RAS Mutations in Advanced Colorectal Cancer: Mechanisms, Clinical Implications, and Novel Therapeutic Approaches

 , , and
, , and
Abstract

1. Introduction
1.1. RAS Mutations: Function and Impact on Treatment Resistance
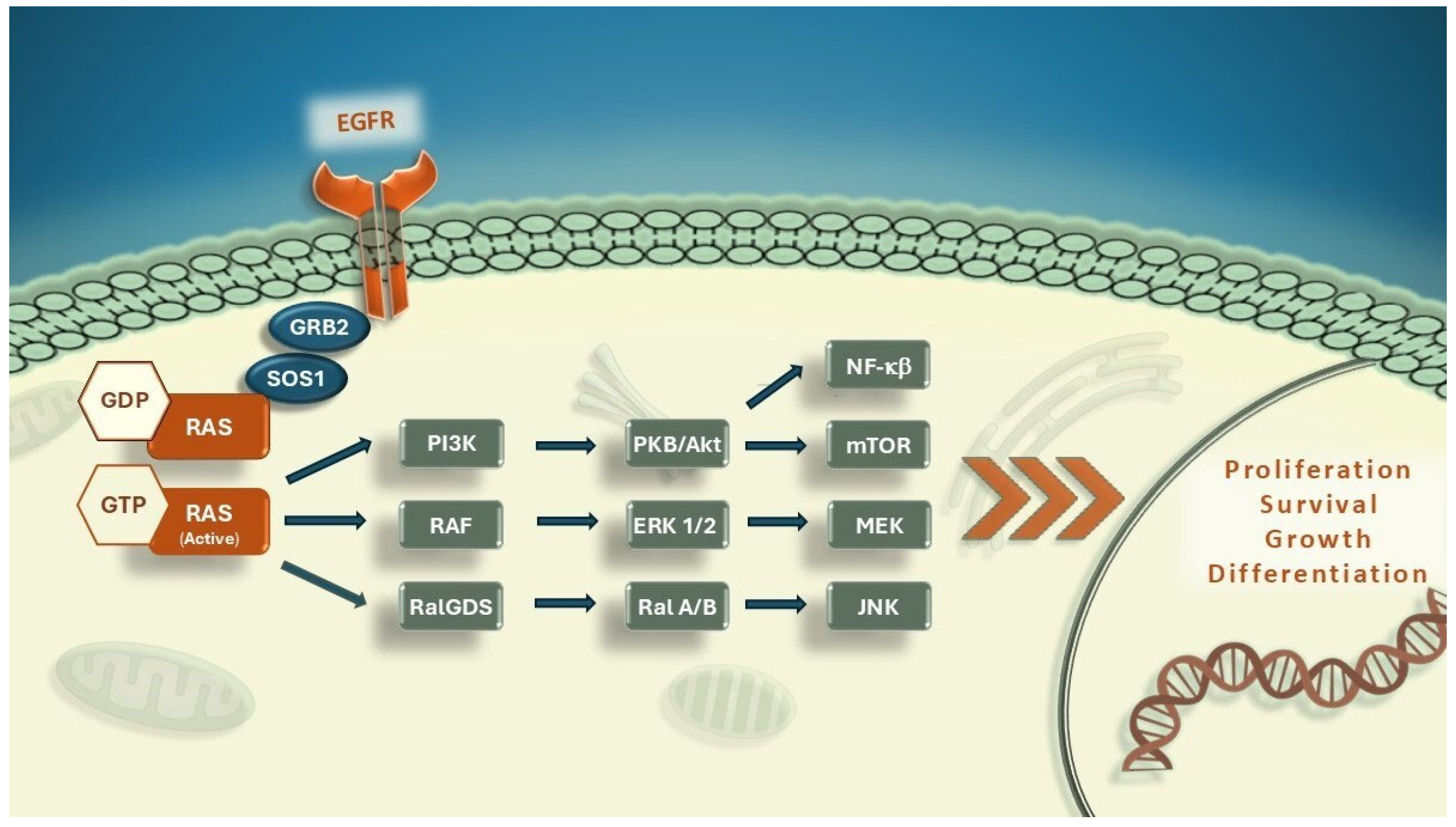
1.1.1. RAS Function in Colorectal Carcinoma
1.1.2. The Function of RAS Mutations in Anti-Cancer Therapy Resistance
1.1.3. Resistance Mechanisms and Evolution of Targeted Therapies in RAS-Mutant Colorectal Cancer
1.2. Prognostic and Predictive Role of RAS Mutation in Patients Diagnosed with mCRC
1.2.1. First-Line Therapy: Anti-EGFR and Anti-VEGF Trials
1.2.2. Second-Line Therapy: Angiogenesis Inhibitors
Bevacizumab
Aflibercept
Ramucirumab
1.3. Differential Benefits of Single-Agent Bevacizumab Maintenance Therapy: Subgroup Analysis Insights
1.4. Third-Line and Beyond: KRAS G12C Inhibitors and Novel Agents
1.4.1. Regorafenib
1.4.2. TAS 102
1.5. Trifluridine/Tipiracil Plus Bevacizumab
Frequintinib
1.6. NEORAS Status and Potential Use of Anti-EGFR Agents
1.7. Efficacy of KRAS G12C Inhibitors in mCRC
1.8. Alternative Therapeutic Strategies in RAS-Mutant Colorectal Cancer Beyond Direct RAS Inhibition
1.8.1. SOS1 Inhibitors
1.8.2. SHP2 Inhibitors
1.8.3. RAF-MEK-ERK Inhibitors
1.9. Immunotherapy
1.9.1. Microsatellite Instable Colorectal Cancers
1.9.2. Microsatellite Stable Colorectal Cancers
1.10. Future Directions and Potential Research Areas
1.10.1. Emerging Therapies Targeting KRASG12C Resistance: RMC-6291 and FMC-376
1.10.2. KRAS G12D Mutation: High Oncogenic Potential and Innovative Therapeutic Approaches in Colorectal Cancer
1.10.3. A Novel Pan-RAS Inhibitor for Targeting KRAS Mutations in Colorectal Cancer
1.11. Future Perspectives: Genetic Engineering, Immunotherapy, and Vaccination Strategies Targeting RAS-Mutant CRC
1.11.1. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
1.11.2. Adoptive Cell Therapy
1.11.3. Anti-RAS Vaccines
2. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Fakih, M.G.; Salvatore, L.; Esaki, T.; Modest, D.P.; Lopez-Bravo, D.P.; Taieb, J.; Karamouzis, M.V.; Ruiz-Garcia, E.; Kim, T.-W.; Kuboki, Y. Sotorasib plus panitumumab in refractory colorectal cancer with mutated, KRAS G12C. N. Engl. J. Med. 2023, 389, 2125–2139. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A. Adagrasib with or without cetuximab in colorectal cancer with mutated, KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing complexity of Ras signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, C.P.; ZoBell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef]
- Yildirim, H.C.; Gunenc, D.; Almuradova, E.; Sutcuoglu, O.; Yalcin, S. A narrative review of RAS mutations in early-stage colorectal cancer: Mechanisms and clinical implications. Medicina 2025, 61, 408. [Google Scholar] [CrossRef]
- Zenonos, K.; Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J. Gastrointest. Oncol. 2013, 5, 97. [Google Scholar] [CrossRef]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.-E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Modest, D.P.; Ricard, I.; Heinemann, V.; Hegewisch-Becker, S.; Schmiegel, W.; Porschen, R.; Stintzing, S.; Graeven, U.; Arnold, D.; Von Weikersthal, L. Outcome according to KRAS-, NRAS-and BRAF-mutation as well as KRAS mutation variants: Pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann. Oncol. 2016, 27, 1746–1753. [Google Scholar] [CrossRef]
- Zocche, D.M.; Ramirez, C.; Fontao, F.M.; Costa, L.D.; Redal, M.A. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Front. Genet. 2015, 6, 116. [Google Scholar] [CrossRef]
- Lievre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Maughan, T.S.; Adams, R.A.; Smith, C.G.; Meade, A.M.; Seymour, M.T.; Wilson, R.H.; Idziaszczyk, S.; Harris, R.; Fisher, D.; Kenny, S.L. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 2011, 377, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H. Association of KRAS p. G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef]
