A Comparative Study on the Multidimensional Features of Hereditary and Sporadic Medullary Thyroid Carcinoma Patients: A Single-Center Retrospective Study

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Setting
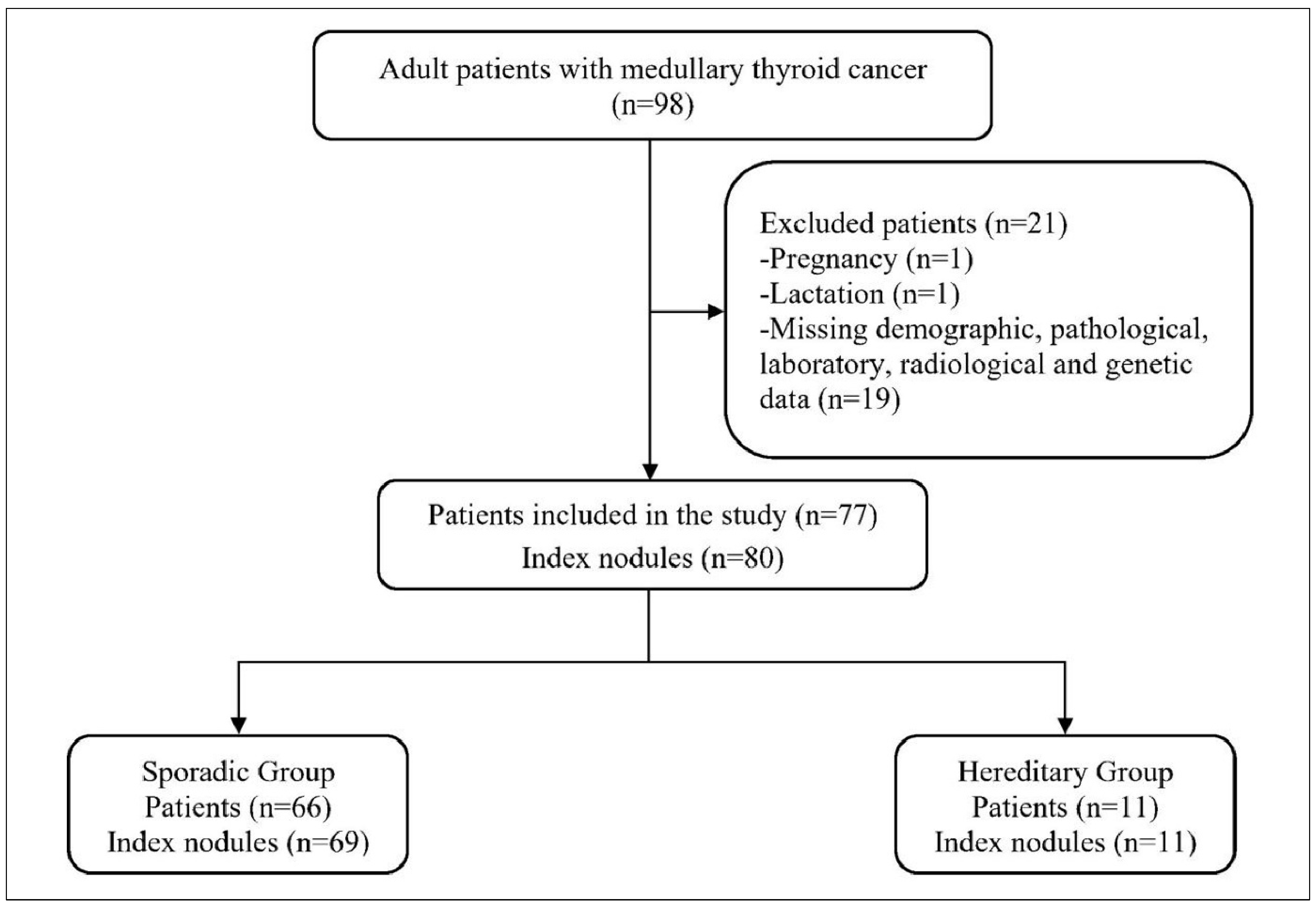
2.2. Study Population
Sporadic vs. Hereditary Stratification
2.3. Data and Variables
2.4. Surgical Management
2.5. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lasolle, H.; Do Cao, C.; Lamartina, L.; Al Ghuzlan, A.; Drui, D.; Buffet, C.; Leboulleux, S.; Hescot, S.; Godbert, Y.; Zerdoud, S.; et al. ENDOCAN-TUTHYREF network consensus recommendations. Refractory medullary thyroid cancer. Ann. Endocrinol. 2025, 86, 101733. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.; Hussein, M.; Anker, A.; Baah, S.; Pinion, D.; Jishu, J.; Sadakkadulla, S.; Case, M.; LaForteza, A.; Moroz, K.; et al. Survival Outcomes of Medullary Thyroid Cancer with and Without Amyloid Deposition. Endocr. Pract. 2024, 30, 311–318. [Google Scholar] [CrossRef]
- Tao, Z.; Deng, X.; Guo, B.; Ding, Z.; Fan, Y. Subgroup analysis of steadily increased trends in medullary thyroid carcinoma incidence and mortality in the USA, 2000–2020: A population-based retrospective cohort study. Endocr. Relat. Cancer 2024, 31, e230319. [Google Scholar] [CrossRef]
- Green, K.; Hintze, J.; O’Neill, J.P. Surgical aspects and controversies in the management of medullary thyroid cancer. Ir. J. Med. Sci. 2022, 191, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Matrone, A.; Gambale, C.; Prete, A.; Elisei, R. Sporadic Medullary Thyroid Carcinoma: Towards a Precision Medicine. Front. Endocrinol. 2022, 13, 864253. [Google Scholar] [CrossRef] [PubMed]
- Kaliszewski, K.; Ludwig, M.; Ludwig, B.; Mikuła, A.; Greniuk, M.; Rudnicki, J. Update on the Diagnosis and Management of Medullary Thyroid Cancer: What Has Changed in Recent Years? Cancers 2022, 14, 3643. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Trimboli, P.; Mian, C.; Piccardo, A.; Treglia, G. Diagnostic tests for medullary thyroid carcinoma: An umbrella review. Endocrine 2023, 81, 183–193. [Google Scholar] [CrossRef]
- Gambardella, C.; Offi, C.; Patrone, R.; Clarizia, G.; Mauriello, C.; Tartaglia, E.; Di Capua, F.; Di Martino, S.; Romano, R.M.; Fiore, L.; et al. Calcitonin negative Medullary Thyroid Carcinoma: A challenging diagnosis or a medical dilemma? BMC Endocr. Disord. 2019, 19, 45. [Google Scholar] [CrossRef]
