

Normal Haemostasis, Inherited Bleeding Disorders and Surgery: What Does the Anaesthesiologist Need to Know?

 , , and
, , and
Abstract
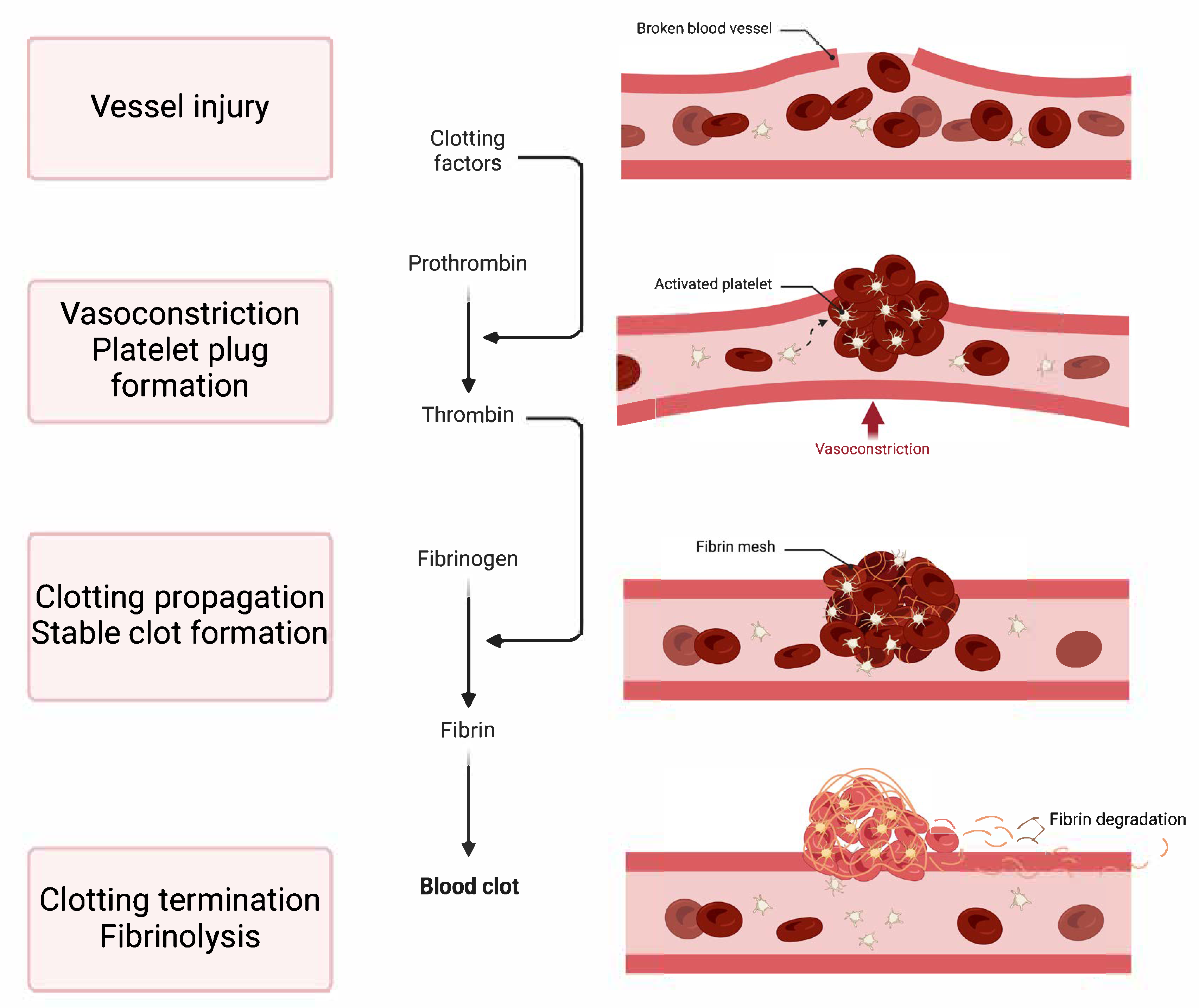
1. Introduction to Haemostasis
- Vasoconstriction: This is the immediate narrowing of the blood vessel, which reduces blood flow to the injured area.
- Platelet Plug Formation: At the injury site, platelets stick, become activated, and clump together to form a provisional plug that closes off small tears in the damaged vessel.
