Induction of Apoptosis by Sinulariolide from Soft Coral through Mitochondrial-Related and p38MAPK Pathways on Human Bladder Carcinoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
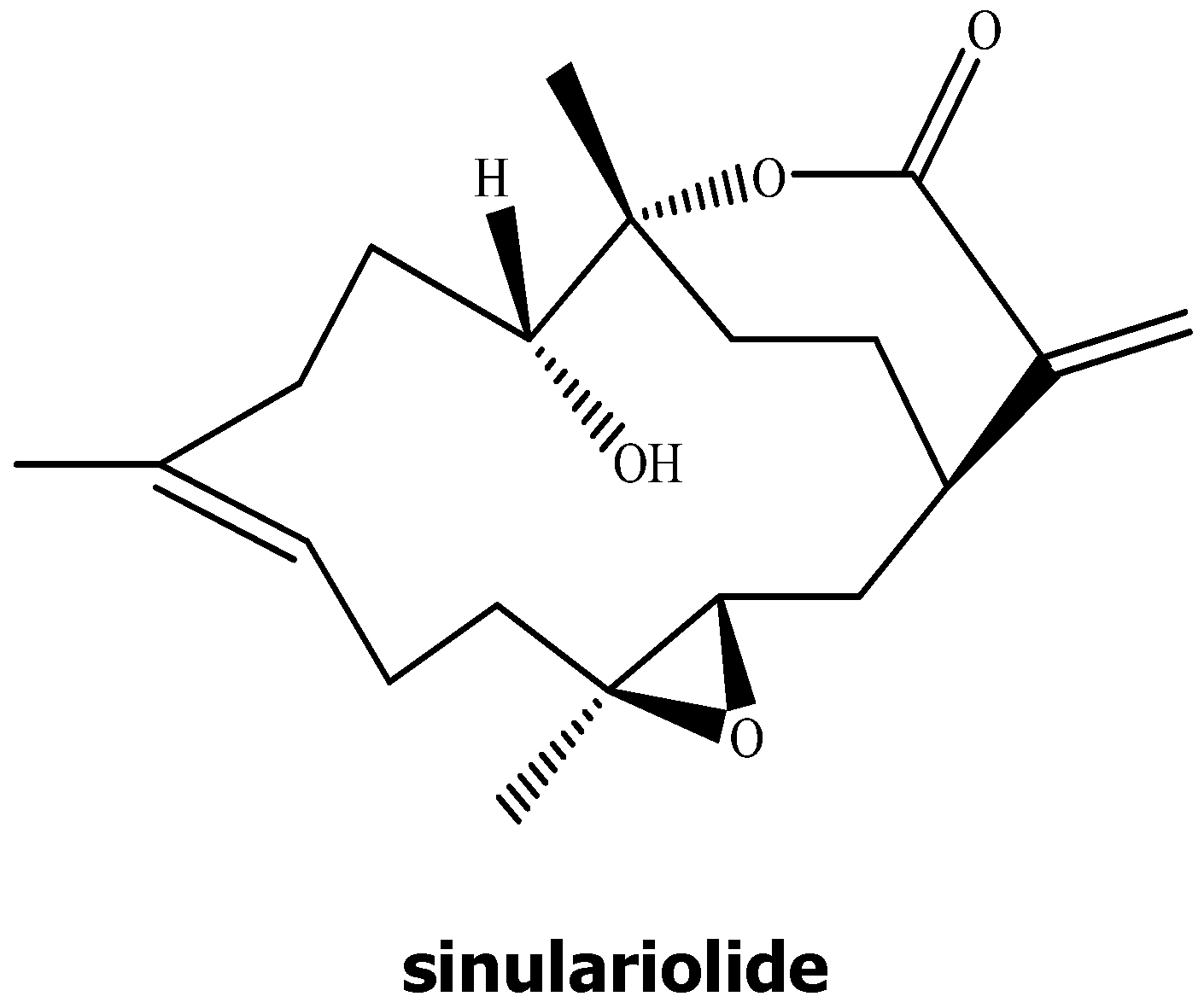
2. Results
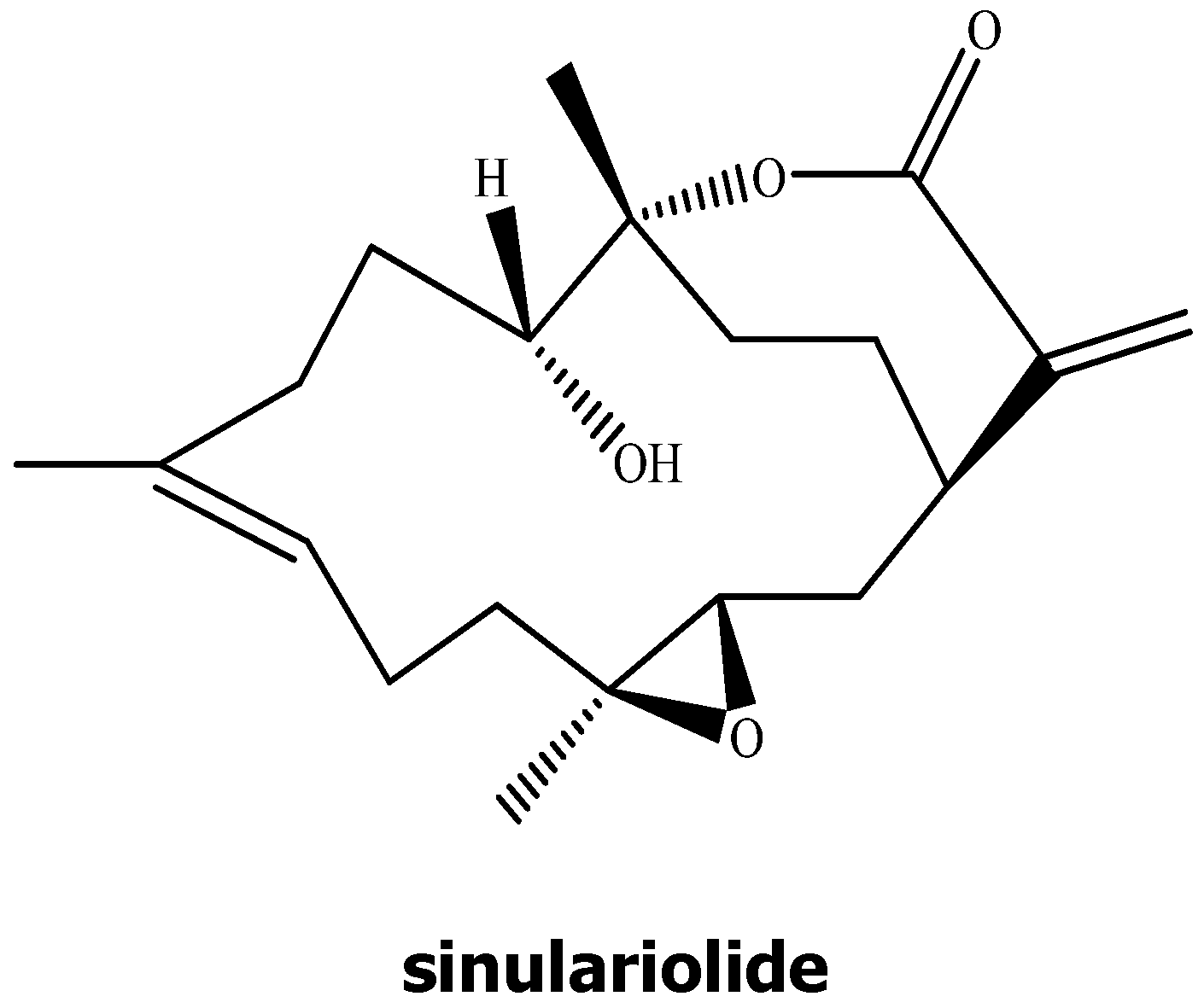
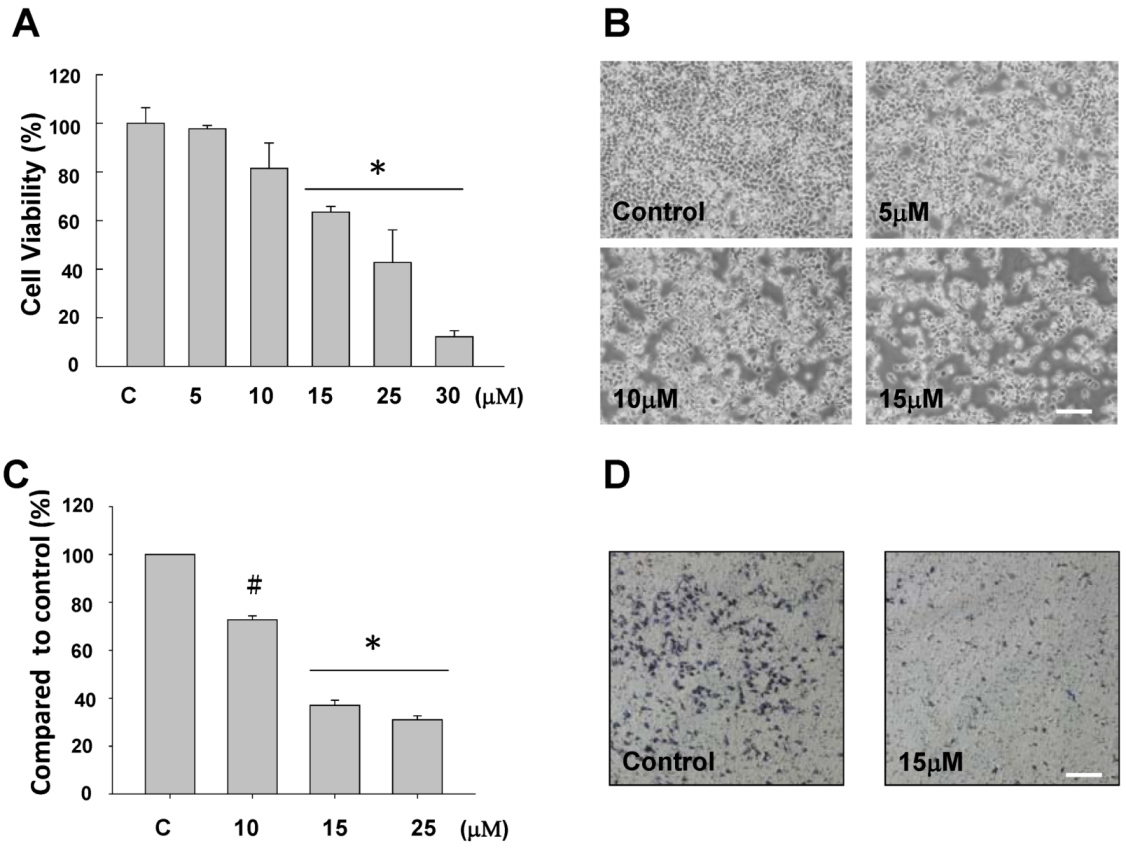
2.1. The Anti-Proliferative and Anti-Migratory Effect of Sinulariolide on TSGH Cells
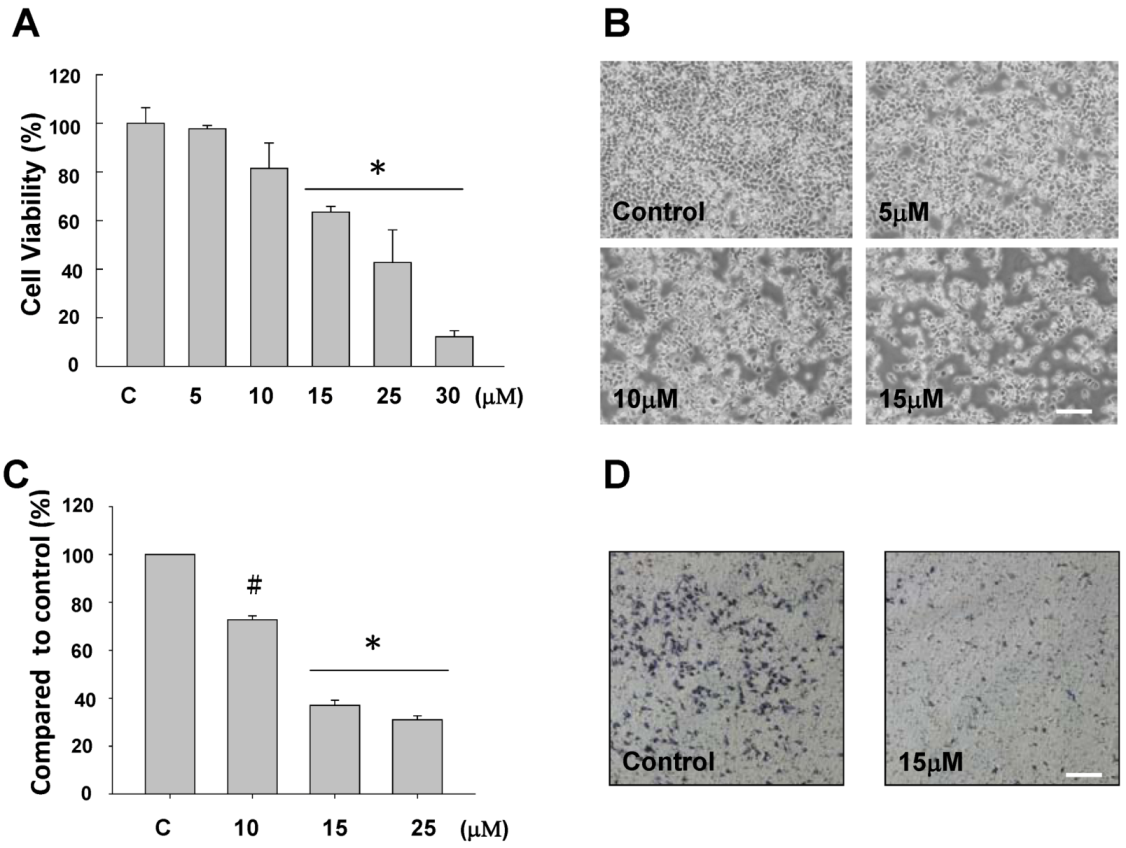
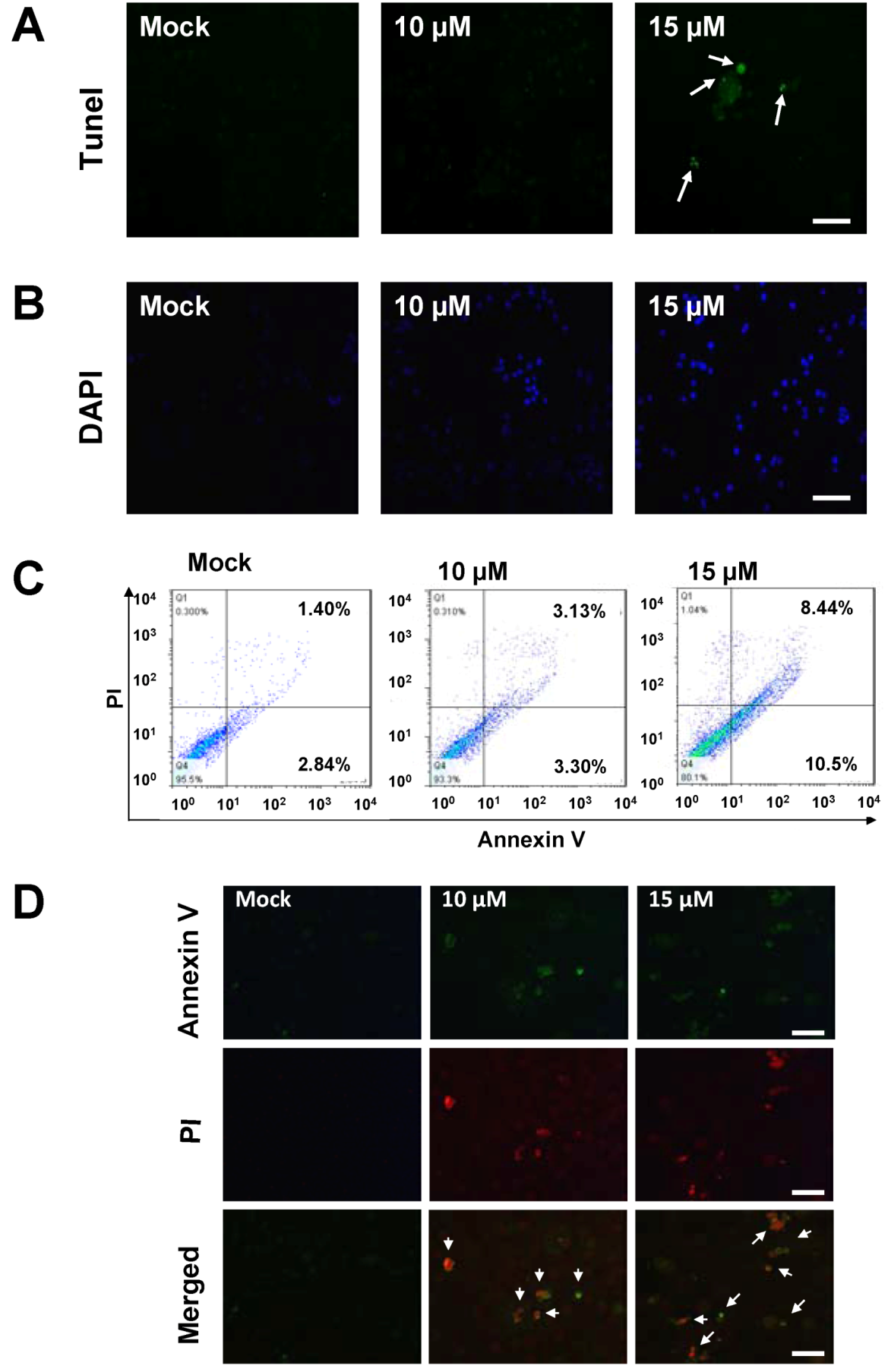
2.2. Sinulariolide-Treated TSGH Cells Adopt Apoptosis Characteristics
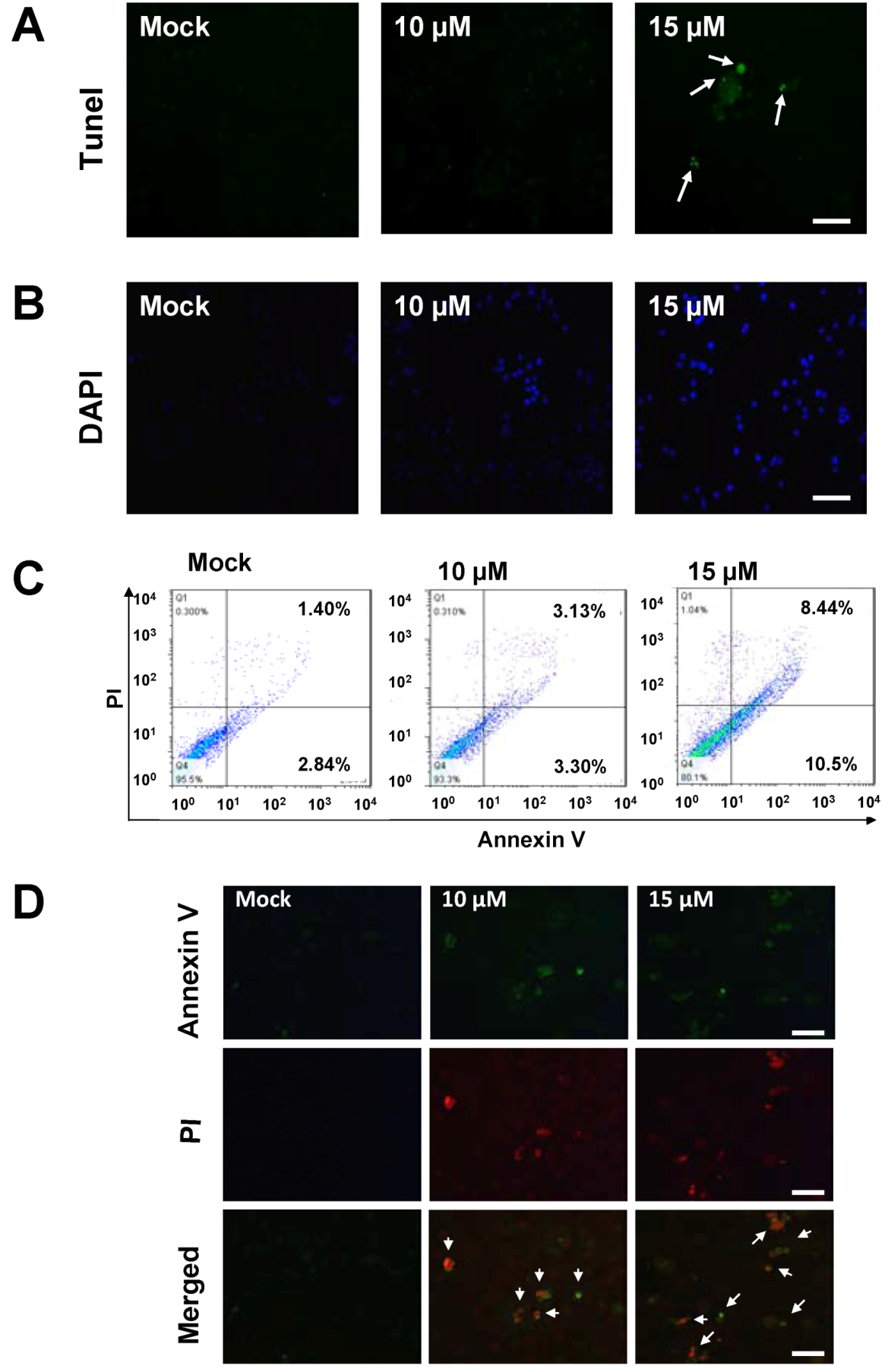
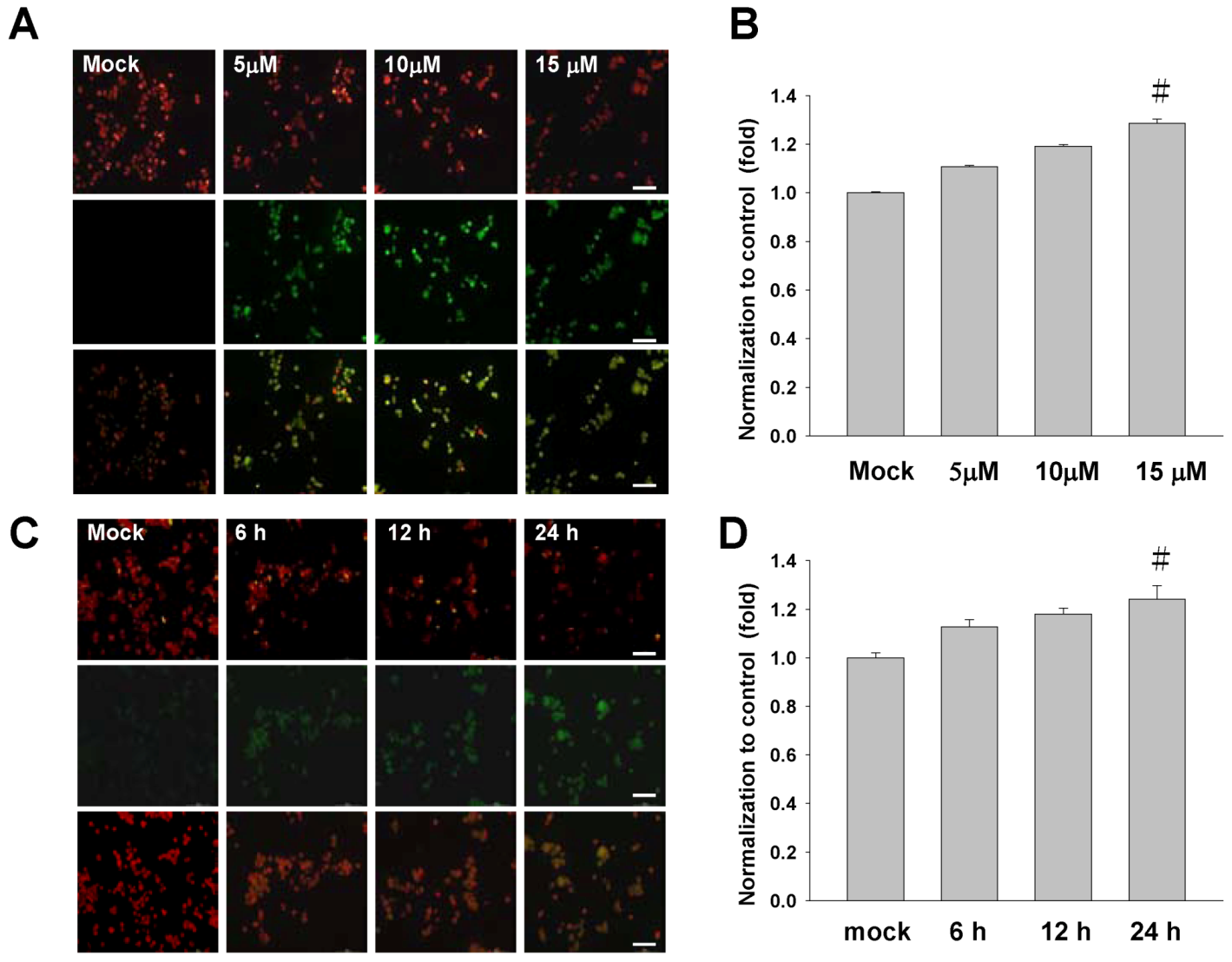
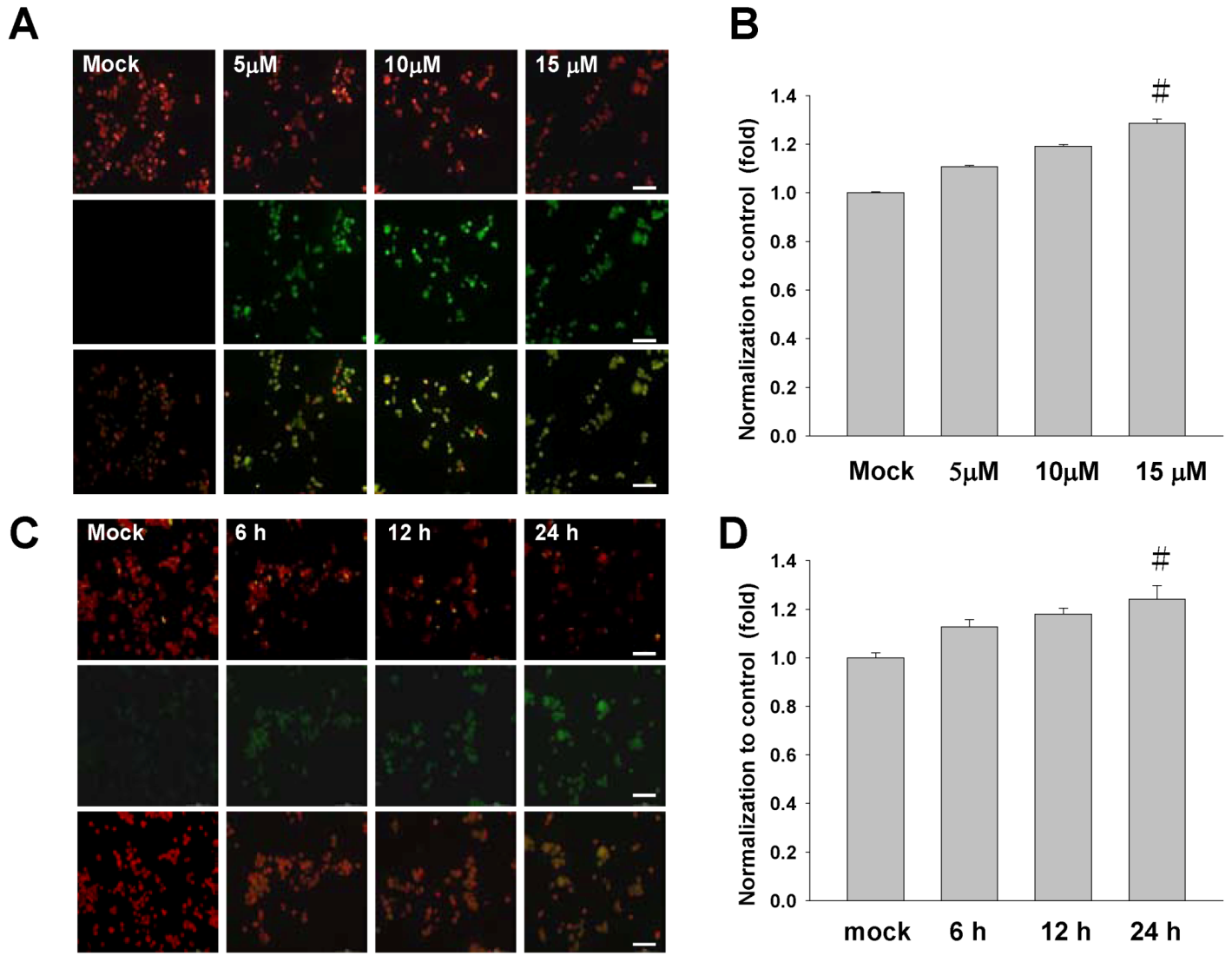
2.3. Treatment of Sinulariolide Causes the Mitochondrial Depolarization in TSGH Cells
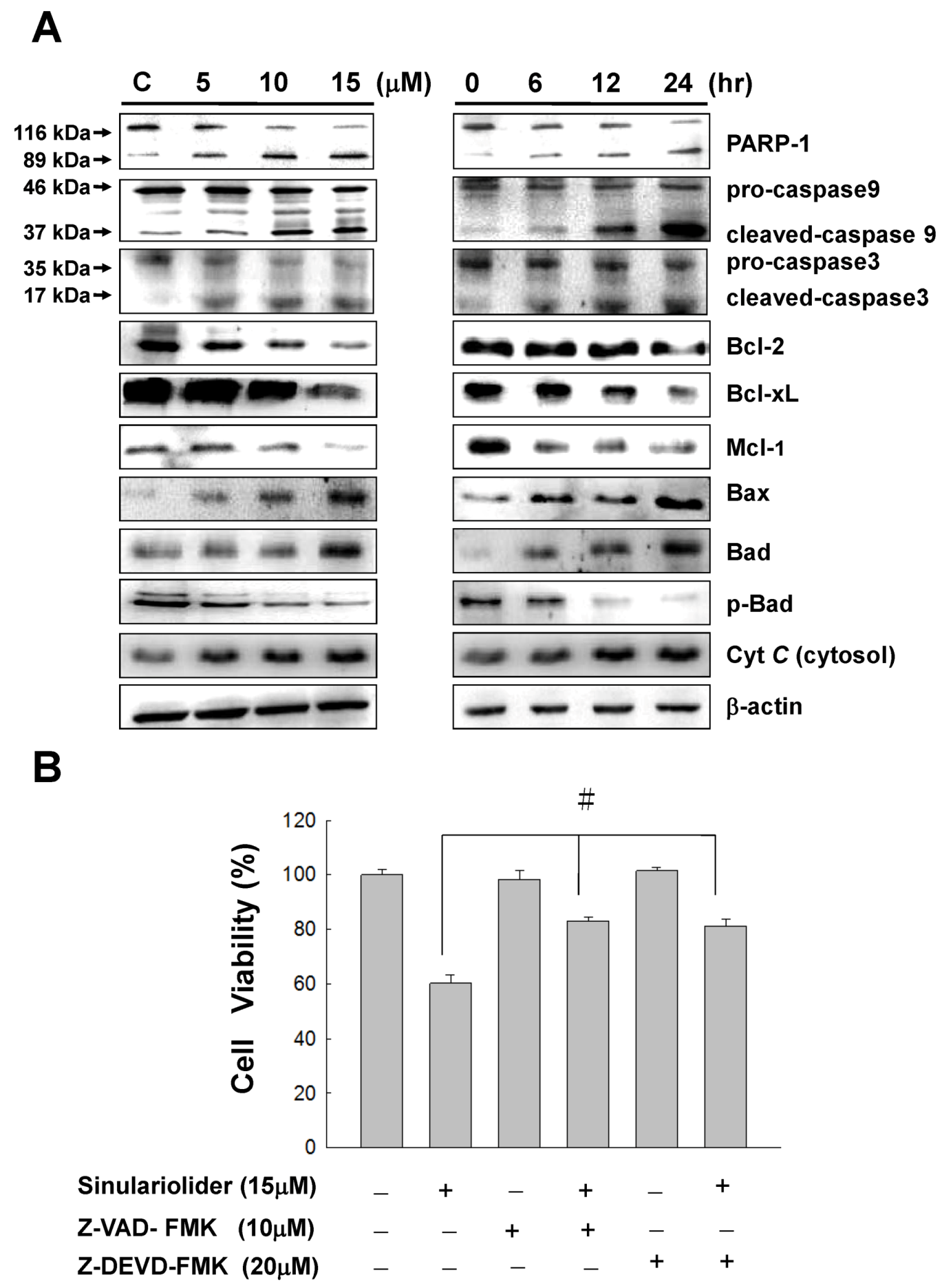
2.4. Sinulariolide Activates the Caspase-Dependent Pathway Resulting in Cell Apoptosis
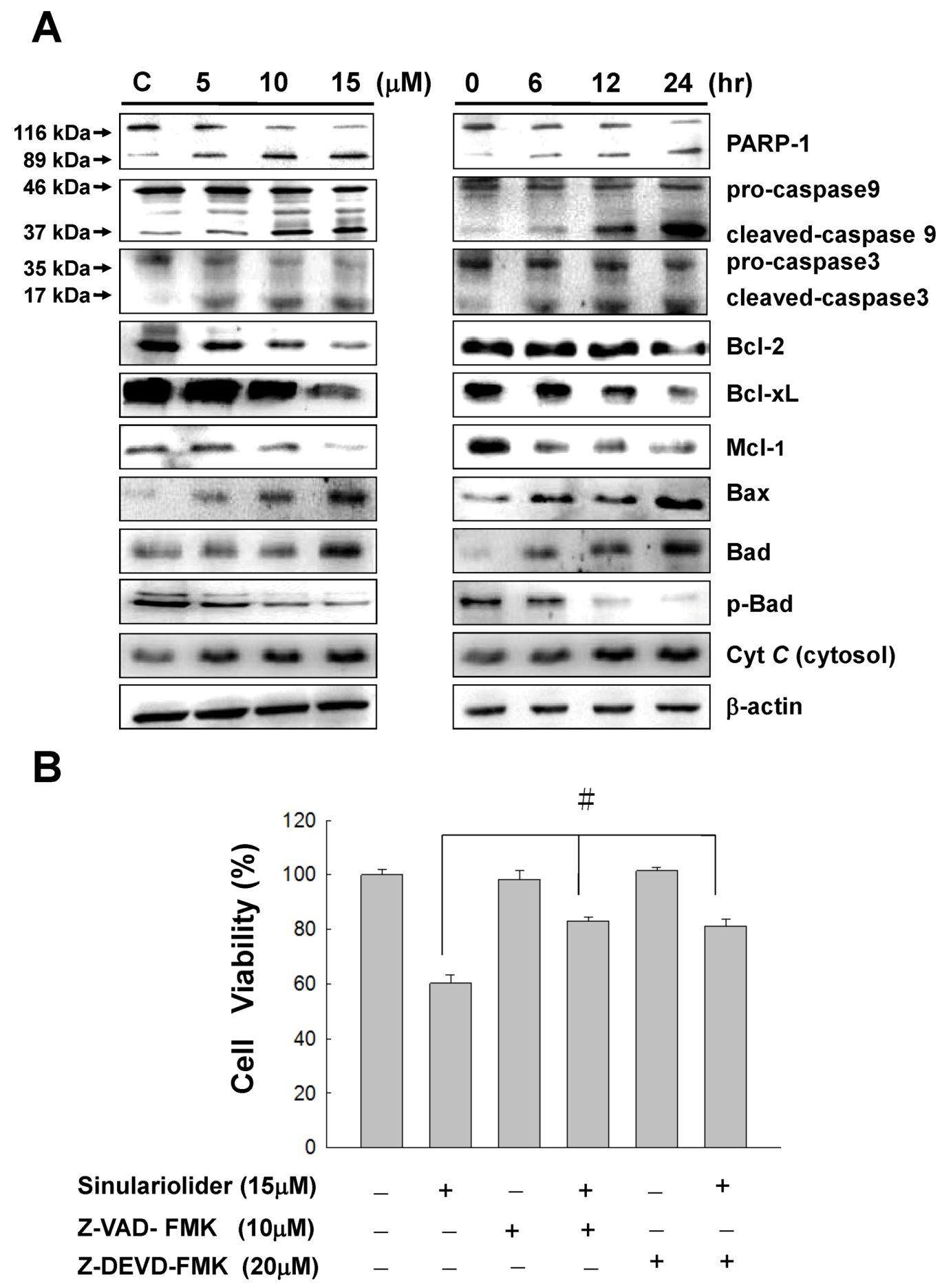
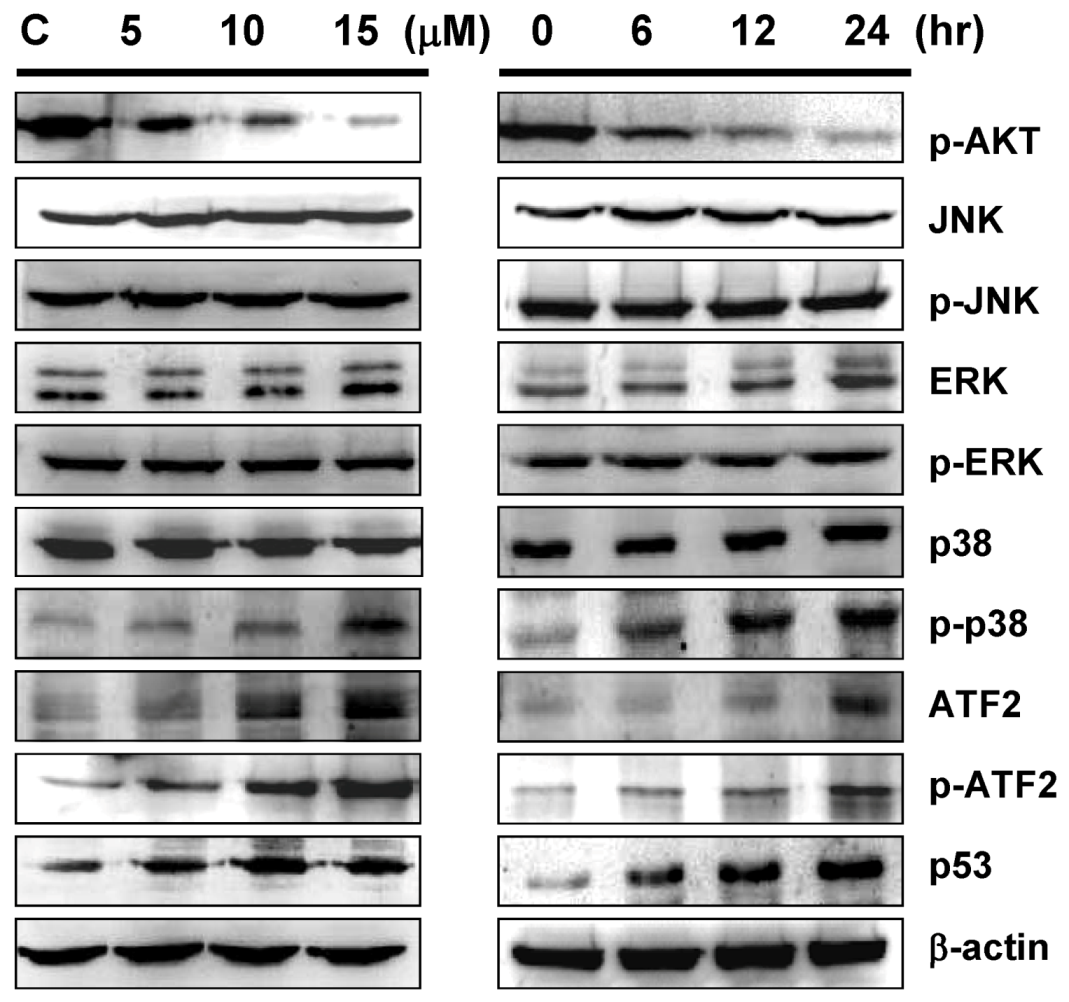
2.5. Sinulariolide-Induced the Activation of p38MAPK-ATF2 Pathway
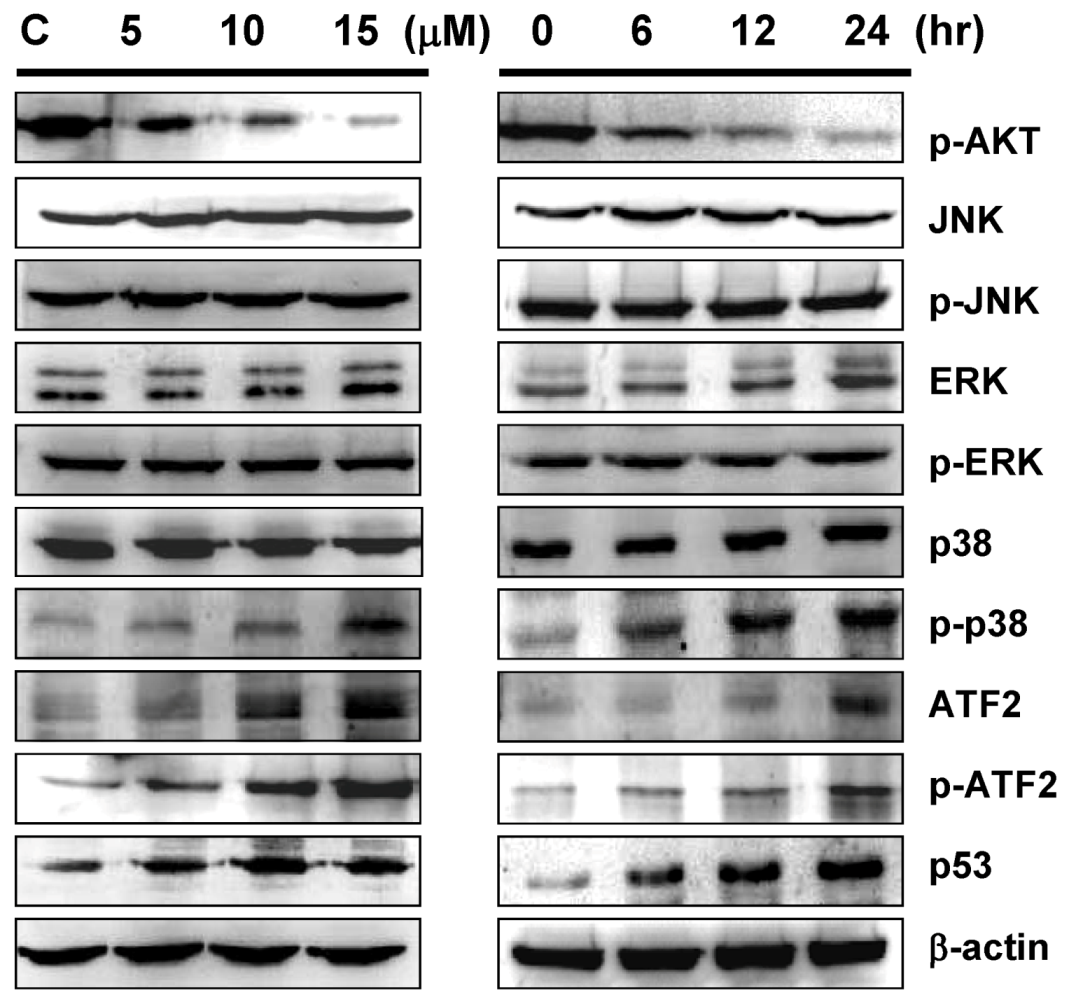
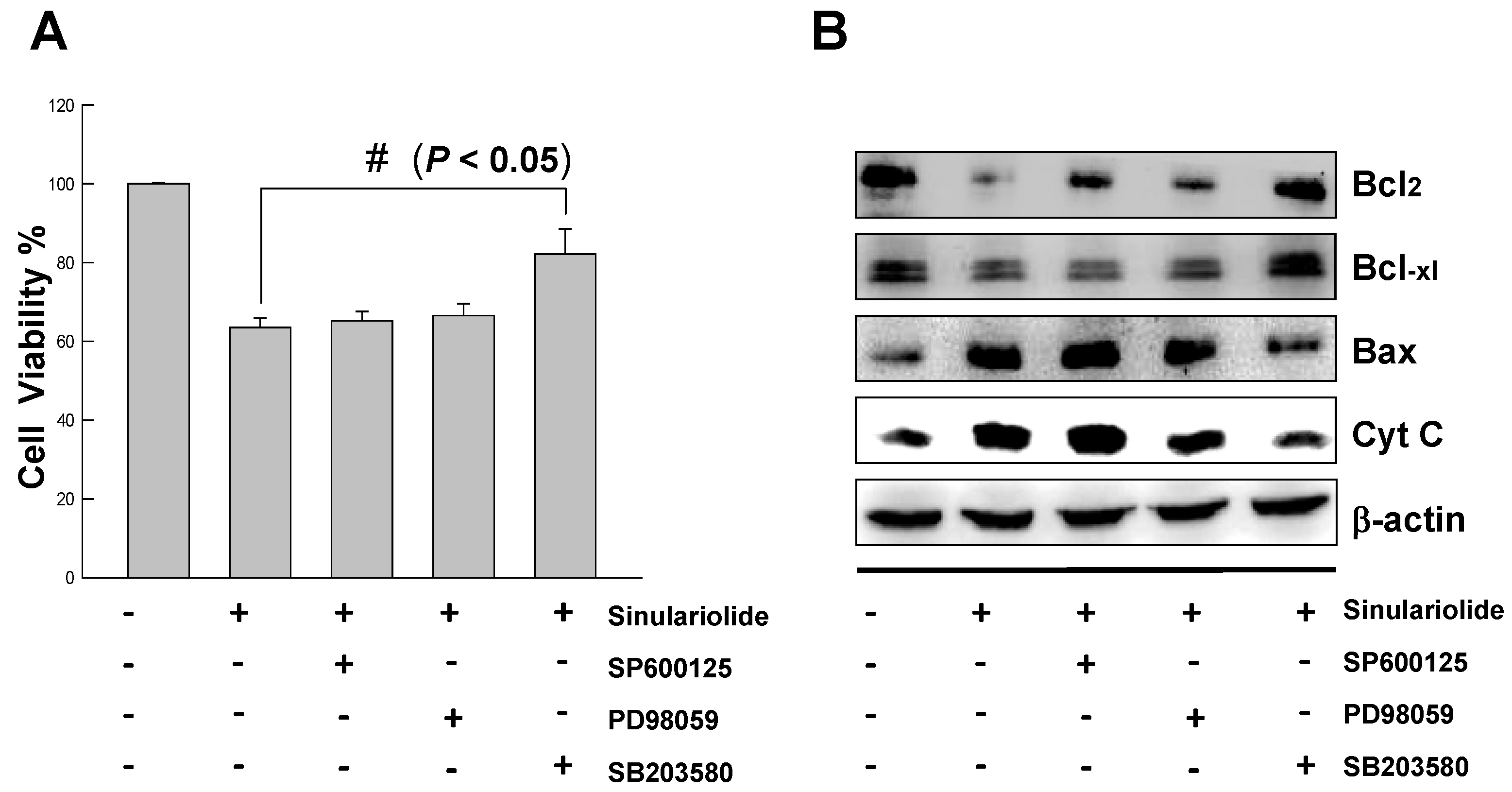
2.6. Inhibition of p38MAPK Activity Rescued the Cell Cytotoxicity of TSGH Cells by Sinulariolide

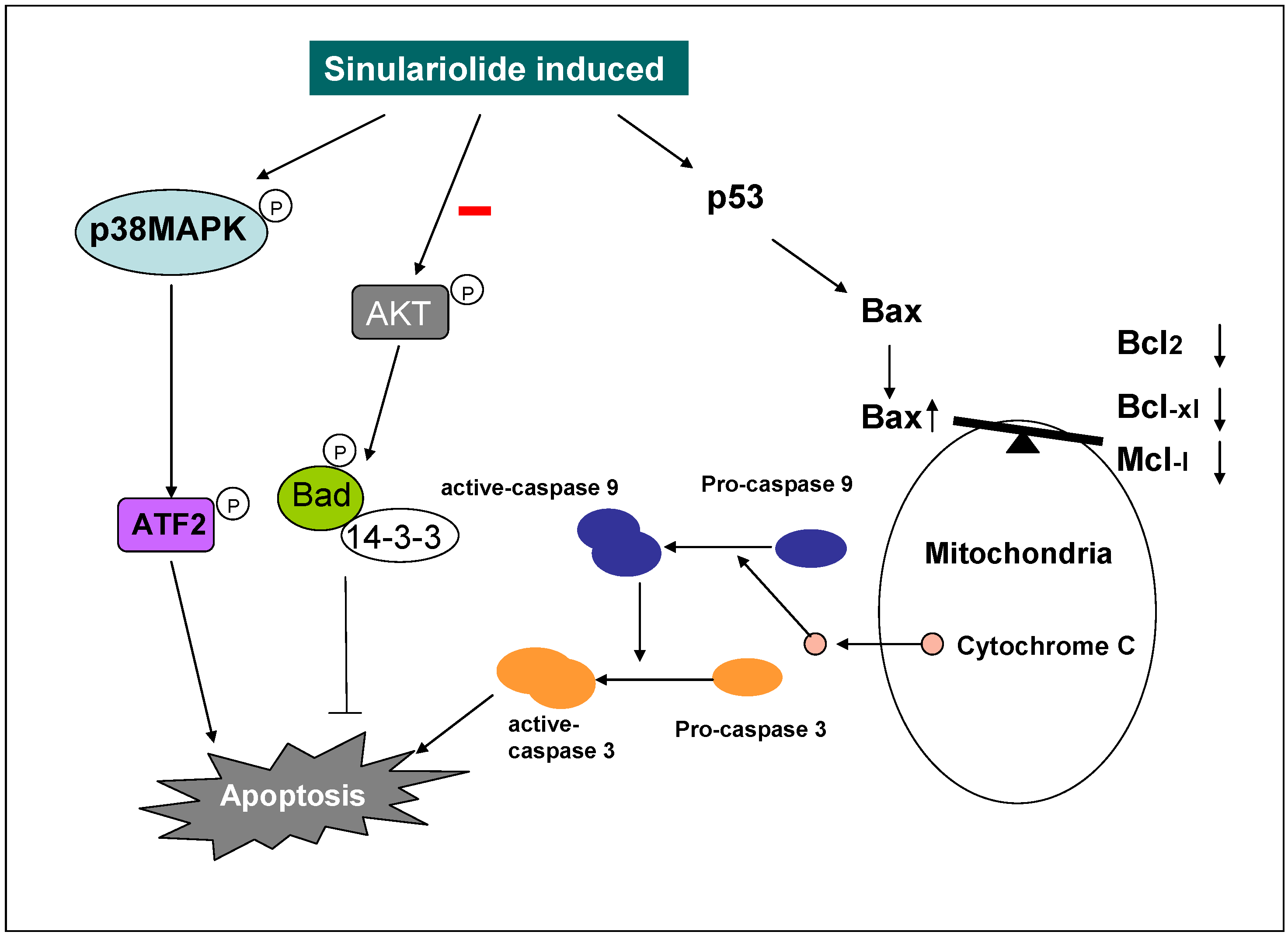
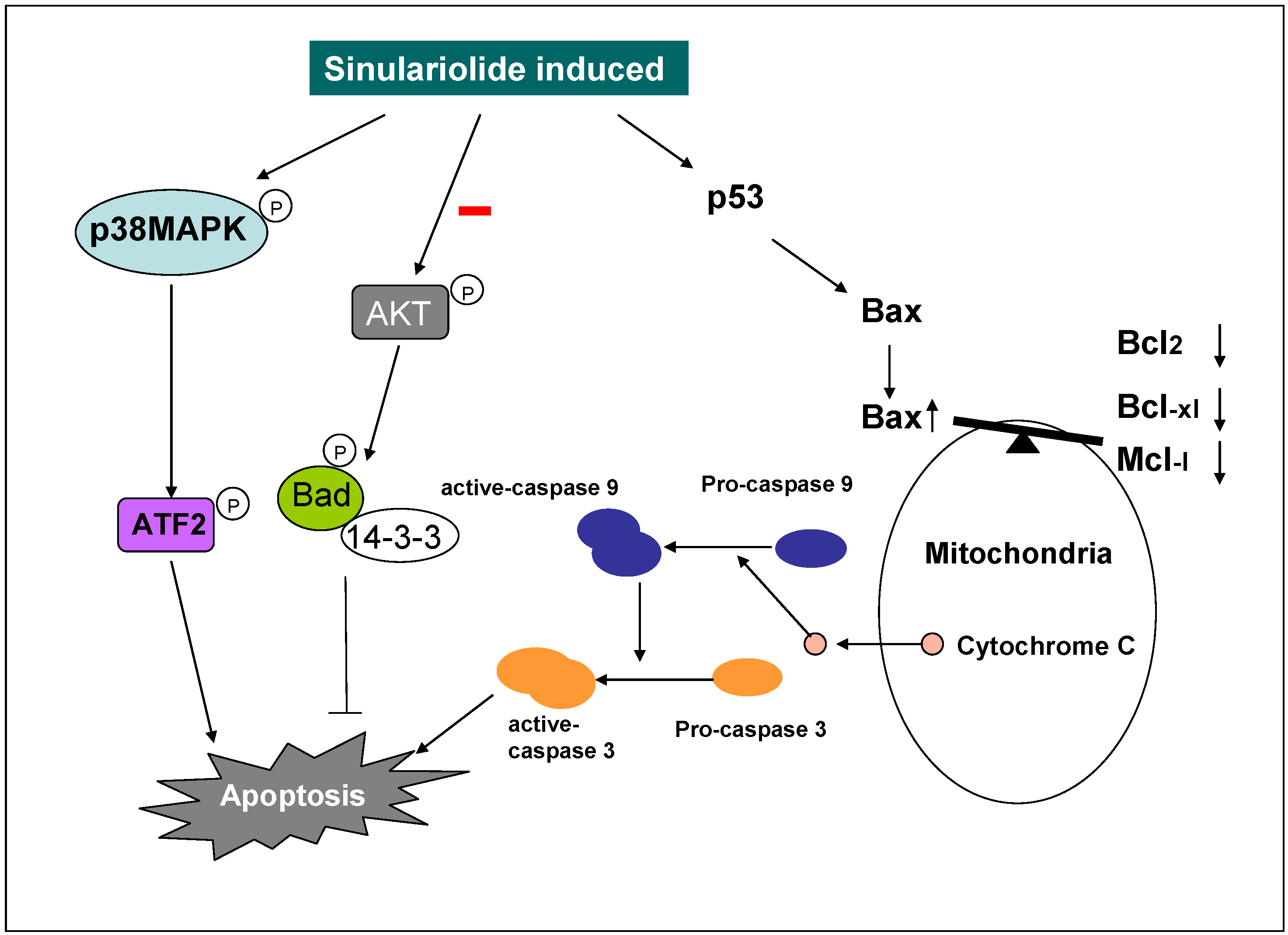
3. Discussion
3.1. Sinulariolide Induces Apoptosis on the TSGH Cells
3.2. Sinulariolide Induces Apoptosis and Causes the Mitochondria Dysfunction on TSGH Cells
4. Materials and Methods
4.1. Materials and Chemical Reagents
4.2. Cell Culture and the Treatment with Sinulariolide
4.3. Cell Viability Assay
4.4. Cell Migration Assay
4.5. Analysis Apoptosis by Flow Cytometry
4.6. Immunofluorescence Microscopy
4.7. Mitochondrial Membrane Potential (ΔΨm) Assay Using Fluorescence Microscopy
