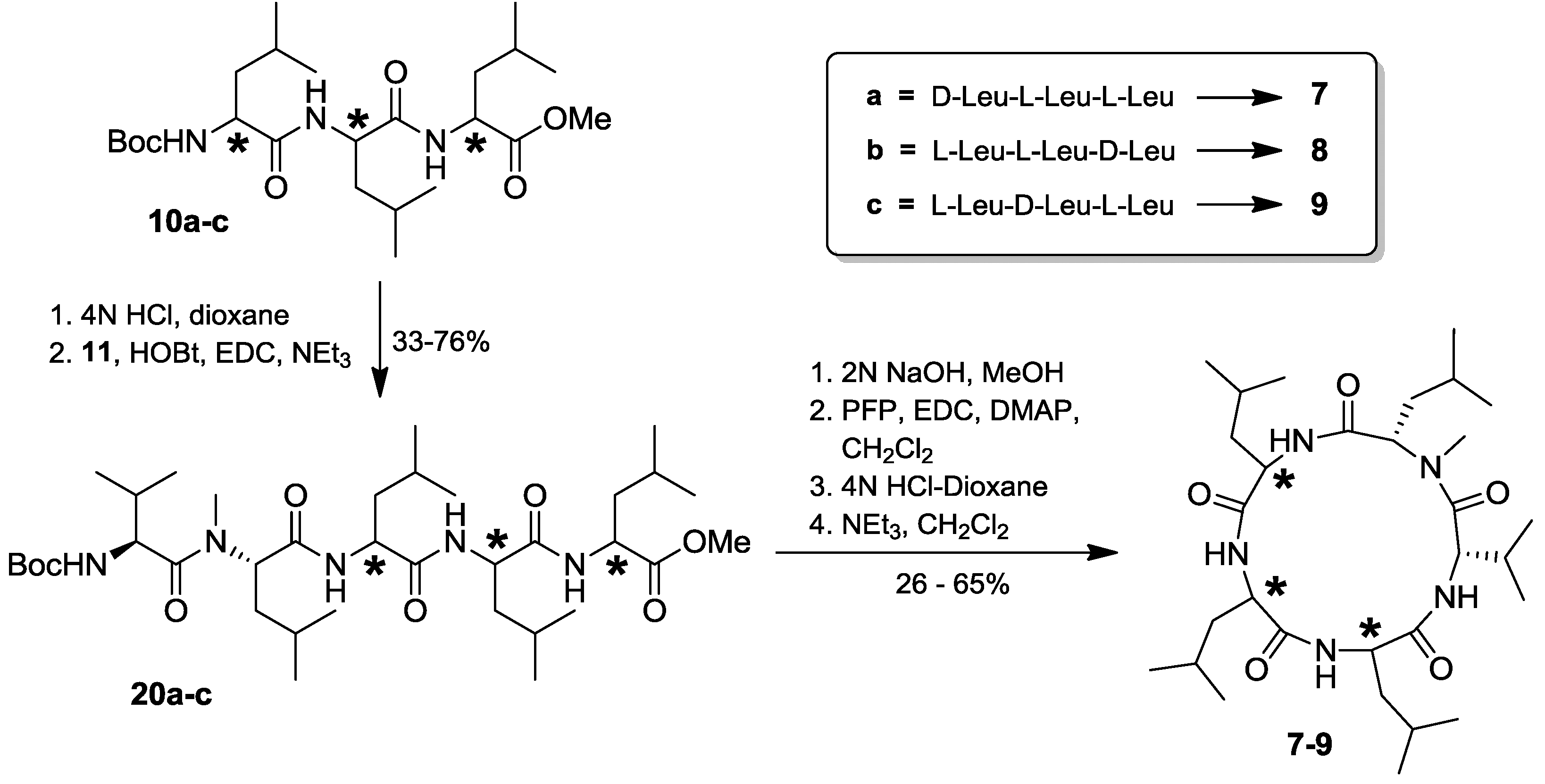
2.4. Total Synthesis of Natural Lajollamide A (1) and of Its Diastereomeric Analogs B–D (7–9)
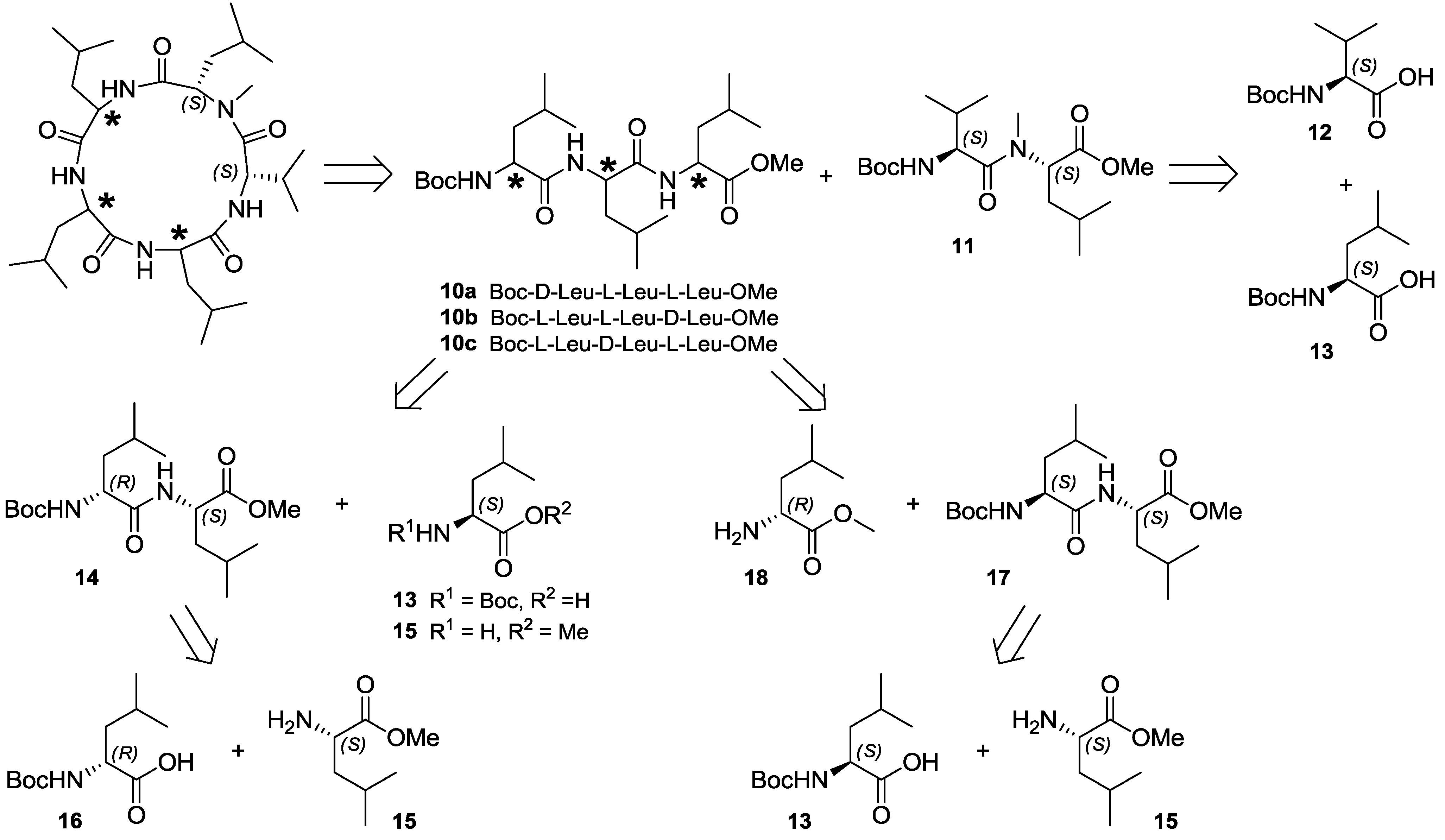
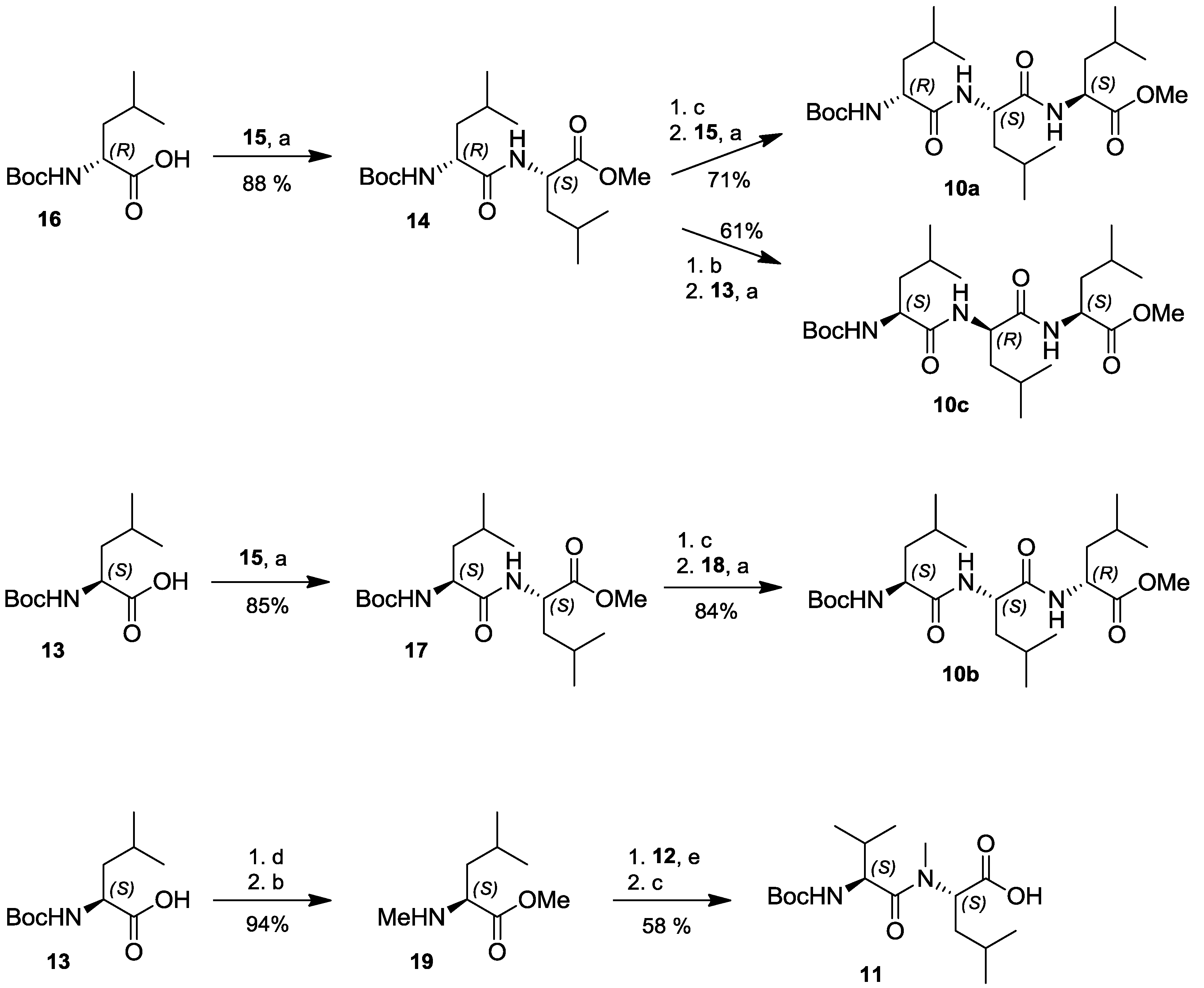
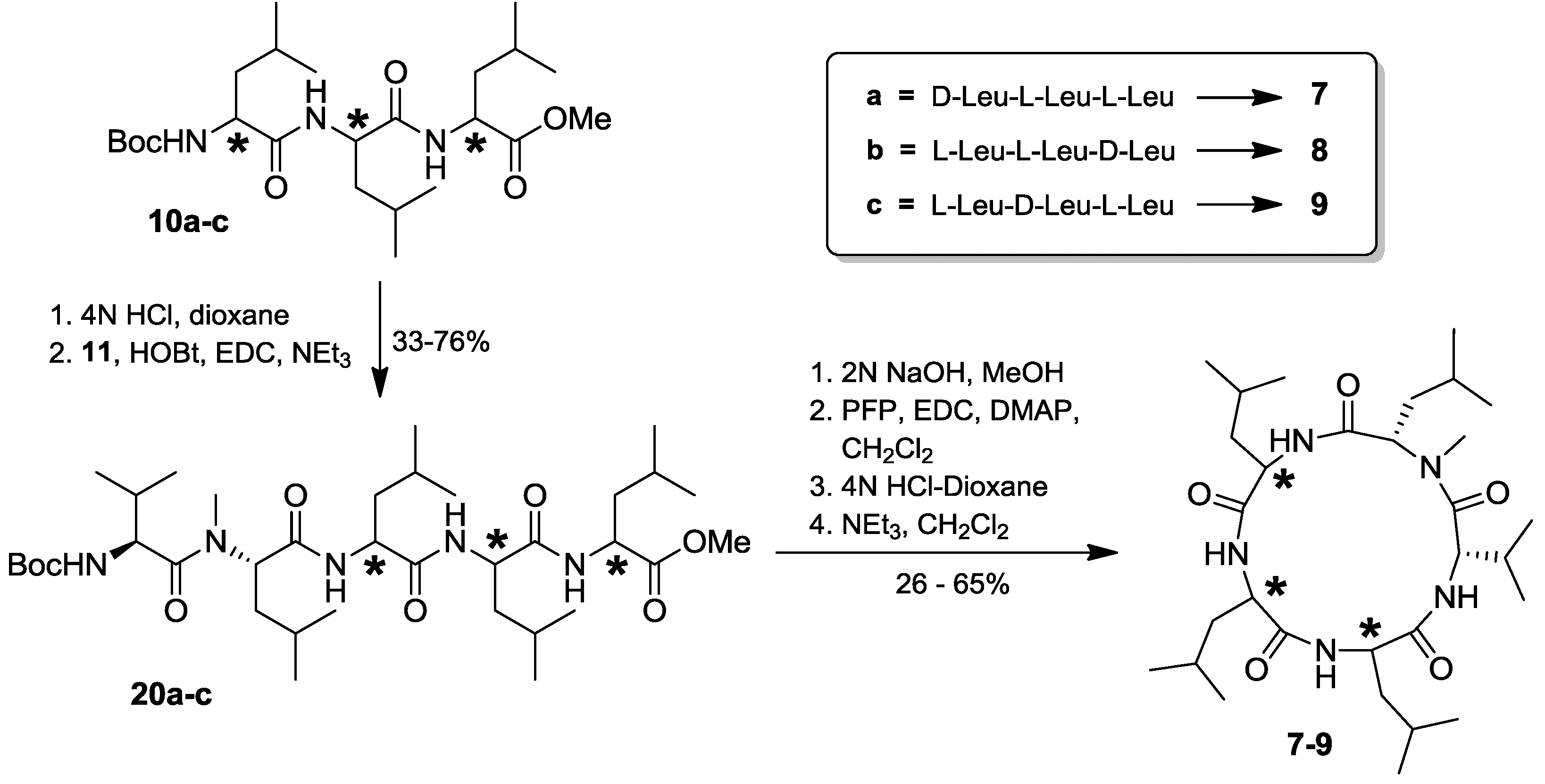

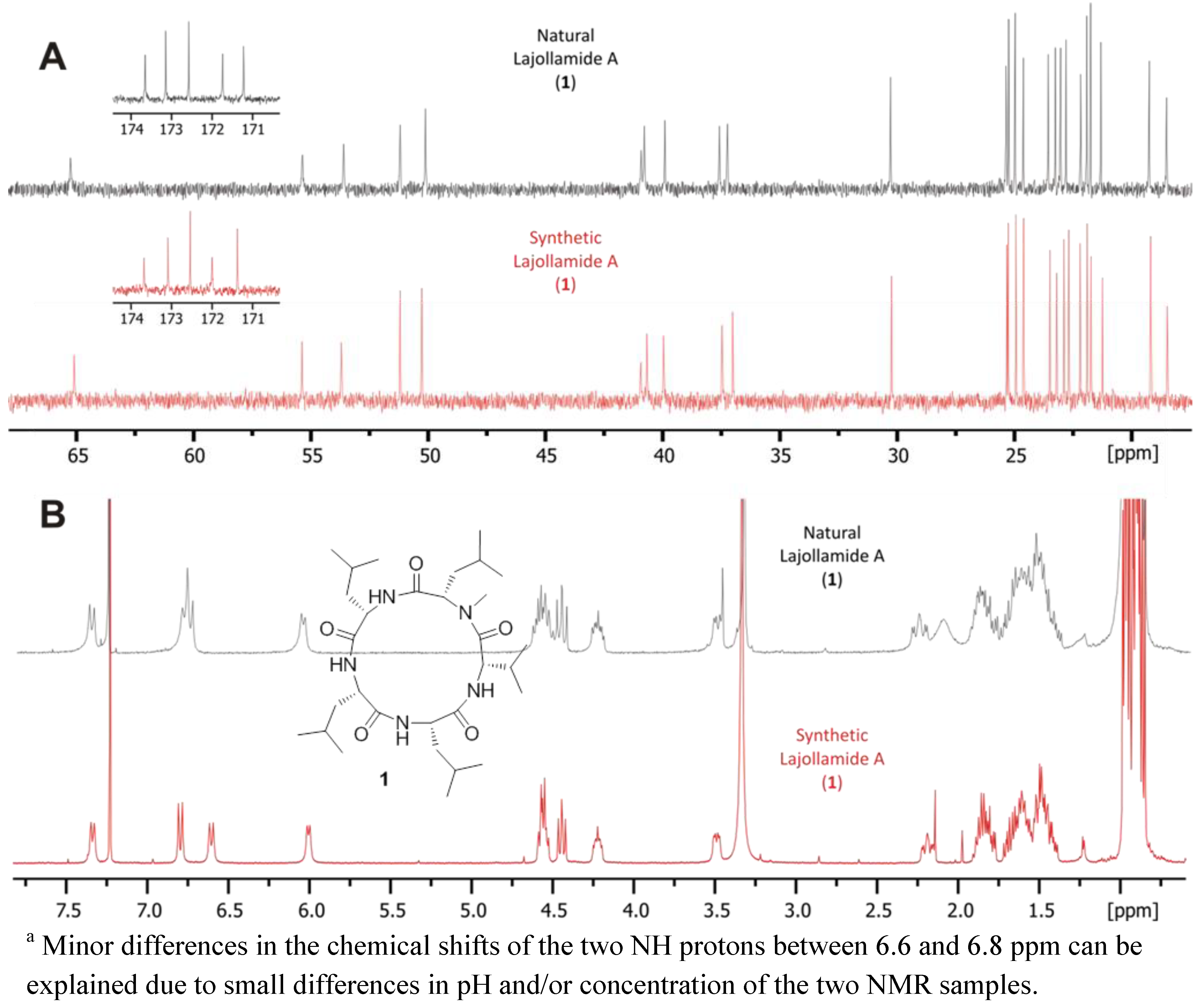
2.5. Biological Activities of Compounds 1–9
3. Experimental Section
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Screening of Secondary Metabolites Biosynthesized under Different Conditions (OSMAC Aproach)
3.4. Scaled-Up Cultivation, Extraction and Isolation of Metabolites of A. cruciatus
3.5. Amino Acid Analysis by Chiral HPLC
3.6. General Synthetic Protocols for the Preparation of Lajollamides A–D (1, 7–9)
3.6.1. Peptide Coupling (Protocol A)
3.6.2. N-Deprotection (Protocol B)
3.6.2.1. Deprotection with Acetyl Chloride in Methanol
3.6.2.2. Deprotection with 4 M HCl∙Dioxane
3.6.3. Saponification (Protocol C)
3.6.4. Macrolactonization (Protocol D)
3.7. Total Synthesis of Lajollamides A–D (1, 7–9)
3.7.1. N-Boc-L-Leu-L-Leu-OMe (17)
3.7.2. N-Boc-D-Leu-L-Leu-OMe (14)
3.7.3. N-Boc-D-Leu-L-Leu-L-Leu-OMe (10a)
3.7.4. N-Boc-L-Leu-L-Leu-D-Leu-OMe (10b)
3.7.5. N-Boc-L-Leu-D-Leu-L-Leu-OMe (10c)
3.7.6. N-Boc-L-Leu-L-Leu-l-Leu-OMe (10d)
3.7.7. N-Me-L-Leu-OMe (19)
3.7.8. N-Boc-L-Val-N-Me-L-Leu (11)
3.7.9. N-Boc-L-Val-N-Me-L-Leu-D-Leu-L-Leu-L-Leu-OMe (20a)
3.7.10. N-Boc-L-Val-N-Me-L-Leu-L-Leu-L-Leu-D-Leu-OMe (20b)
