
Characteristics of 21 Patients with Secondary Hemophagocytic Lymphohistiocytosis—Insights from a Single-Center Retrospective Study




Abstract
1. Introduction
2. Patients and Methods
2.1. Study Population
2.2. Statistical Elaboration
3. Results
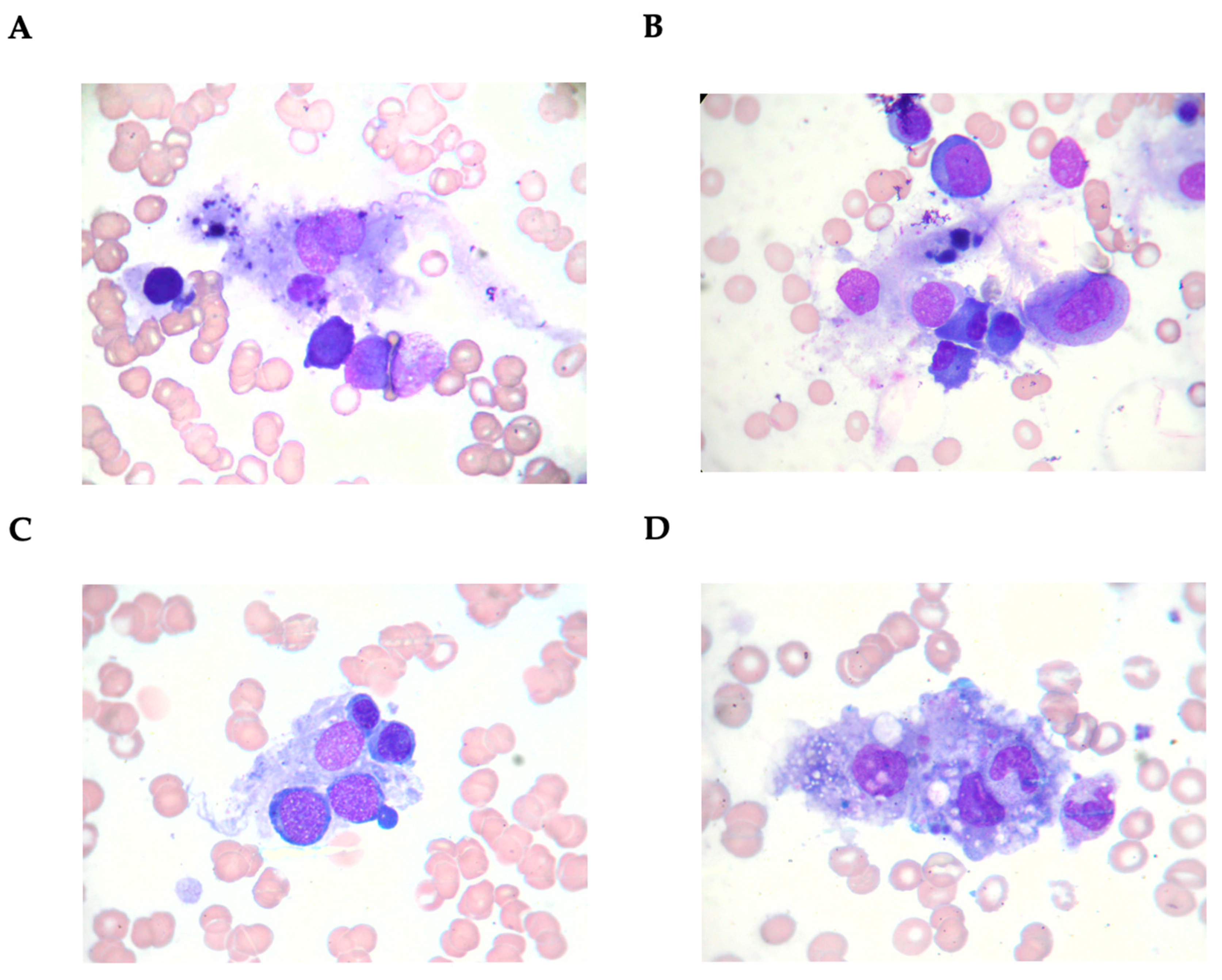
3.1. Characteristics of Patients with Secondary Hemophagocytic Lymphohistiocytosis
3.2. Characteristics of Patients with Hemophagocytic Lymphohistiocytosis Deceased During Hospitalization
3.3. Characteristics of Patients with Macrophage Activation Syndrome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skinner, J.; Yankey, B.; Shelton, B.K. Hemophagocytic Lymphohistiocytosis. AACN Adv. Crit. Care 2019, 30, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.F.; Mackenzie, S.; Low, R.; Brown, M.; Sanchez, E.; Carr, A.; Carpenter, B.; Bishton, M.; Duncombe, A.; Akpabio, A.; et al. Diagnosis and Investigation of Suspected Haemophagocytic Lymphohistiocytosis in Adults: 2023 Hyperinflammation and HLH Across Speciality Collaboration (HiHASC) Consensus Guideline. Lancet Rheumatol. 2024, 6, e51–e62. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult Haemophagocytic Syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Brisse, E.; Wouters, C.H.; Matthys, P. Advances in the Pathogenesis of Primary and Secondary Haemophagocytic Lymphohistiocytosis: Differences and Similarities. Br. J. Haematol. 2016, 174, 203–217. [Google Scholar] [CrossRef]
- Canna, S.W.; Marsh, R.A. Pediatric Hemophagocytic Lymphohistiocytosis. Blood 2020, 135, 1332–1343. [Google Scholar] [CrossRef]
- Sepulveda, F.E.; De Saint Basile, G. Hemophagocytic Syndrome: Primary Forms and Predisposing Conditions. Curr. Opin. Immunol. 2017, 49, 20–26. [Google Scholar] [CrossRef]
- Bracaglia, C.; De Graaf, K.; Pires Marafon, D.; Guilhot, F.; Ferlin, W.; Prencipe, G.; Caiello, I.; Davì, S.; Schulert, G.; Ravelli, A.; et al. Elevated Circulating Levels of Interferon-γ and Interferon-γ-Induced Chemokines Characterise Patients with Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis. Ann. Rheum. Dis. 2017, 76, 166–172. [Google Scholar] [CrossRef]
- Filipovich, A.; McClain, K.; Grom, A. Histiocytic Disorders: Recent Insights into Pathophysiology and Practical Guidelines. Biol. Blood Marrow Transplant. 2010, 16, S82–S89. [Google Scholar] [CrossRef]
- Chen, C.; Fahmy, L.M.; Schreidah, C.M.; Magro, C.M.; Geskin, L.J. Febrile Ulceronecrotic Mucha-Habermann Disease Associated with Hemophagocytic Lymphohistiocytosis: A Case Report and Review of the Literature. Am. J. Dermatopathol. 2024, 46, 238–242. [Google Scholar] [CrossRef]
- Morimoto, A.; Nakazawa, Y.; Ishii, E. Hemophagocytic Lymphohistiocytosis: Pathogenesis, Diagnosis, and Management. Pediatr. Int. 2016, 58, 817–825. [Google Scholar] [CrossRef]