4.8. Protein Extraction and Protein Concentration Determination
4.9. Mitochondria and Cytosol Fractionation
4.10. Western Blotting Analysis
4.11. Inhibitor Assessment
5. Conclusion
Acknowledgements
References
- Toll, A.D.; Epstein, J.I. Invasive low-grade papillary urothelial carcinoma: A clinicopathologic analysis of 41 cases. Am. J. Surg. Pathol. 2012, 36, 1081–1086. [Google Scholar] [CrossRef]
- Gotoh, A.; Nagaya, H.; Kanno, T.; Nishizaki, T. Antitumor action of α(1)-adrenoceptor blockers on human bladder, prostate and renal cancer cells. Pharmacology 2012, 90, 242–246. [Google Scholar] [CrossRef]
- Kim, J.J. Recent advances in treatment of advanced urothelial carcinoma. Curr. Urol. Rep. 2012, 13, 147–152. [Google Scholar] [CrossRef]
- Azemar, M.D.; Audouin, M.; Revaux, A.; Misrai, V.; Comperat, E.; Bitker, M.O.; Chartier-Kastler, E.; Richard, F.; Cussenot, O.; Roupret, M. Primary upper urinary tract tumors and subsequent location in the bladder. Prog. Urol. 2009, 19, 583–588. [Google Scholar] [CrossRef]
- Dzombeta, T.; Krajacic-Jagarcec, G.; Tomas, D.; Kraus, O.; Ruzic, B.; Kruslin, B. Urothelial carcinoma with an inverted growth pattern: A report of 4 cases. Acta Med. Croatica 2010, 64, 47–50. [Google Scholar]
- Ruiz, E.; Alarcon Caba, M.; Toselli, L.; Moldes, J.; Ormaechea, M.; de Badiola, F.; Christiansen, S. Transitional cell carcinoma of the bladder in adolescents: A diagnosis to bear in mind. Arch. Argent. Pediatr. 2009, 107, 49–52. [Google Scholar]
- Hsieh, J.L.; Wu, C.L.; Lai, M.D.; Lee, C.H.; Tsai, C.S.; Shiau, A.L. Gene therapy for bladder cancer using E1B-55 kD-deleted adenovirus in combination with adenoviral vector encoding plasminogen kringles 1–5. Br. J. Cancer 2003, 88, 1492–1499. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, K.; Zhang, J.; Chen, J.; Zhang, N.; Xu, Z. Relationship between patient age and superficial transitional cell carcinoma characteristics. Urology 2008, 71, 1186–1190. [Google Scholar] [CrossRef]