- Price, T.J.; Bruhn, M.A.; Lee, C.K.; Hardingham, J.E.; Townsend, A.R.; Mann, K.P.; Simes, J.; Weickhardt, A.; Wrin, J.W.; Wilson, K. Correlation of extended RAS and PIK3CA gene mutation status with outcomes from the phase III AGITG MAX STUDY involving capecitabine alone or in combination with bevacizumab plus or minus mitomycin C in advanced colorectal cancer. Br. J. Cancer 2015, 112, 963–970. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Yi, J.; Ince, W.; Novotny, W.F.; Rosen, O. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: Analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist 2009, 14, 22–28. [Google Scholar] [CrossRef]
- Hecht, J.R.; Mitchell, E.; Chidiac, T.; Scroggin, C.; Hagenstad, C.; Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 672–680. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.-J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N. Effect of first-line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal cancer: A randomized clinical trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Tveit, K.M.; Guren, T.; Glimelius, B.; Pfeiffer, P.; Sorbye, H.; Pyrhonen, S.; Sigurdsson, F.; Kure, E.; Ikdahl, T.; Skovlund, E. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: The NORDIC-VII study. J. Clin. Oncol. 2012, 30, 1755–1762. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.-J.; Köhne, C.-H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; Van Krieken, J.H. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Fischer von Weikersthal, L.; Decker, T.; Vehling-Kaiser, U.; Uhlig, J.; Schenk, M.; Freiberg-Richter, J.; Peuser, B.; Denzlinger, C.; Peveling genannt Reddemann, C. Sequential versus combination therapy of metastatic colorectal Cancer using Fluoropyrimidines, irinotecan, and bevacizumab: A randomized, controlled study—XELAVIRI (AIO KRK0110). J. Clin. Oncol. 2019, 37, 22–32. [Google Scholar] [CrossRef]
- Richman, S.D.; Southward, K.; Chambers, P.; Cross, D.; Barrett, J.; Hemmings, G.; Taylor, M.; Wood, H.; Hutchins, G.; Foster, J.M. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 2016, 238, 562–570. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-label, phase IIa multiple basket study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Strickler, J.H.; Cercek, A.; Siena, S.; André, T.; Ng, K.; Van Cutsem, E.; Wu, C.; Paulson, A.S.; Hubbard, J.M.; Coveler, A.L. Tucatinib plus trastuzumab for chemotherapy-refractory, HER2-positive, RAS wild-type unresectable or metastatic colorectal cancer (MOUNTAINEER): A multicentre, open-label, phase 2 study. Lancet Oncol. 2023, 24, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Wainberg, Z.; Elez, E. Final results of DESTINY-CRC01 investigating trastuzumab deruxtecan in patients with HER2-expressing metastatic colorectal cancer. Nat. Commun. 2023, 14, 3332. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patane, D.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef]
- Fernández Montes, A.; Martínez Lago, N.; Covela Rúa, M.; de la Cámara Gómez, J.; González Villaroel, P.; Méndez Méndez, J.C.; Jorge Fernández, M.; Salgado Fernández, M.; Reboredo López, M.; Quintero Aldana, G. Efficacy and safety of FOLFIRI/aflibercept in second-line treatment of metastatic colorectal cancer in a real-world population: Prognostic and predictive markers. Cancer Med. 2019, 8, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Kubicka, S.; Greil, R.; André, T.; Bennouna, J.; Sastre, J.; Van Cutsem, E.; Von Moos, R.; Österlund, P.; Reyes-Rivera, I.; Müller, T. Bevacizumab plus chemotherapy continued beyond first progression in patients with metastatic colorectal cancer previously treated with bevacizumab plus chemotherapy: ML18147 study KRAS subgroup findings. Ann. Oncol. 2013, 24, 2342–2349. [Google Scholar] [CrossRef]
- Bennouna, J.; Sastre, J.; Arnold, D.; Österlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Viéitez, J.M.; Bouché, O.; Borg, C. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Antoniotti, C.; Rossini, D.; Lonardi, S.; Loupakis, F.; Pietrantonio, F.; Bordonaro, R.; Latiano, T.P.; Tamburini, E.; Santini, D. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020, 21, 497–507. [Google Scholar] [PubMed]