- Shakoory, B.; Geerlinks, A.; Wilejto, M.; Kernan, K.; Hines, M.; Romano, M.; Piskin, D.; Ravelli, A.; Sinha, R.; Aletaha, D.; et al. The 2022 EULAR/ACR Points to Consider at the Early Stages of Diagnosis and Management of Suspected Haemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS). Ann. Rheum. Dis. 2023, 82, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Filipovich, A.H. Hemophagocytic Lymphohistiocytosis (HLH) and Related Disorders. Hematology 2009, 2009, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Ponnatt, T.S.; Lilley, C.M.; Mirza, K.M. Hemophagocytic Lymphohistiocytosis. Arch. Pathol. Lab. Med. 2022, 146, 507–519. [Google Scholar] [CrossRef]
- Mehta, R.S.; Smith, R.E. Hemophagocytic Lymphohistiocytosis (HLH): A Review of Literature. Med. Oncol. 2013, 30, 740. [Google Scholar] [CrossRef]
- Yildiz, H.; Castanares-Zapatero, D.; d’Abadie, P.; Bailly, S.; Yombi, J.C. Hemophagocytic Lymphohistiocytosis in Adults: A Retrospective Study in a Belgian Teaching Hospital. Int. J. Gen. Med. 2022, 15, 8111–8120. [Google Scholar] [CrossRef]
- Kumar, G.; Hererra, M.; Patel, D.; Nanchal, R.; Guddati, A.K. Outcomes of Adult Critically Ill Patients with Hemophagocytic Lymphohistiocytosis in United States-Analysis from an Administrative Database from 2007 to 2015. Am. J. Blood Res. 2020, 10, 330–338. [Google Scholar] [PubMed]
- Lehmberg, K.; Ehl, S. Diagnostic Evaluation of Patients with Suspected Haemophagocytic Lymphohistiocytosis. Br. J. Haematol. 2013, 160, 275–287. [Google Scholar] [CrossRef]
- Otrock, Z.K.; Eby, C.S. Clinical Characteristics, Prognostic Factors, and Outcomes of Adult Patients with Hemophagocytic Lymphohistiocytosis. Am. J. Hematol. 2015, 90, 220–224. [Google Scholar] [CrossRef]
- Giemza-Stokłosa, J.; Islam, M.A.; Kotyla, P.J. Hyperferritinaemia: An Iron Sword of Autoimmunity. Curr. Pharm. Des. 2019, 25, 2909–2918. [Google Scholar] [CrossRef]
- Karakike, E.; Giamarellos-Bourboulis, E.J. Macrophage Activation-Like Syndrome: A Distinct Entity Leading to Early Death in Sepsis. Front. Immunol. 2019, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Popko, K.; Górska, E.; Wołowiec, M.; Malinowska, I. Disturbances in NK Cells in Various Types of Hemophagocytic Lymphohistiocytosis in a Population of Polish Children. J. Pediatr. Hematol./Oncol. 2019, 41, e277–e283. [Google Scholar] [CrossRef]
- Gupta, S.; Weitzman, S. Primary and Secondary Hemophagocytic Lymphohistiocytosis: Clinical Features, Pathogenesis and Therapy. Expert Rev. Clin. Immunol. 2010, 6, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, L.; Zhou, D.; Li, L.; Xie, W.; Tan, Y.; Ye, X. Risk Factors and Prognosis of Early Death in Secondary Hemophagocytic Lymphohistiocytosis. Ann. Hematol. 2023, 102, 2301–2308. [Google Scholar] [CrossRef]