- Eichhorn, J.H.; Young, R.H. Transitional cell carcinoma of the ovary: A morphologic study of 100 cases with emphasis on differential diagnosis. Am. J. Surg. Pathol. 2004, 28, 453–463. [Google Scholar] [CrossRef]
- Amling, C.L. Diagnosis and management of superficial bladder cancer. Curr. Probl. Cancer 2001, 25, 219–278. [Google Scholar]
- Babjuk, M.; Dvoracek, J. Diagnosis and therapy of superficial tumors of the urinary bladder. Cas. Lek. Cesk 2002, 141, 723–728. [Google Scholar]
- Wang, Z.; Lu, T.; Du, L.; Hu, Z.; Zhuang, Q.; Li, Y.; Wang, C.Y.; Zhu, H.; Ye, Z. Plasmacytoid urothelial carcinoma of the urinary bladder: A clinical pathological study and literature review. Int. J. Clin. Exp. Pathol. 2012, 5, 601–608. [Google Scholar]
- Ichigo, S.; Takagi, H.; Matsunami, K.; Murase, T.; Ikeda, T.; Imai, A. Transitional cell carcinoma of the ovary (Review). Oncol. Lett. 2012, 3, 3–6. [Google Scholar]
- Liu, C.I.; Wang, R.Y.; Lin, J.J.; Su, J.H.; Chiu, C.C.; Chen, J.C.; Chen, J.Y.; Wu, Y.J. Proteomic profiling of the 11-dehydrosinulariolide-treated oral carcinoma cells Ca9–22: Effects on the cell apoptosis through mitochondrial-related and ER stress pathway. J. Proteomics 2012, 75, 5578–5589. [Google Scholar] [CrossRef]
- Denicourt, C.; Dowdy, S.F. Medicine. Targeting apoptotic pathways in cancer cells. Science 2004, 305, 1411–1413. [Google Scholar] [CrossRef]