3.7.11. N-Boc-L-Val-N-Me-L-Leu-L-Leu-d-Leu-L-Leu-OMe (20c)
3.7.12. N-Boc-L-Val-N-Me-L-Leu-L-Leu-L-Leu-L-Leu-OMe (20d)
3.7.13. Lajollamide B (7)
3.7.14. Lajollamide C (8)
3.7.15. Lajollamide D (9)
3.7.16. Lajollamide A (1)
3.8. Biological Activity
4. Conclusions
Acknowledgments
Supplementary Files
Supplementary File 1References
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef]
- Greve, H.; Mohamed, I.E.; Pontius, A.; Kehraus, S.; Gross, H.; König, G.M. Fungal metabolites: Structural diversity as incentive for anticancer drug development. Phytochem. Rev. 2010, 9, 537–545. [Google Scholar] [CrossRef]
- Saleem, M.; Ali, M.S.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar] [CrossRef]
- Bhadury, P.; Mohammad, B.T.; Wright, P.C. The current status of natural products from marine fungi and their potential as anti-infective agents. J. Ind. Microbiol. Biotechnol. 2006, 33, 325–337. [Google Scholar]
- Van den Berg, M.A.; Albang, R.; Albermann, K.; Badger, J.H.; Daran, J.-M.; Driessen, A.J.M.; Garcia-Estrada, C.; Fedorova, N.D.; Harris, D.M.; Heijne, W.H.M.; et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 2008, 26, 1161–1168. [Google Scholar] [CrossRef]
- Sanchez, J.F.; Somoza, A.D.; Keller, N.P.; Wang, C.C.C. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat. Prod. Rep. 2012, 29, 351–371. [Google Scholar] [CrossRef]