- Ardern-Jones, M.R.; Stammers, M.; Phan, H.T.; Borca, F.; Koutalopoulou, A.; Teo, Y.; Batchelor, J.; Smith, T.; Duncombe, A.S. Secondary Haemophagocytic Lymphohistiocytosis in Hospitalised COVID-19 Patients as Indicated by a Modified HScore Is Infrequent and High Scores Do Not Associate with Increased Mortality. Clin. Med. 2021, 21, e543–e547. [Google Scholar] [CrossRef]
- Zoref-Lorenz, A.; Witzig, T.E.; Cerhan, J.R.; Jordan, M.B. Malignancy-Associated HLH: Mechanisms, Diagnosis, and Treatment of a Severe Hyperinflammatory Syndrome. Leuk. Lymphoma 2024, 66, 628–636. [Google Scholar] [CrossRef]
- Gualdoni, G.A.; Hofmann, G.A.; Wohlfarth, P.; Winkler, H.-M.; Winkler, S.; Haslacher, H.; Thalhammer, R.; Makristathis, A.; Ratzinger, F.; Burgmann, H. Prevalence and Outcome of Secondary Hemophagocytic Lymphohistiocytosis Among SIRS Patients: Results from a Prospective Cohort Study. J. Clin. Med. 2019, 8, 541. [Google Scholar] [CrossRef] [PubMed]
- Baldo, F.; Erkens, R.G.A.; Mizuta, M.; Rogani, G.; Lucioni, F.; Bracaglia, C.; Foell, D.; Gattorno, M.; Jelusic, M.; Anton, J.; et al. Current Treatment in Macrophage Activation Syndrome Worldwide: A Systematic Literature Review to Inform the METAPHOR Project. Rheumatology 2025, 64, 32–44. [Google Scholar] [CrossRef]
- Henter, J.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Summerlin, J.; Wells, D.A.; Anderson, M.K.; Halford, Z. A Review of Current and Emerging Therapeutic Options for Hemophagocytic Lymphohistiocytosis. Ann. Pharmacother. 2023, 57, 867–879. [Google Scholar] [CrossRef]
- Sen, E.S.; Clarke, S.L.N.; Ramanan, A.V. Macrophage Activation Syndrome. Indian. J. Pediatr. 2016, 83, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S. Macrophage Activation Syndrome: A Potentially Fatal Complication of Rheumatic Disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gioia, C.; Paroli, M.; Izzo, R.; Di Sanzo, L.; Rossi, E.; Pignatelli, P.; Accapezzato, D. Pathogenesis of Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome: A Case Report and Review of the Literature. Int. J. Mol. Sci. 2024, 25, 5921. [Google Scholar] [CrossRef] [PubMed]
- Vilaiyuk, S.; Sirachainan, N.; Wanitkun, S.; Pirojsakul, K.; Vaewpanich, J. Recurrent Macrophage Activation Syndrome as the Primary Manifestation in Systemic Lupus Erythematosus and the Benefit of Serial Ferritin Measurements: A Case-Based Review. Clin. Rheumatol. 2013, 32, 899–904. [Google Scholar] [CrossRef]
- Gilboa, M.; Bornstein, G.; Ben-Zvi, I.; Grossman, C. Macrophage Activation Syndrome Complicating Rheumatic Diseases in Adults: Case-Based Review. Rheumatol. Int. 2020, 40, 663–669. [Google Scholar] [CrossRef]
- Zhu, D.; Ying, S.; Yang, C.; Li, S.; Tang, S.; Sun, C.; Fang, H.; Qiao, J. Clinical Features of Macrophage Activation Syndrome in Adult Dermatomyositis: A Single-center Retrospective Case-control Study. Immun. Inflam. Amp; Dis. 2024, 12, e1141. [Google Scholar] [CrossRef]