- Barletta, J.A.; Nosé, V.; Sadow, P.M. Genomics and Epigenomics of Medullary Thyroid Carcinoma: From Sporadic Disease to Familial Manifestations. Endocr. Pathol. 2021, 32, 35–43. [Google Scholar] [CrossRef]
- Bai, Y.; Niu, D.; Yao, Q.; Lin, D.; Kakudo, K. Updates in the advances of sporadic medullary thyroid carcinoma: From the molecules to the clinic. Gland Surg. 2020, 9, 1847–1856. [Google Scholar] [CrossRef]
- Elisei, R.; Bottici, V.; Cappagli, V.; Ramone, T.; Tacito, A.; Ciampi, R.; Romei, C. Clinical utility of genetic diagnosis for sporadic and hereditary medullary thyroid carcinoma. Ann. Endocrinol. 2019, 80, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Accardo, G.; Conzo, G.; Esposito, D.; Gambardella, C.; Mazzella, M.; Castaldo, F.; Di Donna, C.; Polistena, A.; Avenia, N.; Colantuoni, V.; et al. Genetics of medullary thyroid cancer: An overview. Int. J. Surg. 2017, 41 (Suppl S1), S2–S6. [Google Scholar] [CrossRef]
- Pacini, F.; Castagna, M.G.; Cipri, C.; Schlumberger, M. Medullary thyroid carcinoma. Clin. Oncol. (R. Coll. Radiol.) 2010, 22, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J. Oncol. 2012, 2012, 705036. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Pacini, F.; Robinson, B.G.; Santoro, M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: An update. J. Clin. Endocrinol. Metab. 2013, 98, 3149–3164. [Google Scholar] [CrossRef]
- Raue, F.; Bruckner, T.; Frank-Raue, K. Similar Stage-dependent Survival and Outcome in Sporadic and Hereditary Medullary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2021, 106, e3582–e3591. [Google Scholar] [CrossRef]
- Machens, A.; Lorenz, K.; Weber, F.; Dralle, H. Oncological features of sporadic vs. hereditary pediatric medullary thyroid cancer. Endocrine 2024, 85, 1091–1095. [Google Scholar] [CrossRef]
- Machens, A.; Lorenz, K.; Weber, F.; Dralle, H. Dissection of RET p.M918T-driven progression of hereditary vs. sporadic medullary thyroid cancer. Eur. J. Surg. Oncol. 2025, 51, 109549. [Google Scholar] [CrossRef]
- Gammons, S.; Hu, M.I.; Zafereo, M.E.; Busaidy, N.L.; Perrier, N.D.; Bassett, R.L.; Hyde, S.M.; Grubbs, E.G.; Waguespack, S.G. MON-LB015 Sporadic MTC in Children: Characterization of a Rare Disease. J. Endocr. Soc. 2020, 4, 276–277. [Google Scholar] [CrossRef]
- Kihara, M.; Miyauchi, A.; Yoshioka, K.; Oda, H.; Nakayama, A.; Sasai, H.; Yabuta, T.; Masuoka, H.; Higashiyama, T.; Fukushima, M.; et al. Germline RET mutation carriers in Japanese patients with apparently sporadic medullary thyroid carcinoma: A single institution experience. Auris Nasus Larynx 2016, 43, 551–555. [Google Scholar] [CrossRef]