- Gross, H. Genomic mining—A concept for the discovery of new bioactive natural products. Curr. Opin. Drug Discov. Dev. 2009, 12, 207–219. [Google Scholar]
- Hennebert, G.L. Wardomyces and Asteromyces. Can. J. Bot. 1962, 40, 1203–1216. [Google Scholar] [CrossRef]
- Shin, J.; Fenical, W. Isolation of gliovictin from the marine deuteromycete Asteromyces cruciatus. Phytochemistry 1987, 26, 3347. [Google Scholar] [CrossRef]
- Höller, U.; König, G.M.; Wright, A.D. A new tyrosine kinase inhibitor from a marine isolate of Ulocladium botrytis and new metabolites from the marine fungi Asteromyces cruciatus and Varicosporina ramulosa. Eur. J. Org. Chem. 1999, 1999, 2949–2955. [Google Scholar] [CrossRef]
- Mesry, R. Isolierung und Strukturaufklärung von Sekundärmetaboliten aus den Marinen Pilzen Asteromyces cruciatus und Stagonospora sp. Diploma Thesis, University of Bonn, Germany, 2008. [Google Scholar]
- Bode, H.B.; Bethe, B.; Höfs, A.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Pimenta, E.F.; Vita-Marques, A.M.; Tininis, A.; Seleghim, M.H.R.; Sette, L.D.; Veloso, K.; Ferreira, A.G.; Williams, D.E.; Patrick, B.O.; Dalisay, D.S.; et al. Use of experimental design for the optimization of the production of new secondary metabolites by two Penicillium species. J. Nat. Prod. 2010, 73, 1821–1832. [Google Scholar] [CrossRef]