- Van Cutsem, E.; Joulain, F.; Hoff, P.M.; Mitchell, E.; Ruff, P.; Lakomý, R.; Prausová, J.; Moiseyenko, V.M.; Van Hazel, G.; Cunningham, D. Aflibercept plus FOLFIRI vs. placebo plus FOLFIRI in second-line metastatic colorectal cancer: A post hoc analysis of survival from the phase III VELOUR study subsequent to exclusion of patients who had recurrence during or within 6 months of completing adjuvant oxaliplatin-based therapy. Target. Oncol. 2016, 11, 383–400. [Google Scholar]
- Wirapati, P.; Pomella, V.; Vandenbosch, B.; Kerr, P.; Maiello, E.; Grahame, M.J.; Curca, R.-O.D.; Karthaus, M.; Bridgewater, J.A.; Mihailov, A.C. Velour trial biomarkers update: Impact of RAS, BRAF, and sidedness on aflibercept activity. Ann. Oncol. 2017, 28, iii151–iii152. [Google Scholar] [CrossRef]
- Chen, C.-P.; Ke, T.-W.; Cheng, R.; Wang, J.-Y. Ramucirumab in the second-line treatment of metastatic colorectal cancer: A narrative review of literature from clinical trials. Transl. Cancer Res. 2020, 9, 5645. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.-E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [PubMed]
- Hegewisch-Becker, S.; Graeven, U.; Lerchenmüller, C.A.; Killing, B.; Depenbusch, R.; Steffens, C.-C.; Al-Batran, S.-E.; Lange, T.; Dietrich, G.; Stoehlmacher, J. Maintenance strategies after first-line oxaliplatin plus fluoropyrimidine plus bevacizumab for patients with metastatic colorectal cancer (AIO 0207): A randomised, non-inferiority, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 1355–1369. [Google Scholar] [CrossRef]
- Aparicio, T.; Ghiringhelli, F.; Boige, V.; Le Malicot, K.; Taieb, J.; Bouché, O.; Phelip, J.-M.; François, E.; Borel, C.; Faroux, R. Bevacizumab maintenance versus no maintenance during chemotherapy-free intervals in metastatic colorectal cancer: A randomized phase III trial (PRODIGE 9). J. Clin. Oncol. 2018, 36, 674–681. [Google Scholar] [CrossRef]
- Salvatore, L.; Bria, E.; Sperduti, I.; Hinke, A.; Hegewisch-Becker, S.; Aparicio, T.; Le Malicot, K.; Boige, V.; Koeberle, D.; Baertschi, D. Bevacizumab as maintenance therapy in patients with metastatic colorectal cancer: A meta-analysis of individual patients’ data from 3 phase III studies. Cancer Treat. Rev. 2021, 97, 102202. [Google Scholar] [CrossRef]
- Stahler, A.; Heinemann, V.; Ricard, I.; von Einem, J.C.; Giessen-Jung, C.; Westphalen, C.B.; Michl, M.; Heinrich, K.; Miller-Phillips, L.; Jelas, I. Current treatment options in RAS mutant metastatic colorectal cancer patients: A meta-analysis of 14 randomized phase III trials. J. Cancer Res. Clin. Oncol. 2020, 146, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Van Cutsem, E.; Falcone, A.; Yoshino, T.; Garcia-Carbonero, R.; Mizunuma, N.; Yamazaki, K.; Shimada, Y.; Tabernero, J.; Komatsu, Y. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl. J. Med. 2015, 372, 1909–1919. [Google Scholar] [CrossRef]
- Huang, F.; Yang, H.; Bao, W.; Bin, Y.; Zhou, S.; Wang, M.; Lv, X. Efficacy and safety of trifluridine/tipiracil (TAS-102) in patients with metastatic colorectal cancer: A systematic review and meta-analysis. Clin. Transl. Oncol. 2024, 26, 468–476. [Google Scholar] [CrossRef]
- Prager, G.W.; Taieb, J.; Fakih, M.; Ciardiello, F.; Van Cutsem, E.; Elez, E.; Cruz, F.M.; Wyrwicz, L.; Stroyakovskiy, D.; Pápai, Z.; et al. Trifluridine-Tipiracil and Bevacizumab in Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2023, 388, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Taieb, J.; Fakih, M.; Prager, G.; Van Cutsem, E.; Ciardiello, F.; Mayer, R.; Amellal, N.; Skanji, D.; Calleja, E. Impact of KRASG12 mutations on survival with trifluridine/tipiracil plus bevacizumab in patients with refractory metastatic colorectal cancer: Post hoc analysis of the phase III SUNLIGHT trial. ESMO Open 2024, 9, 102945. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qin, S.; Xu, R.-H.; Shen, L.; Xu, J.; Bai, Y.; Yang, L.; Deng, Y.; Chen, Z.D.; Zhong, H.; et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: The FRESCO randomized clinical trial. JAMA 2018, 319, 2486–2496. [Google Scholar] [CrossRef]