- Clotting Propagation: This phase involves strengthening the platelet plug with fibrin, which produces a stable clot. This step occurs through the coagulation cascade, a series of enzyme-driven reactions that end with the creation of fibrin, an insoluble protein.
- Clotting Termination and Fibrinolysis: The haemostatic process is intricately controlled by opposing mechanisms that inhibit excessive clotting (thrombosis) and guarantee the disintegration of clots as tissue repair progresses (fibrinolysis). Imbalances in haemostasis can lead to either haemorrhage or abnormal clot formation.
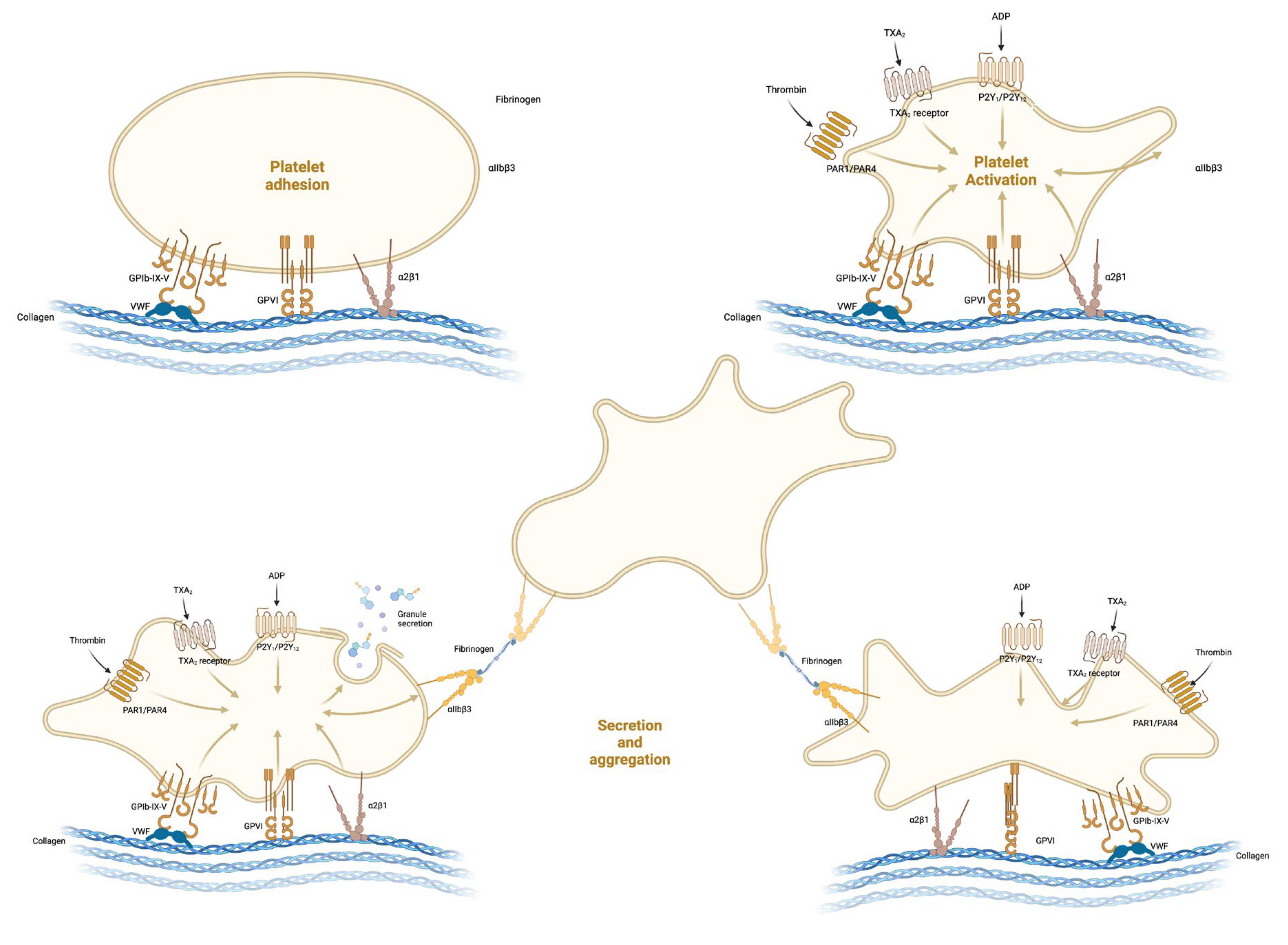
2. Formation of the Platelet Plug
2.1. Platelet Adhesion
2.2. Activation and Shape Change
2.3. Platelet Secretion
- Recruitment: Compounds such as ADP and serotonin attract additional platelets to the injury site, encouraging their adherence and activation.