- Bazan-Socha, S.; Zolcinski, M.; Szostek, M.; Jurczyszyn, A.; Rucinska, M.; Demczuk, S.; Musial, J. A Fatal Case of Acquired Hemophagocytic Lymphohistiocytosis (Macrophage Activation Syndrome) in the Initial Course of Dermatomyositis with Anti-J O-1 Antibody. Int. J. Rheum. Dis. 2017, 20, 2171–2174. [Google Scholar] [CrossRef]
- Bin, Q.; Gao, J.-H.; Luo, J.-M. Prognostic Factors of Early Outcome in Pediatric Hemophagocytic Lymphohistiocytosis: An Analysis of 116 Cases. Ann. Hematol. 2016, 95, 1411–1418. [Google Scholar] [CrossRef]
- Luo, Z.-B.; Chen, Y.-Y.; Xu, X.-J.; Zhao, N.; Tang, Y.-M. Prognostic Factors of Early Death in Children with Hemophagocytic Lymphohistiocytosis. Cytokine 2017, 97, 80–85. [Google Scholar] [CrossRef]
- Harnchoowong, S.; Soponkanaporn, S.; Vilaiyuk, S.; Lerkvaleekul, B.; Pakakasama, S. Central Nervous System Involvement and Thrombocytopenia as Predictors of Mortality in Children with Hemophagocytic Lymphohistiocytosis. Front. Pediatr. 2022, 10, 941318. [Google Scholar] [CrossRef]
- Trottestam, H.; Berglöf, E.; Horne, A.; Onelöv, E.; Beutel, K.; Lehmberg, K.; Sieni, E.; Silfverberg, T.; Aricò, M.; Janka, G.; et al. Risk Factors for Early Death in Children with Haemophagocytic Lymphohistiocytosis. Acta Paediatr. 2012, 101, 313–318. [Google Scholar] [CrossRef] [PubMed]
- La Marle, S.; Richard-Colmant, G.; Fauvernier, M.; Ghesquières, H.; Hot, A.; Sève, P.; Jamilloux, Y. Mortality and Associated Causes in Hemophagocytic Lymphohistiocytosis: A Multiple-Cause-of-Death Analysis in France. J. Clin. Med. 2023, 12, 1696. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Logan, A.C. Diagnosis and Management of Adult Malignancy-Associated Hemophagocytic Lymphohistiocytosis. Cancers 2023, 15, 1839. [Google Scholar] [CrossRef] [PubMed]
- Poto, R.; Cristinziano, L.; Criscuolo, G.; Strisciuglio, C.; Palestra, F.; Lagnese, G.; Di Salvatore, A.; Marone, G.; Spadaro, G.; Loffredo, S.; et al. The JAK1/JAK2 Inhibitor Ruxolitinib Inhibits Mediator Release from Human Basophils and Mast Cells. Front. Immunol. 2024, 15, 1443704. [Google Scholar] [CrossRef]
- Schuster, F.S.; Nyvlt, P.; Heeren, P.; Spies, C.; Adam, M.F.; Schenk, T.; Brunkhorst, F.M.; Janka, G.; La Rosée, P.; Lachmann, C.; et al. Differential Diagnosis of Hyperferritinemia in Critically Ill Patients. J. Clin. Med. 2022, 12, 192. [Google Scholar] [CrossRef]

{kind=link}
| Parameter | Values |
|---|---|
| 1. Fever | Body temperature ≥ 38.5 °C |
| 2. Organomegaly (splenomegaly/lymphadenopathy) | Present or absent |
| 3. Bicytopenia or pancytopenia | Neutrophils < 1000/µL |
| Hemoglobin < 9 g/dL | |
| Platelet count < 100 × 103/µL | |