- Dasari, A.; Lonardi, S.; Garcia-Carbonero, R.; Elez, E.; Yoshino, T.; Sobrero, A.; Yao, J.; García-Alfonso, P.; Kocsis, J.; Gracian, A.C. Fruquintinib versus placebo in patients with refractory metastatic colorectal cancer (FRESCO-2): An international, multicentre, randomised, double-blind, phase 3 study. Lancet 2023, 402, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, W.; Ying, J.; Zhang, Y.; Pan, Y.; Qiu, W.; Su, W.; Tan, P.; Yin, H.; Wang, Y. Preliminary results of a phase 1b study of fruquintinib plus sintilimab in advanced colorectal cancer. J. Clin. Oncol. 2021, 39, 2514. [Google Scholar] [CrossRef]
- Osumi, H.; Takashima, A.; Ooki, A.; Yoshinari, Y.; Wakatsuki, T.; Hirano, H.; Nakayama, I.; Okita, N.; Sawada, R.; Ouchi, K. A multi-institutional observational study evaluating the incidence and the clinicopathological characteristics of NeoRAS wild-type metastatic colorectal cancer. Transl. Oncol. 2023, 35, 101718. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nakamura, Y.; Denda, T.; Ohta, T.; Esaki, T.; Shiozawa, M.; Yamaguchi, K.; Yamazaki, K.; Sunakawa, Y.; Kato, T. Clinical validation of plasma-based genotyping for RAS and BRAF V600E mutation in metastatic colorectal cancer: SCRUM-Japan GOZILA substudy. JCO Precis. Oncol. 2023, 7, e2200688. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Belardinilli, F.; Vestri, A.; Magri, V.; De Renzi, G.; De Meo, M.; Caponnetto, S.; Di Nicolantonio, F.; Cortesi, E.; Giannini, G. RAS mutation conversion in bevacizumab-treated metastatic colorectal cancer patients: A liquid biopsy based study. Cancers 2022, 14, 802. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S. KRASG12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.-W.; Heinemann, V.; Muro, K. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef]
- Kuboki, Y.; Fakih, M.; Strickler, J.; Yaeger, R.; Masuishi, T.; Kim, E.J.; Bestvina, C.M.; Kopetz, S.; Falchook, G.S.; Langer, C. Sotorasib with panitumumab in chemotherapy-refractory KRAS G12C-mutated colorectal cancer: A phase 1b trial. Nat. Med. 2024, 30, 265–270. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRAS G12C solid tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef]
- Sacher, A.; LoRusso, P.; Patel, M.R.; Miller, W.H., Jr.; Garralda, E.; Forster, M.D.; Santoro, A.; Falcon, A.; Kim, T.W.; Paz-Ares, L. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N. Engl. J. Med. 2023, 389, 710–721. [Google Scholar] [CrossRef]
- Desai, J.; Alonso, G.; Kim, S.H.; Cervantes, A.; Karasic, T.; Medina, L.; Shacham-Shmueli, E.; Cosman, R.; Falcon, A.; Gort, E. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: A phase 1b trial. Nat. Med. 2024, 30, 271–278. [Google Scholar] [CrossRef]
- Xu, R.; Xu, Y.; Yan, D.; Munster, P.; Ruan, D.; Deng, Y.; Pan, H.; Underhill, C.; Richardson, G.; Nordman, I. 550O Safety and efficacy of D-1553 in combination with cetuximab in KRAS G12C mutated colorectal cancer (CRC): A phase II study. Ann. Oncol. 2023, 34, S410–S411. [Google Scholar] [CrossRef]
- Yuan, Y.; Deng, Y.; Jin, Y.; Pan, Y.; Wang, C.; Wang, Z.; Zhang, Z.; Meng, X.; Hu, Y.; Zhao, M. 106P Efficacy and safety of IBI351 (GFH925) monotherapy in metastatic colorectal cancer harboring KRASG12C mutation: Updated results from a pooled analysis of two phase I studies. Ann. Oncol. 2023, 34, S1512. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.C.; Sun, Q.; Daniels, R.N.; Camper, D.; Kennedy, J.P.; Phan, J.; Olejniczak, E.T.; Lee, T.; Waterson, A.G.; Rossanese, O.W. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. USA 2014, 111, 3401–3406. [Google Scholar] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef]
- Plangger, A.; Rath, B.; Stickler, S.; Hochmair, M.; Lang, C.; Weigl, L.; Funovics, M.; Hamilton, G. Cytotoxicity of combinations of the pan-KRAS SOS1 inhibitor BAY-293 against pancreatic cancer cell lines. Discov. Oncol. 2022, 13, 84. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.-Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.; Herrera, S.G. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, Z.; Song, P.; Sun, Y.; Liu, P.; Yue, L.; Lv, K.; Wang, X.; Shen, Y.; Si, J. SHP2 inhibition mitigates adaptive resistance to MEK inhibitors in KRAS-mutant gastric cancer through the suppression of KSR1 activity. Cancer Lett. 2023, 555, 216029. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Park, H.; Tolcher, A.W.; Ou, S.-H.I.; Garon, E.B.; George, B.; Janne, P.A.; Moody, S.E.; Tan, E.Y.; Sen, S.K. Krystal-2: A phase i/ii trial of adagrasib (mrtx849) in combination with tno155 in patients with advanced solid tumors with Kras g12c mutation. J. Clin. Oncol. 2021, 39, TPS146. [Google Scholar] [CrossRef]