- Paranagama, P.A.; Wijeratne, E.M.K.; Gunatilaka, A.A.L. Uncovering biosynthetic potential of plant-associated fungi: Effect of culture conditions on metabolite production by Paraphaeosphaeria quadriseptata and Chaetomium chiversii. J. Nat. Prod. 2007, 70, 1939–1945. [Google Scholar] [CrossRef]
- Williams, R.B.; Henrikson, J.C.; Hoover, A.R.; Lee, A.E.; Cichewicz, R.H. Epigenetic remodeling of the fungal secondary metabolome. Org. Biomol. Chem. 2008, 6, 1895–1897. [Google Scholar] [CrossRef]
- Michel, K.H.; Chaney, M.O.; Jones, N.D.; Hoehn, M.M.; Nagarajan, R. Epipolythiopiperazinedione antibiotics from Penicillium turbatum. J. Antibiot. 1974, 27, 57–64. [Google Scholar] [CrossRef]
- Dorn, F.; Arigoni, D. Gliovictin, ein neuer Metabolit von Helminthosporium victoriae. Experientia 1974, 30, 134–135. [Google Scholar] [CrossRef]
- Isaka, M.; Palasarn, S.; Rachtawee, P.; Vimuttipong, S.; Kongsaeree, P. Unique diketopiperazine dimers from the insect pathogenic fungus Verticillium hemipterigenum BCC 1449. Org. Lett. 2005, 7, 2257–2260. [Google Scholar] [CrossRef]
- Prachyawarakorn, V.; Mahidol, C.; Sureram, S.; Sangpetsiripan, S.; Wiyakrutta, S.; Ruchirawat, S.; Kittakoop, P. Diketopiperazines and phthalides from a marine-derived fungus of the order Pleosporales. Planta Med. 2008, 74, 69–72. [Google Scholar] [CrossRef]
- Kirby, G.W.; Rao, G.V.; Robins, D.J. New co-metabolites of gliotoxin in Gliocladium virens. J. Chem. Soc. Perkin Trans. I 1988, 301–304. [Google Scholar] [CrossRef]