- Machens, A.; Lorenz, K.; Weber, F.; Dralle, H. Origin and impact of multifocal growth in sporadic vs. hereditary medullary thyroid cancer. Eur. J. Surg. Oncol. 2024, 50, 108625. [Google Scholar] [CrossRef] [PubMed]
- Hensley, S.G.; Hu, M.I.; Bassett, R.L.; Ying, A.K.; Zafereo, M.E.; Perrier, N.D.; Busaidy, N.L.; Hyde, S.M.; Grubbs, E.G.; Waguespack, S.G. Pediatric Medullary Thyroid Carcinoma: Clinical Presentations and Long-Term Outcomes in 144 Patients Over 6 Decades. J. Clin. Endocrinol. Metab. 2024, 109, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Edge, S.B.; Greene, F.L.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C. AJCC Cancer Staging Manual, 8th ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, A.; Erickson, D.; Geske, J.R.; Hay, I.D.; Castro, M.R. Predicting Outcomes in Sporadic and Hereditary Medullary Thyroid Carcinoma over Two Decades. Thyroid 2021, 31, 616–626. [Google Scholar] [CrossRef]
- Bongiovanni, M.; Spitale, A.; Faquin, W.C.; Mazzucchelli, L.; Baloch, Z.W. The Bethesda System for Reporting Thyroid Cytopathology: A meta-analysis. Acta Cytol. 2012, 56, 333–339. [Google Scholar] [CrossRef]
- Vardarli, I.; Weber, M.; Weidemann, F.; Führer, D.; Herrmann, K.; Görges, R. Diagnostic accuracy of routine calcitonin measurement for the detection of medullary thyroid carcinoma in the management of patients with nodular thyroid disease: A meta-analysis. Endocr. Connect. 2021, 10, 358–370. [Google Scholar] [CrossRef]
- Broecker-Preuss, M.; Simon, D.; Fries, M.; Kornely, E.; Weber, M.; Vardarli, I.; Gilman, E.; Herrmann, K.; Görges, R. Update on Calcitonin Screening for Medullary Thyroid Carcinoma and the Results of a Retrospective Analysis of 12,984 Patients with Thyroid Nodules. Cancers 2023, 15, 2333. [Google Scholar] [CrossRef]
- Moura, M.M.; Cabrera, R.A.; Esteves, S.; Cavaco, B.M.; Soares, P.; Leite, V. Correlation of molecular data with histopathological and clinical features in a series of 66 patients with medullary thyroid carcinoma. J. Endocrinol. Investig. 2021, 44, 1837–1846. [Google Scholar] [CrossRef]
- Verbeek, H.H.; de Groot, J.W.B.; Sluiter, W.J.; Muller Kobold, A.C.; van den Heuvel, E.R.; Plukker, J.T.; Links, T.P. Calcitonin testing for detection of medullary thyroid cancer in people with thyroid nodules. Cochrane Database Syst. Rev. 2020, 3, CD010159. [Google Scholar] [CrossRef]