- Zimmer, L.; Barlesi, F.; Martinez-Garcia, M.; Dieras, V.; Schellens, J.H.; Spano, J.-P.; Middleton, M.R.; Calvo, E.; Paz-Ares, L.; Larkin, J. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS–RAF mutations. Clin. Cancer Res. 2014, 20, 4251–4261. [Google Scholar] [CrossRef]
- Blumenschein, G., Jr.; Smit, E.; Planchard, D.; Kim, D.-W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.-J.; Hanna, N. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A.; Dorsch, M.; Yao, Y.-M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Alsina, M.; Soares, H.P.; Braña, I.; Britten, C.D.; Del Conte, G.; Ezeh, P.; Houk, B.; Kern, K.A.; Leong, S. A multi-arm phase I study of the PI3K/mTOR inhibitors PF-04691502 and gedatolisib (PF-05212384) plus irinotecan or the MEK inhibitor PD-0325901 in advanced cancer. Target. Oncol. 2017, 12, 775–785. [Google Scholar] [CrossRef]
- Brummelen, E.M.; Huijberts, S.; Herpen, C.; Desar, I.; Opdam, F.; Geel, R.; Marchetti, S.; Steeghs, N.; Monkhorst, K.; Thijssen, B. Phase I Study of Afatinib and Selumetinib in Patients with KRAS-Mutated Colorectal, Non-Small Cell Lung, and Pancreatic Cancer. Oncologist 2021, 26, 290-e545. [Google Scholar] [CrossRef]
- van Geel, R.M.; van Brummelen, E.M.; Eskens, F.A.; Huijberts, S.C.; de Vos, F.Y.; Lolkema, M.P.; Devriese, L.A.; Opdam, F.L.; Marchetti, S.; Steeghs, N. Phase 1 study of the pan-HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD-0325901 in patients with KRAS-mutation-positive colorectal, non-small-cell lung and pancreatic cancer. Br. J. Cancer 2020, 122, 1166–1174. [Google Scholar] [CrossRef]
- Bendell, J.; Won, K.T.; Ean, C.C.; Yung-Jue, B.; Lee, C.; Desai, J.; Lewin, J.; Wallin, J.; Thakur, M.D.; Mwawasi, G. LBA-01 safety and efficacy of cobimetinib (cobi) and atezolizumab (atezo) in a Phase 1b study of metastatic colorectal cancer (mCRC). Ann. Oncol. 2016, 27, ii140. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Yaeger, R.; Delord, J.-P.; Tabernero, J.; Siu, L.L.; Ducreux, M.; Siena, S.; Elez, E.; Kasper, S.; Zander, T. Phase Ib/II Study of the Efficacy and Safety of Binimetinib (MEK162) Plus Panitumumab for Mutant or Wild-Type RAS Metastatic Colorectal Cancer. Oncologist 2023, 28, e1209–e1218. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595. [Google Scholar] [CrossRef]
- Diaz, L.A.; Le, D.T.; Yoshino, T.; Andre, T.; Bendell, J.C.; Koshiji, M.; Zhang, Y.; Kang, S.P.; Lam, B.; Jaeger, D. Phase 3, open-label, randomized study of first-line pembrolizumab (pembro) vs. investigator-choice chemotherapy for mismatch repair-deficient (dMMR) or microsatellite instability-high (MSI-H) metastatic colorectal carcinoma (mCRC): KEYNOTE-177. J. Clin. Oncol. 2017, 35, TPS3618. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Andre, T.; Elez, E.; Van Cutsem, E.; Jensen, L.H.; Bennouna, J.; Mendez, G.; Schenker, M.; De La Fouchardiere, C.; Limon, M.L.; Yoshino, T. Nivolumab (NIVO) plus ipilimumab (IPI) vs chemotherapy (chemo) as first-line (1L) treatment for microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC): First results of the CheckMate 8HW study. J. Clin. Oncol. 2024, 42, LBA768. [Google Scholar] [CrossRef]
- Shiu, K.-K.; Andre, T.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P. KEYNOTE-177: Phase III randomized study of pembrolizumab versus chemotherapy for microsatellite instability-high advanced colorectal cancer. J. Clin. Oncol. 2021, 39, 6. [Google Scholar] [CrossRef]