- Amplification: ADP’s release is particularly critical for the enhancement of platelet activation. It binds to P2Y1 and P2Y12 receptors on platelets, prompting a shape change and further granule release, thereby creating a positive feedback loop.
- Aggregation Enhancement: Thromboxane A2, synthesised by platelets, not only promotes platelet aggregation but also causes vasoconstriction, thereby diminishing blood flow to the injured area.
- Stabilisation: Proteins such as thrombospondin aid in solidifying the platelet mass by binding to fibrinogen and other extracellular matrix elements.
- Vessel Repair and Remodelling: Growth factors promote the healing and rebuilding of the vessel wall.
2.4. Platelet Aggregation
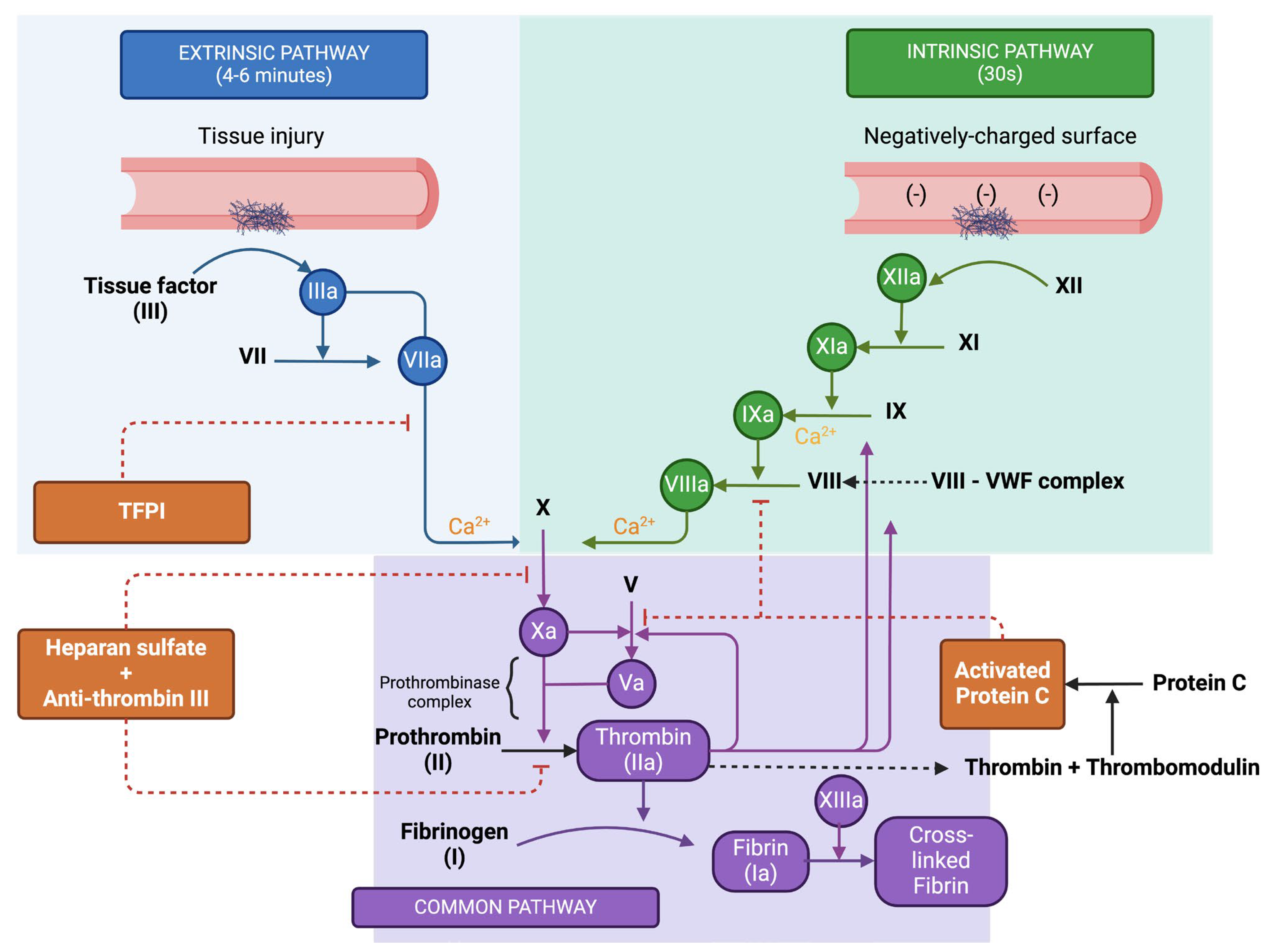
3. The Coagulation Cascade and Resolution of Clotting
- Factor XII, known as Hageman factor, is converted to its active form XIIa.