- Venkatasubbaiah, P.; Chilton, W.S. Toxins produced by the dogwood anthracnose fungus Discula sp. J. Nat. Prod. 1991, 54, 1293–1297. [Google Scholar] [CrossRef]
- Evidente, A.; Superchi, S.; Cimmino, A.; Mazzeo, G.; Mugnai, L.; Rubiales, D.; Andolfi, A.; Villegas-Fernández, A.M. Regiolone and isosclerone, two enantiomeric phytotoxic naphthalenone pentaketides: Computational assignment of absolute configuration and its relationship with phytotoxic activity. Eur. J. Org. Chem. 2011, 2011, 5564–5570. [Google Scholar]
- Höller, U. Isolation, Biological Activity, and Secondary Metabolite Investigations of Marine-Derived Fungi and Selected Host Sponges. Ph.D. Thesis, Braunschweig University of Technology, Germany, 1999. [Google Scholar]
- Höller, U.; König, G.M.; Wright, A.D. Three new metabolites from marine-derived fungi of the genera Coniothyrium and Microsphaeropsis. J. Nat. Prod. 1999, 62, 114–118. [Google Scholar] [CrossRef]
- Höller, U.; Wright, A.D.; Matthée, G.F.; König, G.M.; Draeger, S.; Aust, H.-J.; Schulz, B. Fungi from marine sponges: Diversity, biological activity and secondary metabolites. Mycol. Res. 2000, 104, 1354–1365. [Google Scholar] [CrossRef]
- Thom, C.; Raper, K.B. A Manual of the Aspergilli; Williams and Wilkins Co.: Baltimore, MD, USA, 1945. [Google Scholar]