- Liao, C.T.; Chang, J.T.; Wang, H.M.; Ng, S.H.; Hsueh, C.; Lee, L.Y.; Lin, C.H.; Chen, I.H.; Huang, S.F.; Cheng, A.J.; et al. Analysis of risk factors of predictive local tumor control in oral cavity cancer. Ann. Surg. Oncol. 2008, 15, 915–922. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Thornberry, N.A. Apoptosis. Life and death decisions. Science 2003, 299, 214–215. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Naekyung Kim, C.; Liu, L.; Huang, Y.; Perkins, C.L.; Green, D.R.; Bhalla, K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood 1998, 91, 1700–1705. [Google Scholar]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2002, 19, 1–48. [Google Scholar]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49. [Google Scholar] [CrossRef]
- Ojika, M.; Islam, M.K.; Shintani, T.; Zhang, Y.; Okamoto, T.; Sakagami, Y. Three new cytotoxic acylspermidines from the soft coral, Sinularia sp. Biosci. Biotechnol. Biochem. 2003, 67, 1410–1412. [Google Scholar] [CrossRef]
- Hsieh, P.W.; Chang, F.R.; McPhail, A.T.; Lee, K.H.; Wu, Y.C. New cembranolide analogues from the formosan soft coral Sinularia flexibilis and their cytotoxicity. Nat. Prod. Res. 2003, 17, 409–418. [Google Scholar] [CrossRef]
- Chiang, P.C.; Chien, C.L.; Pan, S.L.; Chen, W.P.; Teng, C.M.; Shen, Y.C.; Guh, J.H. Induction of endoplasmic reticulum stress and apoptosis by a marine prostanoid in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 679–686. [Google Scholar] [CrossRef]
- Chiang, P.C.; Kung, F.L.; Huang, D.M.; Li, T.K.; Fan, J.R.; Pan, S.L.; Shen, Y.C.; Guh, J.H. Induction of Fas clustering and apoptosis by coral prostanoid in human hormone-resistant prostate cancer cells. Eur. J. Pharmacol. 2006, 542, 22–30. [Google Scholar] [CrossRef]
- Su, C.C.; Su, J.H.; Lin, J.J.; Chen, C.C.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. An investigation into the cytotoxic effects of 13-acetoxysarcocrassolide from the soft coral Sarcophyton crassocaule on bladder cancer cells. Mar. Drugs 2011, 9, 2622–2642. [Google Scholar] [CrossRef]