| 4. Hypertriglyceridemia | ≥ 265 mg/dL |
| 5. Hypofibrinogenemia | ≤ 1.8 g/L |
| 6. Hemophagocytosis in bone marrow, liver, spleen, or lymph node | Present or absent |
| 7. Hyperferritinemia | ≥ 500 ng/mL |
| 8. Elevated soluble interleukin-2 receptor 1 | ≥ 2400 U/mL |
| 9. Absent or decreased natural killer cell function | < 80 cells/µL |
| Parameter | Criteria with Exact Points |
|---|---|
| Known underlying immunosuppression (human immunodeficiency virus-positive or receiving long-term immunosuppressive therapy [i.e., glucocorticosteroids, cyclosporine, azathioprine]) | No → 0 Yes → 18 |
| Temperature, °C | < 38.4 → 0 38.4–39.4 → 33 > 39.4 → 49 |
| Organomegaly | No → 0 Hepatomegaly or splenomegaly → 23 Hepatomegaly and splenomegaly → 38 |
| Number of cytopenias (defined as hemoglobin ≤ 9.2 g/dL and/or white blood cells ≤ 5000/mm3 and/or platelets ≤ 110,000/mm3) | 1 lineage → 0 2 lineages → 24 3 lineages → 34 |
| Ferritin, ng/mL | < 2000 → 0 2000–6000 → 35 > 6000 → 50 |
| Triglycerides, mmol/L | < 1.5 → 0 1.5–4 → 44 > 4 → 64 |
| Fibrinogen, g/L | > 2.5 → 0 ≤ 2.5 → 30 |
| Aspartate transaminase, U/L | < 30 → 0 ≥ 30 → 19 |
| Hemophagocytosis features on bone marrow aspirate | No → 0 Yes → 35 |
| Criteria | Number of the HLH Case | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HLH-2009 Criteria | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| At least 3 of 4: | |||||||||||||||||||||
| a. fever | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| b. splenomegaly | N | Y | Y | Y | N | Y | Y | N | Y | Y | Y | N | N | N | Y | N | Y | Y | N | Y | Y |
| c. cytopenias | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| d. hepatitis | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Additionally, at least 1 of 4: | |||||||||||||||||||||
| a. hemophagocytosis | Y | N | N | Y | Y | N | ND | Y | Y | Y | ND | Y | N | ND | Y | N | Y | Y | Y | Y | N |
| b. elevated ferritin | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| c. sIL2Ra (age-based) | not done | ||||||||||||||||||||
| d. NK dysfunction | ND | Y | Y | N | Y | ND | Y | Y | Y | Y | ND | N | ND | Y | ND | ND | ND | N | ND | Y | ND |
| Other results for diagnosis: | |||||||||||||||||||||
| a. hypertriglyceridemia | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ND | N | Y | N | Y |
| b. hypofibrinogenemia | Y | N | Y | N | Y | Y | N | Y | N | Y | N | Y | Y | N | Y | N | ND | Y | Y | Y | Y |
| c. hyponatremia | N | N | Y | Y | Y | Y | N | N | Y | Y | N | N | N | Y | N | Y | N | Y | N | N | Y |
| HScore | 219 | 208 | 218 | 255 | 230 | 269 | 261 | 263 | 223 | 304 | 231 | 282 | 271 | 214 | 271 | 244 | 211 | 258 | 281 | 209 | 217 |
| Other clinical data | |||||||||||||||||||||
| Age at the time of HLH, years | 35 | 64 | 62 | 25 | 56 | 20 | 25 | 53 | 23 | 19 | 22 | 38 | 36 | 44 | 30 | 67 | 32 | 19 | 39 | 29 | 54 |