- André, T.; Elez, E.; Lenz, H.-J.; Jensen, L.H.; Touchefeu, Y.; Van Cutsem, E.; Garcia-Carbonero, R.; Tougeron, D.; Mendez, G.A.; Schenker, M.; et al. Nivolumab plus ipilimumab versus nivolumab in microsatellite instability-high metastatic colorectal cancer (CheckMate 8HW): A randomised, open-label, phase 3 trial. Lancet 2025, 405, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Elez, E.; Van Cutsem, E.; Jensen, L.H.; Bennouna, J.; Mendez, G.; Schenker, M.; de la Fouchardiere, C.; Limon, M.L.; Yoshino, T. Nivolumab plus Ipilimumab in Microsatellite-Instability–High Metastatic Colorectal Cancer. N. Engl. J. Med. 2024, 391, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Personeni, N.; Pietrantonio, F.; Germano, G.; Amatu, A.; Bonoldi, E.; Valtorta, E.; Barault, L.; Di Nicolantonio, F. Pembrolizumab in MMR-proficient metastatic colorectal cancer pharmacologically primed to trigger dynamic hypermutation status: The ARETHUSA trial. J. Clin. Oncol. 2019, 37, TPS2659. [Google Scholar] [CrossRef]
- Morano, F.; Raimondi, A.; Pagani, F.; Lonardi, S.; Salvatore, L.; Cremolini, C.; Murgioni, S.; Randon, G.; Palermo, F.; Antonuzzo, L. Temozolomide followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite-stable, O6-methylguanine–DNA methyltransferase–silenced metastatic colorectal cancer: The MAYA trial. J. Clin. Oncol. 2022, 40, 1562–1573. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Chibaudel, B.; Taieb, J.; Bennouna, J.; Martin-Babau, J.; Fonck, M.; Borg, C.; Cohen, R.; Thibaudin, M.; Limagne, E. Durvalumab and tremelimumab in combination with FOLFOX in patients with RAS-mutated, microsatellite-stable, previously untreated metastatic colorectal cancer (MCRC): Results of the first intermediate analysis of the phase Ib/II MEDETREME trial. J. Clin. Oncol. 2020, 38, 3006. [Google Scholar] [CrossRef]
- Fang, X.; Zhu, N.; Zhong, C.; Wang, L.; Li, J.; Weng, S.; Hu, H.; Dong, C.; Li, D.; Song, Y. Sintilimab plus bevacizumab, oxaliplatin and capecitabine as first-line therapy in RAS-mutant, microsatellite stable, unresectable metastatic colorectal cancer: An open-label, single-arm, phase II trial. EClinicalMedicine 2023, 62, 102123. [Google Scholar] [CrossRef]
- Jänne, P.A.; Bigot, F.; Papadopoulos, K.; Eberst, L.; Sommerhalder, D.; Lebellec, L.; Voon, P.J.; Pellini, B.; Kalinka, E.; Arbour, K. Abstract PR014: Preliminary safety and anti-tumor activity of RMC-6291, a first-in-class, tri-complex KRASG12C (ON) inhibitor, in patients with or without prior KRASG12C (OFF) inhibitor treatment. Mol. Cancer Ther. 2023, 22, PR014. [Google Scholar] [CrossRef]
- Wang, Y.; Neve, R.M.; Staunton, J.; Webster, K.R. FMC-376 a dual inhibitor of ON and OFF states of KRASG12C is broadly active in PDX models of resistance. Cancer Res. 2024, 84, 5948. [Google Scholar] [CrossRef]
- Pellatt, A.J.; Bhamidipati, D.; Subbiah, V. Ready, Set, Go: Setting Off on the Mission to Target KRAS in Colorectal Cancer. JCO Oncol. Pract. 2024, 20, 1289–1292. [Google Scholar] [CrossRef]
- McGregor, L.M.; Jenkins, M.L.; Kerwin, C.; Burke, J.E.; Shokat, K.M. Expanding the scope of electrophiles capable of targeting K-Ras oncogenes. Biochemistry 2017, 56, 3178–3183. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 2021, 65, 3123–3133. [Google Scholar] [CrossRef]
- Tajiknia, V.; El-Deiry, W.S.; Schwermann, M.; Huntington, K.; Zhou, L.; Srinivasan, P. Combination of 5-FU plus KRAS G12D inhibitor MRTX1133 against human colorectal and pancreatic cancer cells and the affects on inhibition of pERK and immune-stimulatory cytokine patterns in in KRAS G12D and KRAS G12V tumor cells. J. Clin. Oncol. 2023, 41, e16301. [Google Scholar] [CrossRef]
- Strickler, J.H.; Yoshino, T.; Stevinson, K.; Eichinger, C.S.; Giannopoulou, C.; Rehn, M.; Modest, D.P. Prevalence of KRAS G12C Mutation and Co-mutations and Associated Clinical Outcomes in Patients With Colorectal Cancer: A Systematic Literature Review. Oncologist 2023, 28, e981–e994. [Google Scholar] [CrossRef]
- Foote, J.B.; Mattox, T.E.; Keeton, A.B.; Chen, X.; Smith, F.T.; Berry, K.L.; Holmes, T.; Wang, J.; Huang, C.-H.; Ward, A.B. A novel Pan-RAS inhibitor with a unique mechanism of action blocks tumor growth in mouse models of GI cancer. bioRxiv 2023, 2023.2005.2017.541233. [Google Scholar]
- Keeton, A.B.; Chen, X.; Valiyaveettil, J.; Huang, C.-H.; Mattox, T.E.; Fadlalla, K.; Foote, J.B.; Buchsbaum, D.J.; Berry, K.L.; Nurmemmedov, E. ADT-007 binds RAS and inhibits RAS signaling. Cancer Res. 2023, 83, 1658. [Google Scholar] [CrossRef]