- Prinzi, A.; Vella, V.; Bosco, A.; Mirone, A.; Russo, M.; Piticchio, T.; Di Benedetto, G.; Bartoloni, G.; Frasca, F.; Malandrino, P. Sporadic and Familial Medullary Thyroid Carcinoma: A Retrospective Single Center Study on Presentation and Outcome. Endocr. Res. 2024, 49, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kaserer, K.; Scheuba, C.; Neuhold, N.; Weinhäusel, A.; Haas, O.A.; Vierhapper, H.; Niederle, B. Sporadic versus familial medullary thyroid microcarcinoma: A histopathologic study of 50 consecutive patients. Am. J. Surg. Pathol. 2001, 25, 1245–1251. [Google Scholar] [CrossRef]
- Zhang, D.; Liang, N.; Sun, H.; Frattini, F.; Sui, C.; Yang, M.; Wang, H.; Dionigi, G. Critically evaluated key points on hereditary medullary thyroid carcinoma. Front. Endocrinol. 2024, 15, 1412942. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Variables | Sporadic Group (n = 66) | Hereditary Group (n = 11) | p |
|---|---|---|---|
| Age (year), Median (IQR) | 55.0 (47.0–64.0) | 35.0 (28.0–56.0) | 0.021 |
| Gender, n (%) | |||
| Male | 24 (36.4) | 5 (45.5) | 0.738 a |
| Female | 42 (63.6) | 6 (54.5) | |
| Family history of thyroid cancer, n (%) | 2 (3.0) | 7 (63.6) | <0.001 |
| Thyroid function status, n (%) | |||
| Hyperthyroidism | 3 (4.5) | 0 (0.0) | 0.999 a |
| Hypothyroidism | 7 (10.6) | 1 (9.1) | |
| Euthyroidism | 56 (84.8) | 10 (90.9) | |
| Family history of other tumors n (%) | 6 (9.1) | 3 (27.2) | 0.082 |
| Pheochromocytoma, n (%) | 0 (0.0) | 2 (18.2) | 0.019 a |
| Free T3 (pg/mL), Median (IQR) | 3.35 (3.13–3.71) | 3.55 (3.18–4.05) | 0.095 |
| Free T4 (pg/mL), Median (IQR) | 1.16 (1.08–1.26) | 1.23 (0.95–1.50) | 0.620 |
| TSH (μIU/mL), Median (IQR) | 1.49 (1.05–2.60) | 1.90 (0.61–2.46) | 0.959 |
| Thyroglobulin (ng/mL), Median (IQR) | 14.5 (6.1–40.5) | 15.0 (6.2–26.0) | 0.808 |
| Anti-TG (IU/mL), Median (IQR) | 2.20 (0.90–15.55) | 1.00 (0.60–1.30) | 0.081 |
| Anti-TPO (IU/mL), Median (IQR) | 41.0 (28.0–68.2) | 62.0 (28.0–65.0) | 0.575 |
| Calcitonin (pg/mL), Median (IQR) | 217.5 (52.7–858.0) | 223.0 (21.0–3202.0) | 0.838 |
| CEA (ng/mL), Median (IQR) | 5.30 (1.84–28.42) | 3.10 (1.49–42.70) | 0.738 |
| Type of surgery, n (%) | |||
| TT | 19 (28.8) | 1 (9.1) | 0.137 a |
| TT + CLND | 26 (39.4) | 3 (27.3) | |
| TT + CLND + LLND | 21 (31.8) | 7 (63.6) |
| Variables | Sporadic Group (n = 66) | Hereditary Group (n = 11) | p |
|---|---|---|---|
| Tumor size (mm), Median (IQR) | 13.0 (7.0–22.2) | 13.0 (5.0–20.0) | 0.749 |
| Variants, n (%) | |||
| Isolated | 47 (71.2) | 10 (90.9) | 0.581 a |
| Composite | 4 (6.1) | 0 (0.0) | |
| Collision | 15 (22.7) | 1 (9.1) | |
| Right-lobe localization, n (%) | 36 (54.5) | 6 (54.5) | 0.999 b |