| Sex (female/male) | M | M | F | M | F | M | F | F | F | M | F | F | M | F | F | F | F | M | F | M | M |
| Etiology of the secondary HLH | SLE | lymphoma | cytomegalovirus infection | hepatitis B virus | Epstein Barr virus | septic shock | SLE | IIM | unknown | unknown | SLE | hepatitis C virus | SLE | microscopic polyangiitis | SLE | lymphoma | juvenile idiopathic arthritis | unknown | psoriatic arthritis | aceruloplasminemia | adult-onset Still’s disease |
| Outcome during the HLH | S | D | S | S | D | S | S | S | S | D | S | S | S | S | S | D | S | S | S | S | S |
| Parameter | Secondary HLH Patients n = 21 |
|---|---|
| Demographic factors | |
| Age at the secondary HLH diagnosis, years | 35.0 (24.0–53.5) |
| Sex, female, n (%) | 12 (57.1%) |
| Clinical characteristics | |
| Age at the accompanying illness diagnosis, years | 32.5 (24.8–53.8) |
| Time from illness to secondary HLH diagnosis, years | 0.0 (0.0–4.3) |
| Splenomegaly, n (%) | 13 (61.9%) |
| Lymphadenopathy, n (%) | 9 (42.9%) |
| Neuropsychiatric symptoms, n (%) | 8 (38.1%) |
| Fever, n (%) | 21 (100.0%) |
| Disseminated intravascular coagulation, n (%) | 1 (4.8%) |
| Hemophagocytosis 1, n (%) | 12 (66.7%) |
| Hypofibrinogenemia 2, n (%) | 13 (65.0%) |
| Death due to HLH, n (%) | 4 (19.0%) |
| HScore, points | 244 (218–270) |
| Treatment pattern | |
| Glucocorticosteroids, n (%) | 21 (100.0%) |
| Immunoglobulin, n (%) | 11 (52.4%) |
| Rituximab, n (%) | 2 (9.5%) |
| Cyclosporin, n (%) | 12 (57.1%) |
| Etoposide, n (%) | 2 (9.5%) |
| Antibiotics, n (%) | 20 (95.2%) |
| Antiviral drugs, n (%) | 11 (52.4%) |
| Antifungal drugs, n (%) | 12 (57.1%) |
| Parameter | Secondary HLH Patients n = 21 | Reference Ranges |
|---|---|---|
| White blood cells, 103/µL | 2.16 (1.15–5.48) | 4.0–10.0 |
| Neutrophils, 103/µL | 1.11 (0.40–2.92) | 1.8–7.7 |
| Lymphocytes, 103/µL | 0.55 (0.39–1.09) | 1.0–4.5 |
| Lymphocytes NK, 103/µL | 0.047 (0.018–0.089) | 0.080–0.350 |
| Hemoglobin, g/dL | 7.8 (6.1–9.6) | 14.0–18.0 |
| Blood platelets, 103/µL | 81 (32–116) | 140–440 |
| Aspartate transaminase, U/L | 127 (92–289) | 10–50 |
| Alanine transaminase, U/L | 109 (88–263) | 10–50 |
| Lactate dehydrogenase, IU/L | 1372 (591–2009) | 135–225 |
| Albumin, g/L | 26.3 (20.0–31.6) | 35.0–52.0 |
| Triglycerides, mmol/L | 4.53 (3.11–6.76) | <2.26 |
| Fibrinogen, g/L | 1.31 (0.78–2.12) | 1.8–3.5 |
| Ferritin, ng/mL | 14,305 (5632–30,342) | 13–400 |
| D-dimer, mg/L | 3.88 (2.78–35.00) | <0.55 |
| C-reactive protein, mg/L | 224.3 (44.5–277.5) | <5.0 |
| Parameter | Fatal HLH Cases n = 4 | Survived HLH Patients n = 17 | p-Value |
|---|---|---|---|
| Demographic factors | |||
| Age at HLH diagnosis, years | 60.0 (28.3–66.3) | 32.0 (24.0–41.5) | 0.14 |
| Sex, female, n (%) | 2 (50.0%) | 10 (58.8%) | 1.00 |