- This active form, Factor XIIa, then activates Factor XI to XIa.
- Factor XIa proceeds to activate Factor IX to IXa.
- Factor IXa, coupled with its cofactor Factor VIIIa (activated by thrombin), forms a complex that activates Factor X to Xa.
- Tissue factor, a protein found on subendothelial cells outside the blood vessels, associates with Factor VII, resulting in its activation to VIIa.
- The TF-VIIa complex then directly activates Factor X (Xa), bypassing the intrinsic pathway steps.
- Factor Xa, paired with its cofactor Factor Va, assembles into the prothrombinase complex.
- Prothrombinase catalyses the transformation of prothrombin (Factor II) into thrombin (Factor IIa).
- Thrombin, in turn, acts on fibrinogen to convert it into fibrin.
- The resulting fibrin monomers then polymerize, forming a stable meshwork. This meshwork is further solidified by Factor XIIIa, which is activated by thrombin, and creates covalent linkages among the fibrin strands.
3.1. Regulation of the Coagulation Cascade
- Heparan sulphate (HS), which attaches to antithrombin (AT). This interaction induces a structural shift in AT, significantly enhancing its ability to bind with thrombin and Factor Xa, leading to their deactivation.
- Tissue factor pathway inhibitor (TFPI) targets the extrinsic tenase complex, neutralising it and inhibiting its activity.
- Thrombomodulin (TM) connects to thrombin, which is abundantly produced during the coagulation propagation phase. Binding to TM alters thrombin’s structure, which in turn activates protein C (aPC). Once activated, aPC, with protein S as a supporting factor, attaches to Factor Va, rendering it inactive.
3.2. Cell Based Model of Coagulation
- During the initiation phase, coagulation is instigated by vessel damage that exposes tissue factor (TF) in the plasma. This phase unfolds on a TF-bearing cell, leading to the activation of factors V and VII in proximity, which subsequently activate other clotting factors, resulting in the generation of a modest quantity of thrombin. Moreover, platelets passing by become activated by both TF and vWF, as well as by the initial thrombin generated.
- The amplification phase sees the further activation of clotting factors and platelets in readiness for the extensive production of thrombin. Thrombin binds to the GP1b receptors on the platelet surface, activating factors XI, VIII, and V.
- The propagation phase takes place on the surface of an activated platelet, which is primed with activated clotting factors. This platelet acts as a catalyst, leading to the production of substantial amounts of thrombin.
3.3. Resolution of Clotting
4. Inherited Bleeding Disorders
4.1. Disorders of Primary Haemostasis
- Von Willebrand Disease (vWD)
- Type 1 vWD, the most common and mildest form, typically presents with mucocutaneous bleeding, such as epistaxis, menorrhagia, and bleeding after dental extractions.
- Type 2 vWD involves qualitative defects in vWF and presents with a similar but often more severe bleeding phenotype, and has four subtypes (2A, 2B, 2N, 2M).
- Type 3 vWD, the most severe form, results from a near-total lack of vWF and presents with significant bleeding risks.
- Platelet Function Disorders
- Glanzmann Thrombasthenia: A rare condition characterised by the absence or dysfunction of the GPIIb/IIIa receptor, crucial for platelet aggregation. Patients typically present with mucocutaneous bleeding, such as gum bleeding, epistaxis, and menorrhagia, and may experience severe bleeding after surgical procedures.
- Bernard-Soulier Syndrome: Another rare disorder, marked by a deficiency of the GPIb-IX-V complex, essential for platelet adhesion to vWF. This condition presents similarly to Glanzmann Thrombasthenia but can be distinguished by the presence of giant platelets on a blood smear.