| Left-lobe localization, n (%) | 28 (42.4) | 5 (45.5) | 0.999 a |
| Isthmus localization, n (%) | 3 (4.5) | 0 (0.0) | 0.999 a |
| Multicentricity, n (%) | 2 (3.0) | 4 (36.4) | 0.003 a |
| Extrathyroidal extension, n (%) | 9 (9.1) | 3 (27.3) | 0.113 a |
| Surgical margin positivity, n (%) | 5 (7.6) | 2 (18.2) | 0.261 a |
| Capsular invasion, n (%) | 7 (10.6) | 2 (18.2) | 0.608 a |
| Vascular invasion, n (%) | 20 (30.3) | 4 (36.4) | 0.732 a |
| Neural invasion, n (%) | 3 (4.5) | 1 (9.1) | 0.467 a |
| Concurrent PTC, n (%) | 17 (25.8) | 1 (9.1) | 0.441 a |
| Variables | Sporadic Group (n = 66) | Hereditary Group (n = 11) | |
|---|---|---|---|
| TNM | T1aN0M0 | 27 (40.9) | 6 (54.5) |
| T1aN1aM0 | 1 (1.5) | 0 (0.0) | |
| T1aN1bM0 | 2 (3.0) | 0 (0.0) | |
| T1bN0M0 | 11 (16.7) | 0 (0.0) | |
| T1bN1aM0 | 2 (3.0) | 1 (9.1) | |
| T1bN1bM0 | 4 (6.1) | 2 (18.2) | |
| T2N0M0 | 4 (6.1) | 0 (0.0) | |
| T2N1bM0 | 7 (10.6) | 2 (18.2) | |
| T3aN0M0 | 2 (3.0) | 0 (0.0) | |
| T3aN1bM0 | 2 (3.0) | 0 (0.0) | |
| T3N0M0 | 1 (1.5) | 0 (0.0) | |
| T3N1bM0 | 1 (1.5) | 0 (0.0) | |
| T4aN1aM0 | 1 (1.5) | 0 (0.0) | |
| T4aN1bM0 | 1 (1.5) | 0 (0.0) | |
| Anatomical stage, n (%) | 1 | 38 (57.6) | 6 (54.5) |
| 2 | 7 (10.6) | 0 (0.0) | |
| 3 | 3 (4.5) | 1 (9.1) | |
| 4A | 18 (27.3) | 4 (36.4) | |
| Pathological stage, n (%) | 1 | 36 (54.5) | 6 (54.5) |
| 2 | 6 (9.1) | 0 (0.0) | |
| 3 | 9 (13.6) | 1 (9.1) | |
| 4 | 15 (22.7) | 4 (36.4) | |
| Variables | Sporadic Group (n = 69) | Hereditary Group (n = 11) | p |
|---|---|---|---|
| Volume (cc), Median (IQR) | 1.20 (0.30–5.85) | 1.00 (0.20–5.00) | 0.660 |
| Longitudinal diameter (mm), Median (IQR) | 16.0 (10.0–26.6) | 15.0 (10.6–24.0) | 0.706 |
| AP/T | 0.82 (0.70–0.90) | 0.77 (0.66–0.85) | 0.360 |
| Axial localization, n (%) | |||
| Right | 35 (50.7) | 4 (36.4) | 0.584 a |
| Left | 31 (44.9) | 7 (63.6) | |
| Isthmus | 3 (4.3) | 0 (0.0) | |
| Longitudinal localization, n (%) | |||
| Upper third | 8 (11.6) | 2 (18.2) | 0.745 a |
| Upper two-thirds | 2 (2.9) | 0 (0.0) | |
| Middle third | 33 (47.8) | 6 (54.5) | |
| Inferior third | 5 (7.2) | 1 (9.1) | |
| Lower two-thirds | 10 (14.5) | 2 (18.2) | |
| Completely | 11 (15.9) | 0 (0.0) | |
| Echogenicity, n (%) | |||
| Hypoechoic | 26 (37.7) | 2 (18.2) | 0.276 a |
| Isoechoic | 22 (31.9) | 3 (27.3) | |
| Iso-hypoechoic | 21 (30.4) | 6 (54.5) | |
| Calcification, n (%) | |||
| No | 31 (44.9) | 7 (63.6) | 0.742 a |
| Macro | 15 (21.7) | 2 (18.2) | |
| Micro | 16 (23.2) | 1 (9.1) | |
| Micro and macro | 7 (10.1) | 1 (9.1) | |