| Clinical characteristics | |||
| Macrophage activation syndrome, n (%) | 0 (0.0%) | 10 (58.8%) | 0.09 |
| Splenomegaly, n (%) | 2 (50.0%) | 11 (64.7%) | 0.62 |
| Lymphadenopathy, n (%) | 2 (50.0%) | 7 (41.2%) | 1.00 |
| Neuropsychiatric symptoms, n (%) | 3 (75.0%) | 5 (29.4%) | 0.25 |
| Fever, n (%) | 4 (100.0%) | 17 (100.0%) | NA |
| Disseminated intravascular coagulation, n (%) | 0 (0.0%) | 1 (5.9%) | 1.00 |
| Hemophagocytosis, n (%) | 2 (50.0%) | 10 (58.8%) | 1.00 |
| Hscore, points | 237 (214–289) | 255 (218–270) | 0.90 |
| Treatment pattern | |||
| Glucocorticosteroids, n (%) | 4 (100.0%) | 17 (100.0%) | NA |
| Immunoglobulin, n (%) | 2 (50.0%) | 9 (52.9%) | 1.00 |
| Rituximab, n (%) | 0 (0.0%) | 2 (11.8%) | 1.00 |
| Cyclosporin, n (%) | 3 (75.0%) | 9 (52.9%) | 0.60 |
| Etoposide, n (%) | 2 (50.0%) | 0 (0.0%) | 0.029 |
| Antibiotics, n (%) | 4 (100.0%) | 16 (94.1%) | 1.00 |
| Antiviral drugs, n (%) | 3 (75.0%) | 8 (47.1%) | 0.59 |
| Antifungal drugs, n (%) | 4 (100.0%) | 8 (47.1%) | 0.10 |
| Laboratory findings | |||
| White blood cells, 103/µL | 0.77 (0.31–1.64) | 2.69 (1.60–5.87) | 0.031 |
| Neutrophils, 103/µL | 0.23 (0.02–0.78) | 2.09 (0.90–4.70) | 0.020 |
| Lymphocytes, 103/µL | 0.27 (0.20–0.50) | 0.66 (0.42–1.23) | 0.039 |
| Lymphocytes NK, 103/µL | 0.010 (0.002–0.055) | 0.060 (0.033–0.206) | 0.10 |
| Hemoglobin, g/dL | 6.7 (5.5–8.5) | 8.1 (6.6–10.1) | 0.36 |
| Blood platelets, 103/µL | 14 (7–99) | 81 (52–116) | 0.12 |
| Aspartate transaminase, U/L | 147 (52–308) | 121 (92–290) | 0.97 |
| Alanine transaminase, U/L | 248 (91–562) | 105 (88–240) | 0.44 |
| Lactate dehydrogenase, IU/L | 1781 (1414–5352) | 1151 (498–1848) | 0.10 |
| Albumin, g/L | 25.5 (24.3–26.2) | 29.1 (20.0–33.1) | 0.53 |
| Triglycerides, mmol/L | 3.99 (3.20–4.68) | 4.65 (2.56–7.16) | 0.49 |
| Fibrinogen, g/L | 2.00 (1.00–3.83) | 1.31 (0.69–2.07) | 0.44 |
| Ferritin, ng/mL | 9385 (4116–22,657) | 20,133 (7411–34,836) | 0.36 |
| D-dimer, mg/L | 2.60 (2.44–2.60) | 6.52 (2.88–35.10) | 0.15 |
| C-reactive protein, mg/L | 236.1 (111.8–314.4) | 199.0 (21.0–277.5) | 0.49 |
| Parameter | MAS Patients n = 10 | Non-MAS HLH Patients n = 11 | p-Value |
|---|---|---|---|
| Demographic factors | |||
| Age during HLH, years | 35.5 (28.8–46.3) | 29.0 (20.0–62.0) | 0.76 |
| Sex, female, n (%) | 7 (70.0%) | 5 (45.5%) | 0.39 |
| Clinical characteristics | |||
| Death during hospitalization, n (%) | 0 (0.0%) | 4 (36.4%) | 0.09 |
| Splenomegaly, n (%) | 5 (50.0%) | 8 (72.7%) | 0.39 |
| Lymphadenopathy, n (%) | 5 (50.0%) | 4 (36.4%) | 0.67 |
| Neuropsychiatric symptoms, n (%) | 3 (30.0%) | 5 (45.5%) | 0.66 |
| Fever, n (%) | 10 (100.0%) | 11 (100.0%) | NA |
| Disseminated intravascular coagulation, n (%) | 0 (0.0%) | 1 (9.1%) | 1.00 |
| Hemophagocytosis, n (%) | 5 (50.0%) | 7 (63.6%) | 0.67 |