- Dienstmann, R.; Connor, K.; Byrne, A.T.; Fridman, W.; Lambrechts, D.; Sadanandam, A.; Trusolino, L.; Prehn, J.; Tabernero, J.; Kolch, W. Precision therapy in RAS mutant colorectal cancer. Gastroenterology 2020, 158, 806–811. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Kim, H.S.; Song, M.; Cha, Y.H.; Kim, Y.H.; Shin, J.; Lee, E.S.; Joo, Y.; Song, J.J.; et al. Targeting mutant KRAS with CRISPR-Cas9 controls tumor growth. Genome Res. 2018, 28, 374–382. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: Mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 9. [Google Scholar] [CrossRef]
- Bonato, A.; Bomben, R.; Chakraborty, S.; Felician, G.; Martines, C.; Zucchetto, A.; Chiarenza, A.; Del Poeta, G.; Marasca, R.; Tafuri, A. Chronic lymphocytic leukemia cells with mutated nfkbie are positively selected by microenvironmental signals and display reduced sensitivity to ibrutinib treatment. Blood 2021, 138, 248. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.-I.; Restifo, N.P.; Yang, J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef]
- Pant, S.; Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Basturk, O.; VanWyk, H. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: The phase 1 AMPLIFY-201 trial. Nat. Med. 2024, 30, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Cid, R.; Bolívar, J. Platforms for production of protein-based vaccines: From classical to next-generation strategies. Biomolecules 2021, 11, 1072. [Google Scholar] [CrossRef]
- Cohn, A.; Morse, M.A.; O’Neil, B.; Whiting, S.; Coeshott, C.; Ferraro, J.; Bellgrau, D.; Apelian, D.; Rodell, T.C. Whole recombinant Saccharomyces cerevisiae yeast expressing Ras mutations as treatment for patients with solid tumors bearing Ras mutations: Results from a phase 1 trial. J. Immunother. 2018, 41, 141–150. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Trial | Phase | Experimental Arm | Control Arm | PFS (Median Month, HR, Log-Rank p) | OS (Median Month, HR, Log-Rank p) | ||
|---|---|---|---|---|---|---|---|
| Ras Wt | Ras Mut | Ras Wt | Ras Mut | ||||
| [13] Maughan, 2011 (COIN) | III | FOLFOX or XELOX + Cetuximab | FOLFOX or XELOX | 8.6 vs. 8.6 HR: 1.04 p = 0.60 | 6.9 vs. 6.5 HR: 0.94 p = 0.46 | 17.0 vs. 17.9 HR: 1.04 p = 0.960 | 13.6 vs. 14.8 HR: 1.02 p = 0.800 |
| [15] Douillard, 2014 (PRIME) | III | FOLFOX4 + Panitumumab | FOLFOX4 | 10.0 vs. 8.6 HR: 0.80 p = 0.010 | 7.4 vs. 9.2 HR 1.27 p = 0.020 | 23.9 vs. 19.7 HR: 0.88 p = 0.170 | 15.5 vs. 19.2 HR: 1.17 p = 0.140 |
| [22] Tveit, 2012 (NORDIC VII) | III | FLOX + Cetuximab | FLOX | 7.9 vs. 8.7 HR: 1.07 p = 0.660 | 9.2 vs. 7.8 HR: 0.71 p = 0.07 | 20.1 vs. 22 HR: 1.14, p = 0.48 | 21.1 vs. 20.4 HR: 1.03 p = 0.89 |
| [23] Van Kutsem, 2015 (CRYSTAL) | III | FOLFIRI + Cetuximab | FOLFIRI | 11.4 vs. 8.4 HR: 0.56 p < 0.001 | 7.4 vs. 7.5 HR: 1.10 p = 0.470 | 28.4 vs. 20.2 HR: 0.69 p = 0.0024 | 16.4 vs. 17.7 HR:1.05 p = 0.640 |
| [17] Price, 2015 (AGITG MAX) | III | Capecitabine + bevacizumab ± mitomycin | Capecitabine | 8.6 vs. 6.0 HR = 0.69 p = 0.030 | 8.8 vs. 6.2 HR: 0.65 p = 0.007 | 18.9 vs. 20.6 HR = 0.99 p = 0.950 | 20.4 vs. 22.8 HR = 0.91 p = 0.700 |
| [18] Hurwitz, 2009 (AVF-2107) | III | IFL + Bevacizumab | IFL | 13.5 vs. 7.4 HR: 0.44 p < 0.0001 | 9.3 vs. 5.5 HR: 0.41 p = 0.0008 | 27.7 vs. 17.6 HR: 0.58 p = 0.040 | 19.9 vs. 13.6 HR 0.69 p = 0.260 |
| [19] Hecht, 2008 (PACCE) | III | Ox-CT + Bev + Pan | Ox-CT + Bev | 9.8 vs. 11.5 HR: 1.36 | 10.4 vs. 11.0 HR: 1.25 | 20.7 vs. 24.5 HR: 1.89 | 19.3 vs. 19.3 HR: 1.02 |
| Iri-CT + Bev + Pan | Iri-CT + Bev | 10 vs. 12.5 HR: 1.50 | 8.3 vs. 11.9 HR: 1.19 | NE vs. 19.8 HR: 1.28 | 17.8 vs. 20.5 HR: 2.14 | ||
| [20] Cremolini, 2015 (TRIBE) | III | FOLFOXIRI + Bevacizumab | FOLFIRI + Bevacizumab | 12.8 vs. 11.0 HR: 0.84 p = 0.770 | 12.0 vs. 9.5 HR: 0.78 | 26.8 vs. 37.1, HR 0.78, p = 0.66 | 23.9 vs. 27.3, HR 0.88 |
| [24] Modest, 2019 (AIO KRK0110) | III | XELIRI or FOLFIRI + Bevacizumab | FP + Bevacizumab After PD XELIRI or FOLFIRI + Bevacizumab | 11.8 vs. 8.0 HR 0.54 p < 0.001 | 9.3 vs. 8.1 HR 0.87 p = 0.340 | 28.5 vs. 23.5 HR 0.64 p = 0.02 | 23.2 vs. 21.3 HR 0.92 p = 0.62 |
| Trial | Phase | Treatment Line | Experimental Arm | Control Arm | PFS (Median Month, HR, Log-Rank p) | OS (Median Month, HR, Log-Rank p) | ||