| Halo sign, n (%) | 7 (10.1) | 0 (0.0) | 0.585 a |
| EU-TIRADS | |||
| 3 | 16 (23.2) | 4 (36.4) | 0.377 a |
| 4 | 19 (27.5) | 4 (36.4) | |
| 5 | 34 (49.3) | 3 (27.3) | |
| BETHESDA | |||
| 1 | 0 (0.0) | 3 (27.3) | 0.018 a |
| 2 | 3 (4.3) | 0 (0.0) | |
| 3 | 3 (4.3) | 0 (0.0) | |
| 4 | 2 (2.9) | 0 (0.0) | |
| 5 | 22 (31.9) | 4 (36.4) | |
| 6 | 39 (56.5) | 4 (36.4) |
| Variables | Sporadic Group (n = 17) | Hereditary Group (n = 1) | p |
|---|---|---|---|
| PTC variants | |||
| Classic, n (%) | 14 (82.4) | 1 (100.0) | 0.999 a |
| Follicular, n (%) | 5 (29.4) | 0 (0.0) | 0.999 a |
| Tall cell, n (%) | 1 (5.9) | 0 (0.0) | 0.999 a |
| Warthin-like, n (%) | 1 (5.9) | 0 (0.0) | 0.999 a |
| PTC localization | |||
| Right, n (%) | 10 (58.8) | 0 (0.0) | 0.444 a |
| Left, n (%) | 8 (47.1) | 1 (100.0) | 0.999 a |
| Isthmus, n (%) | 4 (23.5) | 0 (0.0) | 0.999 a |
| PTC diameter (mm), Median (IQR) | 7.0 (3.0–13.5) | 1.0 (n.a.-n.a.) | 0.111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deniz, M.S.; Nasiroglu Imga, N.; Tural Balsak, B.O.; Safak Bulut, A.; Savas, F.; Cavdarli, B.; Aydin, C.; Topaloglu, O.; Ersoy, R.; Cakir, B. A Comparative Study on the Multidimensional Features of Hereditary and Sporadic Medullary Thyroid Carcinoma Patients: A Single-Center Retrospective Study. Medicina 2025, 61, 1164. https://doi.org/10.3390/medicina61071164
Deniz MS, Nasiroglu Imga N, Tural Balsak BO, Safak Bulut A, Savas F, Cavdarli B, Aydin C, Topaloglu O, Ersoy R, Cakir B. A Comparative Study on the Multidimensional Features of Hereditary and Sporadic Medullary Thyroid Carcinoma Patients: A Single-Center Retrospective Study. Medicina. 2025; 61(7):1164. https://doi.org/10.3390/medicina61071164
Chicago/Turabian StyleDeniz, Muzaffer Serdar, Narin Nasiroglu Imga, Belma Ozlem Tural Balsak, Asiye Safak Bulut, Furkan Savas, Busranur Cavdarli, Cevdet Aydin, Oya Topaloglu, Reyhan Ersoy, and Bekir Cakir. 2025. "A Comparative Study on the Multidimensional Features of Hereditary and Sporadic Medullary Thyroid Carcinoma Patients: A Single-Center Retrospective Study" Medicina 61, no. 7: 1164. https://doi.org/10.3390/medicina61071164
APA StyleDeniz, M. S., Nasiroglu Imga, N., Tural Balsak, B. O., Safak Bulut, A., Savas, F., Cavdarli, B., Aydin, C., Topaloglu, O., Ersoy, R., & Cakir, B. (2025). A Comparative Study on the Multidimensional Features of Hereditary and Sporadic Medullary Thyroid Carcinoma Patients: A Single-Center Retrospective Study. Medicina, 61(7), 1164. https://doi.org/10.3390/medicina61071164