- Smith, G. An Introduction to Industrial Mycology, 5th ed; Edward Arnold Ltd.: London, UK, 1960. [Google Scholar]
- Demain, A.L. Regulation of secondary metabolism in fungi. Pure Appl. Chem. 1986, 58, 219–226. [Google Scholar] [CrossRef]
- Pettit, R.K. Mixed fermentation for natural product drug discovery. Appl. Microbiol. Biotechnol. 2009, 83, 19–25. [Google Scholar] [CrossRef]
- Howell, C.R.; Stipanovic, R.D. Control of Rhizoctonia solani on cotton seedlings with Pseudomonas fluorescens and with an antibiotic produced by the bacterium. Phytopathology 1979, 69, 480–482. [Google Scholar] [CrossRef]
- Serafimidis, I.; Kay, R.R. New prestalk and prespore inducing signals in Dictyostelium. Dev. Biol. 2005, 282, 432–441. [Google Scholar] [CrossRef]
- Mitova, M.I.; Lang, G.; Wiese, J.; Imhoff, J.F. Subinhibitory concentractions of antibiotics induce phenazine production in a marine Streptomyces sp. J. Nat. Prod. 2008, 71, 824–827. [Google Scholar] [CrossRef]
- Cummins, J.; Reen, F.J.; Baysse, C.; Mooij, M.J.; O’Gara, F. Subinhibitory concentrations of the cationic antimicrobial peptide colistin induce the pseudomonas quinolone signal in Pseudomonas aeruginosa. Microbiology 2009, 155, 2826–2837. [Google Scholar] [CrossRef]
- Schulz, D.; Beese, P.; Ohlendorf, B.; Erhard, A.; Zinecker, H.; Dorador, C.; Imhoff, J.F. Abenquines A–D: Aminoquinone derivatives produced by Streptomyces sp. strain DB634. J. Antibiot. 2011, 64, 763–768. [Google Scholar] [CrossRef]