|---|---|---|---|---|---|---|---|---|
| Ras Wt | Ras Mut | Ras Wt | Ras Mut | |||||
| [32] Bennouna 2013 (ML18147) | III | Second line | FOLFOX or XELOX + Cetuximab | FOLFOX or XELOX | 8.6 vs. 8.6 HR: 1.04 p = 0.60 | 6.9 vs. 6.5 HR: 0.94 p = 0.46 | 17.0 vs. 17.9 HR: 1.04 p = 0.960 | 13.6 vs. 14.8 HR: 1.02 p = 0.800 |
| [37] Tabernero 2015 (RAISE) | III | Second line | FOLFOX4 + Ramucurimab | FOLFOX4 | 10.0 vs. 8.6 HR: 0.80 p = 0.010 | 7.4 vs. 9.2 HR 1.27 p = 0.020 | 23.9 vs. 19.7 HR: 0.88 p = 0.170 | 15.5 vs. 19.2 HR: 1.17 p = 0.140 |
| [42] Grothey 2013 (CORRECT) | II | Later line | FOLFOX4 + Cetuximab | FOLFOX4 | 8.3 vs. 7.2 HR: 0.56 p = 0.0064 | 5.5 vs. 8.6 HR: 1.72 p = 0.015 | 22.8 vs. 18.5 HR: 0.85 p = 0.390 | 13.4 vs. 17.5 HR: 1.29 p = 0.200 |
| [43] Li 2015 (CONCUR) | III | Later line | FLOX + Cetuximab | FLOX | 7.9 vs. 8.7 HR: 1.07 p = 0.660 | 9.2 vs. 7.8 HR: 0.71 p = 0.07 | 20.1 vs. 22 HR: 1.14, p = 0.48 | 21.1 vs. 20.4 HR: 1.03 p = 0.89 |
| [38] Hegewisch-Becker, 2015 (AIOKRK0207) | III | Maintenance | FOLFIRI + Cetuximab | FOLFIRI | 11.4 vs. 8.4 HR: 0.56 p < 0.001 | 7.4 vs. 7.5 HR: 1.10 p = 0.470 | 28.4 vs. 20.2 HR: 0.69 p = 0.0024 | 16.4 vs. 17.7 HR: 1.05 p = 0.640 |
| [39] Aparacio 2018 (PRODIGE 9) | III | Maintenance | Capecitabine plus bevacizumab ± mitomycin | Capecitabine | 8.6 vs. 6.0 HR = 0.69 p = 0.030 | 8.8 vs. 6.2 HR: 0.65 p = 0.007 | 18.9 vs. 20.6 HR = 0.99 p: 0.950 | 20.4 vs. 22.8 HR = 0.91 p: 0.700 |
| [34] Van Cutsem 2016 (VELOUR) | III | Second line | IFL + Bevacizumab | IFL | 13.5 vs. 7.4 HR: 0.44 p < 0.0001 | 9.3 vs. 5.5 HR: 0.41 p = 0.0008 | 27.7 vs. 17.6 HR: 0.58 p = 0.040 | 19.9 vs. 13.6 HR 0.69 p = 0.260 |
| Study Name or ID | Phase | Experimental Drug(s) | ORR/DCR | mPFS | mOS * |
|---|---|---|---|---|---|
| [55] CodeBreaK 100 | Phase I | Sotorasib | 7.1%/73.8% | 6.3 month | 12.5 month |
| [56] CodeBreaK 100 (CRC expansion cohort) | Phase II | Sotorasib 960 mg qd | 9.7%/82.3% | 6.3 month | 12.5 month |
| [57] CodeBreaK 101 | Phase Ib | Sotorasib + panitumumab | 30%/92.5% | 8.2 month | 17.9 month |
| [57] CodeBreaK 101 (subprotocol H) | Phase Ib | Sotorasib 960 mg qd + panitumumab + FOLFİRİ | 58.1%/93.5% | 8.2 month | 17.9 month |
| [2] CodeBreaK 300 | Phase III | Sotorasib 960 mg qd + panitumumab | 26.4%/71.7% | 5.6 month | NR |
| Sotorasib 240 mg qd + panitumumab | 5.7%/67.9% | 3.9 month | 11.9 month | ||
| Standard of care | 0%/46.3% | 2.2 month | 10.3 month | ||
| [58] KRYSTAL-1 | Phase I/Ib | Adagrasib | 50%/- | 11.1 month | -- |
| [3] KRYSTAL-1 | Phase I/II | Adagrasib 600 mg bid | 19%/86% | 5.6 month | -- |
| [3] KRYSTAL-1 | Phase I/II | Adagrasib 600 mg bid + cetuximab | 46%/100% | 6.9 month | 15.9 month |
| [59] NCT04449874 | Phase Ib | Divarasib | 29.1%/– 35.9%/–(400 mg) | 5.6 month | -- |
| [60] NCT04449874 | Phase Ib | Divarasib 400 mg qd + cetuximab | 62.5% | 8 month | -- |
| [61] NCT04585035 | Phase I/II | Garsorasib 600 mg bid + cetuximab | 51.7%/93.1% | 7.5 month | -- |
| [62] NCT05005234, NCT05497336 | Phase Ib Phase III | Fulzerasib 600 mg bid | 43.8%/87.5% | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sütcüoğlu, O.; Yıldırım, H.Ç.; Almuradova, E.; Günenç, D.; Yalçın, Ş. RAS Mutations in Advanced Colorectal Cancer: Mechanisms, Clinical Implications, and Novel Therapeutic Approaches. Medicina 2025, 61, 1202. https://doi.org/10.3390/medicina61071202
Sütcüoğlu O, Yıldırım HÇ, Almuradova E, Günenç D, Yalçın Ş. RAS Mutations in Advanced Colorectal Cancer: Mechanisms, Clinical Implications, and Novel Therapeutic Approaches. Medicina. 2025; 61(7):1202. https://doi.org/10.3390/medicina61071202
Chicago/Turabian StyleSütcüoğlu, Osman, Hasan Çağrı Yıldırım, Elvina Almuradova, Damla Günenç, and Şuayib Yalçın. 2025. "RAS Mutations in Advanced Colorectal Cancer: Mechanisms, Clinical Implications, and Novel Therapeutic Approaches" Medicina 61, no. 7: 1202. https://doi.org/10.3390/medicina61071202
APA StyleSütcüoğlu, O., Yıldırım, H. Ç., Almuradova, E., Günenç, D., & Yalçın, Ş. (2025). RAS Mutations in Advanced Colorectal Cancer: Mechanisms, Clinical Implications, and Novel Therapeutic Approaches. Medicina, 61(7), 1202. https://doi.org/10.3390/medicina61071202