- Liu, C.I.; Chen, C.C.; Chen, J.C.; Su, J.H.; Huang, H.H.; Chen, J.Y.; Wu, Y.J. Proteomic analysis of anti-tumor effects of 11-dehydrosinulariolide on CAL-27 cells. Mar. Drugs 2011, 9, 1254–1272. [Google Scholar] [CrossRef]
- Hassan, H.M.; Khanfar, M.A.; Elnagar, A.Y.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.; Hifnawy, M.S.; El Sayed, K.A. Pachycladins A–E, prostate cancer invasion and migration inhibitory Eunicellin-based diterpenoids from the red sea soft coral Cladiella pachyclados. J. Nat. Prod. 2010, 73, 848–853. [Google Scholar] [CrossRef]
- Su, T.R.; Lin, J.J.; Chiu, C.C.; Chen, J.Y.F.; Su, J.H.; Cheng, Z.J.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. Proteomic investigation of anti-tumor activities exerted by sinularin against A2058 melanoma cells. Electrophoresis 2012, 33, 1139–1152. [Google Scholar] [CrossRef]
- Dean, E.; Greystoke, A.; Ranson, M.; Dive, C. Biomarkers of cell death applicable to early clinical trials. Exp. Cell. Res. 2012, 318, 1252–1259. [Google Scholar] [CrossRef]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar]
- Ribe, E.; Serrano-Saiz, E.; Akpan, N.; Troy, C. Mechanisms of neuronal death in disease: Defining the models and the players. Biochem. J. 2008, 415, 165–182. [Google Scholar] [CrossRef]
- Shankar, S.; Srivastava, R.K. Bax and Bak genes are essential for maximum apoptotic response by curcumin, a polyphenolic compound and cancer chemopreventive agent derived from turmeric, Curcuma longa. Carcinogenesis 2007, 28, 1277–1286. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Choon-Nam, O.; Shen, H.M. Critical role of pro-apoptotic Bcl-2 family members in andrographolide-induced apoptosis in human cancer cells. Biochem. Pharmacol. 2006, 72, 132–144. [Google Scholar]