- Ohlendorf, B.; Schulz, D.; Erhard, A.; Nagel, K.; Imhoff, J.F. Geranylphenazinediol, an acetylcholinesterase inhibitor produced by a Streptomyces species. J. Nat. Prod. 2012, 75, 1400–1404. [Google Scholar] [CrossRef]
- Baki, A.; Bielik, A.; Molnar, L.; Szendrei, G.; Keseru, G.M.A. A high throughput luminescent assay for glycogen synthase kinase-3beta inhibitors. Assay Drug Dev. Technol. 2007, 5, 75–83. [Google Scholar] [CrossRef]
- Schulz, B.; Sucker, J.; Aust, H.J.; Krohn, K.; Ludewig, K.; Jones, P.G.; Döring, D. Biologically active secondary metabolites of the endophytic Pezicula species. Mycol. Res. 1995, 99, 1007–1015. [Google Scholar] [CrossRef]
- Belofsky, B.N.; Jensen, P.R.; Fenical, W. Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999, 40, 2913–2916. [Google Scholar] [CrossRef]
- Mouafo Talontsi, F.; Facey, P.; Kongue Tatong, M.D.; Tofazzal Islam, M.; Frauendorf, H.; Draeger, S.; von Tiedemann, A.; Laatsch, H. Zoosporicidal metabolites from an endophytic fungus Cryptosporiopsis sp. of Zanthoxylum leprieurii. Phytochemistry 2012, 83, 87–94. [Google Scholar]
- Li, H.J.; Lin, Y.C.; Yao, J.H.; Vrijmoed, L.; Jones, G. Two new metabolites from the mangrove endophytic fungus No. 2524. J. Asian Nat. Prod. Res. 2004, 6, 185–191. [Google Scholar] [CrossRef]