| HScore, points | 246 (216–271) | 244 (218–269) | 0.92 |
| Treatment pattern | |||
| Glucocorticosteroids, n (%) | 10 (100.0%) | 11 (100.0%) | NA |
| Immunoglobulin, n (%) | 4 (40.0%) | 7 (63.6%) | 0.40 |
| Rituximab, n (%) | 2 (20.0%) | 0 (0.0%) | 0.21 |
| Cyclosporin, n (%) | 7 (70.0%) | 5 (45.5%) | 0.39 |
| Etoposide, n (%) | 0 (0.0%) | 2 (18.2%) | 0.48 |
| Antibiotics, n (%) | 10 (100.0%) | 10 (90.9%) | 1.00 |
| Antiviral drugs, n (%) | 4 (40.0%) | 7 (63.6%) | 0.40 |
| Antifungal drugs, n (%) | 5 (50.0%) | 7 (63.6%) | 0.67 |
| Laboratory findings | |||
| White blood cells, 103/µL | 3.47 (0.95–6.44) | 1.79 (1.19–2.69) | 0.31 |
| Neutrophils, 103/µL | 2.34 (0.48–4.83) | 1.00 (0.32–1.44) | 0.21 |
| Lymphocytes, 103/µL | 0.53 (0.40–1.53) | 0.56 (0.33–0.90) | 1.00 |
| Lymphocytes NK, n/μL | 0.036 (0.014–0.036) | 0.055 (0.020–0.206) | 0.73 |
| Hemoglobin, g/dL | 7.7 (4.9–10.6) | 7.8 (6.3–9.5) | 0.71 |
| Blood platelets, 103/µL | 81 (64–119) | 57 (13–116) | 0.39 |
| Aspartate transaminase, U/L | 149 (112–253) | 104 (50–362) | 0.31 |
| Alanine transaminase, U/L | 92 (76–222) | 118 (97–375) | 0.30 |
| Lactate dehydrogenase, IU/L | 1483 (1061–2675) | 1027 (469–1700) | 0.28 |
| Albumin, g/L | 22.0 (18.2–28.8) | 29.1 (25.0–34.9) | 0.041 |
| Triglycerides, mmol/L | 4.69 (2.82–7.70) | 4.30 (3.04–5.10) | 0.37 |
| Fibrinogen, g/L | 1.32 (0.59–2.11) | 1.30 (1.00–3.00) | 0.60 |
| Ferritin, ng/mL | 21,682 (11,591–33,937) | 11,708 (4000–25,441) | 0.17 |
| D-dimer, mg/L | 10.03 (3.11–35.20) | 2.83 (1.50–28.30) | 0.08 |
| C-reactive protein, mg/L | 181.0 (12.2–259.2) | 235.0 (71.8–288.0) | 0.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziedzic, R.; Bazan-Socha, S.; Korkosz, M.; Kosałka-Węgiel, J. Characteristics of 21 Patients with Secondary Hemophagocytic Lymphohistiocytosis—Insights from a Single-Center Retrospective Study. Medicina 2025, 61, 977. https://doi.org/10.3390/medicina61060977
Dziedzic R, Bazan-Socha S, Korkosz M, Kosałka-Węgiel J. Characteristics of 21 Patients with Secondary Hemophagocytic Lymphohistiocytosis—Insights from a Single-Center Retrospective Study. Medicina. 2025; 61(6):977. https://doi.org/10.3390/medicina61060977
Chicago/Turabian StyleDziedzic, Radosław, Stanisława Bazan-Socha, Mariusz Korkosz, and Joanna Kosałka-Węgiel. 2025. "Characteristics of 21 Patients with Secondary Hemophagocytic Lymphohistiocytosis—Insights from a Single-Center Retrospective Study" Medicina 61, no. 6: 977. https://doi.org/10.3390/medicina61060977
APA StyleDziedzic, R., Bazan-Socha, S., Korkosz, M., & Kosałka-Węgiel, J. (2025). Characteristics of 21 Patients with Secondary Hemophagocytic Lymphohistiocytosis—Insights from a Single-Center Retrospective Study. Medicina, 61(6), 977. https://doi.org/10.3390/medicina61060977