- Wang, H.L.; Yeh, T.H.; Chou, A.H.; Kuo, Y.L.; Luo, L.J.; He, C.Y.; Huang, P.C.; Li, A.H. Polyglutamine-Expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulating Bax and downregulating Bcl-x(L). Cell. Signal. 2006, 18, 541–552. [Google Scholar] [CrossRef]
- Germain, M.; Winstall, E.; Vodenicharov, M.; Shah, R.G.; Salvesen, G.S.; Poirier, G.G. Caspase-3-Mediated processing of poly (ADP-ribose) glycohydrolase during apoptosis. J. Biol. Chem. 2001, 276, 2935–2942. [Google Scholar]
- Miloso, M.; Scuteri, A.; Foudah, D.; Tredici, G. MAPKs as mediators of cell fate determination: An approach to neurodegenerative diseases. Curr. Med. Chem. 2008, 15, 538–548. [Google Scholar] [CrossRef]
- Opdenakker, K.; Remans, T.; Vangronsveld, J.; Cuypers, A. Mitogen-Activated protein (MAP) kinases in plant metal stress: Regulation and responses in comparison to other biotic and abiotic stresses. Int. J. Mol. Sci. 2012, 13, 7828–7853. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Rego, R.L.; Foster, N.R.; Thibodeau, S.N.; Alberts, S.R.; Windschitl, H.E.; Sargent, D.J. Proapoptotic bad and bid protein expression predict survival in stages II and III colon cancers. Clin. Cancer Res. 2008, 14, 4128–4133. [Google Scholar] [CrossRef]
- Simms, N.; Rajput, A.; Sharratt, E.A.; Ongchin, M.; Teggart, C.A.; Wang, J.; Brattain, M.G. Transforming growth factor-ss suppresses metastasis in a subset of human colon carcinoma cells. BMC Cancer 2012, 12, 221. [Google Scholar] [CrossRef]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef]
- Moffitt, K.L.; Martin, S.L.; Walker, B. From sentencing to execution—The processes of apoptosis. J. Pharm. Pharmacol. 2010, 62, 547–562. [Google Scholar]