- Bode, H.B.; Reimer, D.; Fuchs, S.W.; Kirchner, F.; Dauth, C.; Kegler, C.; Lorenzen, W.; Brachmann, A.O.; Grün, P. Determination of the absolute configuration of peptide natural products by using stable isotope labeling and mass spectrometry. Chem. Eur. J. 2012, 18, 2342–2348. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gulder, T.A.M.; Hong, H.; Correa, J.; Egereva, E.; Wiese, J.; Imhoff, J.F.; Gross, H. Isolation, Structure Elucidation and Total Synthesis of Lajollamide A from the Marine Fungus Asteromyces cruciatus. Mar. Drugs 2012, 10, 2912-2935. https://doi.org/10.3390/md10122912
Gulder TAM, Hong H, Correa J, Egereva E, Wiese J, Imhoff JF, Gross H. Isolation, Structure Elucidation and Total Synthesis of Lajollamide A from the Marine Fungus Asteromyces cruciatus. Marine Drugs. 2012; 10(12):2912-2935. https://doi.org/10.3390/md10122912
Chicago/Turabian StyleGulder, Tobias A. M., Hanna Hong, Jhonny Correa, Ekaterina Egereva, Jutta Wiese, Johannes F. Imhoff, and Harald Gross. 2012. "Isolation, Structure Elucidation and Total Synthesis of Lajollamide A from the Marine Fungus Asteromyces cruciatus" Marine Drugs 10, no. 12: 2912-2935. https://doi.org/10.3390/md10122912
APA StyleGulder, T. A. M., Hong, H., Correa, J., Egereva, E., Wiese, J., Imhoff, J. F., & Gross, H. (2012). Isolation, Structure Elucidation and Total Synthesis of Lajollamide A from the Marine Fungus Asteromyces cruciatus. Marine Drugs, 10(12), 2912-2935. https://doi.org/10.3390/md10122912