- Shimizu, H.; Banno, Y.; Sumi, N.; Naganawa, T.; Kitajima, Y.; Nozawa, Y. Activation of p38 mitogen-activated protein kinase and caspases in UVB-induced apoptosis of human keratinocyte HaCaT cells. J. Invest. Dermatol. 1999, 112, 769–774. [Google Scholar] [CrossRef]
- Borner, C. The Bcl-2 protein family: Sensors and checkpoints for life-or-death decisions. Mol. Immunol. 2003, 39, 615–647. [Google Scholar] [CrossRef]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer Cell 2002, 1, 19–30. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-Family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell. Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-Inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef]
- Su, T.R.; Tsai, F.J.; Lin, J.J.; Huang, H.H.; Chiu, C.C.; Su, J.H.; Yang, Y.T.; Chen, Y.F.; Wong, B.S.; Wu, Y.J. Induction of apoptosiss by 11-dehydrosinulariolide via mitochondrial dysregulation and ER stress pathways in human melanoma cells. Mar. Drugs 2012, 10, 1883–1898. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Yanase, S.; Yasuda, K.; Ishii, N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech. Ageing Dev. 2002, 123, 1579–1587. [Google Scholar] [CrossRef]
- Masmoudi-Kouki, O.; Douiri, S.; Hamdi, Y.; Kaddour, H.; Bahdoudi, S.; Vaudry, D.; Basille, M.; Leprince, J.; Fournier, A.; Vaudry, H.; et al. Pituitary adenylate cyclase-activating polypeptide protects astroglial cells against oxidative stress-induced apoptosis. J. Neurochem. 2011, 117, 403–411. [Google Scholar] [CrossRef]
- Cui, L.; Deng, Y.; Rong, Y.; Lou, W.; Mao, Z.; Feng, Y.; Xie, D.; Jin, D. IRF-2 is over-expressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2012, 33, 247–255. [Google Scholar] [CrossRef]
- Makkinje, A.; Quinn, D.A.; Chen, A.; Cadilla, C.L.; Force, T.; Bonventre, J.V.; Kyriakis, J.M. Gene 33/Mig-6, a transcriptionally inducible adapter protein that binds GTP-Cdc42 and activates SAPK/JNK. J. Biol. Chem. 2000, 275, 17838–17847. [Google Scholar]
- Chiu, C.C.; Chen, J.Y.; Lin, K.L.; Huang, C.J.; Lee, J.C.; Chen, B.H.; Chen, W.Y.; Lo, Y.H.; Chen, Y.L.; Tseng, C.H.; Lin, S.R. P38 MAPK and NF-kappaB pathways are involved in naphtho[1,2-b] furan-4,5-dione induced anti-proliferation and apoptosis of human hepatoma cells. Cancer Lett. 2010, 295, 92–99. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Neoh, C.-A.; Wang, R.Y.-L.; Din, Z.-H.; Su, J.-H.; Chen, Y.-K.; Tsai, F.-J.; Weng, S.-H.; Wu, Y.-J. Induction of Apoptosis by Sinulariolide from Soft Coral through Mitochondrial-Related and p38MAPK Pathways on Human Bladder Carcinoma Cells. Mar. Drugs 2012, 10, 2893-2911. https://doi.org/10.3390/md10122893
Neoh C-A, Wang RY-L, Din Z-H, Su J-H, Chen Y-K, Tsai F-J, Weng S-H, Wu Y-J. Induction of Apoptosis by Sinulariolide from Soft Coral through Mitochondrial-Related and p38MAPK Pathways on Human Bladder Carcinoma Cells. Marine Drugs. 2012; 10(12):2893-2911. https://doi.org/10.3390/md10122893
Chicago/Turabian StyleNeoh, Choo-Aun, Robert Y.-L. Wang, Zhong-Hao Din, Jui-Hsin Su, Yu-Kuei Chen, Feng-Jen Tsai, Shun-Hsiang Weng, and Yu-Jen Wu. 2012. "Induction of Apoptosis by Sinulariolide from Soft Coral through Mitochondrial-Related and p38MAPK Pathways on Human Bladder Carcinoma Cells" Marine Drugs 10, no. 12: 2893-2911. https://doi.org/10.3390/md10122893
APA StyleNeoh, C.-A., Wang, R. Y.-L., Din, Z.-H., Su, J.-H., Chen, Y.-K., Tsai, F.-J., Weng, S.-H., & Wu, Y.-J. (2012). Induction of Apoptosis by Sinulariolide from Soft Coral through Mitochondrial-Related and p38MAPK Pathways on Human Bladder Carcinoma Cells. Marine Drugs, 10(12), 2893-2911. https://doi.org/10.3390/md10122893