- Platelet type Von Willebrand Disease (also known as pseudo vWD) is a distinct entity, characterised by an abnormal interaction between platelets and vWF. This disorder is caused by a gain-of-function mutation in the platelet glycoprotein Ibα, leading to enhanced binding to vWF. Patients present with a clinical phenotype similar to Type 2B vWD, including thrombocytopenia and mucocutaneous bleeding. Diagnosis can be challenging and requires specific laboratory assays to differentiate it from Type 2B vWD, given the similar clinical and laboratory profiles.
- Storage pool disorders (SPDs) are a group of inherited platelet function disorders characterised by deficiencies in platelet granules or their contents. These disorders affect the storage and release of critical haemostatic agents from platelets, leading to bleeding symptoms.
4.2. Disorders of Secondary Haemostasis
- Haemophilia
- Rare Bleeding Disorders
5. Perioperative Anaesthetic Considerations
5.1. Preoperative Evaluation—Bleeding Assessment Tools
5.2. Preoperative Evaluation—Laboratory Tests
5.3. Management Strategies
5.4. Regional Anaesthesia
5.5. Haemostatic Interventions—Substitutions
5.6. Haemostatic Interventions—Desmopressin
5.7. Haemostatic Interventions—Antifibrinolytics
5.8. Haemostatic Interventions—RFVIIa
5.9. Emergency Surgery in Patients with Inherited Bleeding Disorders
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Leung, L.K. Overview of Haemostasis; Mannucci, P., Tirnauer, J., Eds.; Uptodate: Waltham, MA, USA, 2023. [Google Scholar]
- Chambers, D.; Huang, C.; Matthews, G. Chapter 72-Haemostasis. In Basic Physiology for Anaesthetists, 2nd ed.; Cambridge University Press: Cambridge, UK, 2019; pp. 341–349. [Google Scholar]
- Sucker, C.; Zotz, R.B. The Cell-Based Coagulation Model. In Perioperative Hemostasis; Marcucci, C.E., Schoettker, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 3–11. Available online: https://link.springer.com/10.1007/978-3-642-55004-1_1 (accessed on 1 April 2024).
- Hall, J.; Hall, M. (Eds.) Hemostasis and Blood Coagulation. In Guyton and Hall Textbook of Medical Physiology, 14th ed.; Elsevier: Philadephia, PA, USA, 2021; pp. 483–496. [Google Scholar]
- Martinez, M.; Graf, L.; Tsakiris, D.A. Congenital Bleeding Disorders. In Perioperative Hemostasis; Marcucci, C.E., Schoettker, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 71–87. Available online: http://link.springer.com/10.1007/978-3-642-55004-1_6 (accessed on 1 April 2024).
- Shah, U.J.; Narayanan, M.; Graham Smith, J. Anaesthetic considerations in patients with inherited disorders of coagulation. Contin. Educ. Anaesth. Crit. Care Pain 2015, 15, 26–31. [Google Scholar] [CrossRef]
- Loew, G.; Hayward, C. Inherited Platelet Function Disorders; Uptodate: Waltham, MA, USA, 2024. [Google Scholar]
- Orsini, S.; Noris, P.; Bury, L.; Heller, P.G.; Santoro, C.; Kadir, R.A.; Butta, N.C.; Falcinelli, E.; Cid, A.R.; Fabris, F.; et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica 2017, 102, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Hoots, K.H.; Malec, L. Clinical Manifestations and Diagnosis of Hemophilia; Shapiro, A.D., Tirnauer, J.S., Eds.; Uptodate: Waltham, MA, USA, 2023. [Google Scholar]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia 2020, 26, 1–158. [Google Scholar] [CrossRef] [PubMed]
- White, G.C.; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J.; Subcommittee, F.V.A.F.I. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar]
- Atiq, F.; Schütte, L.M.; Looijen, A.E.M.; Boender, J.; Cnossen, M.H.; Eikenboom, J.; de Maat, M.P.M.; Kruip, M.J.H.A.; Leebeek, F.W.G. von Willebrand factor and factor VIII levels after desmopressin are associated with bleeding phenotype in type 1 VWD. Blood Adv. 2019, 3, 4147–4154. [Google Scholar] [CrossRef]
- Menegatti, M.; Peyvandi, F. Treatment of rare factor deficiencies other than hemophilia. Blood 2019, 133, 415–424. [Google Scholar] [CrossRef]
- Mohsenian, S.; Mannucci, P.M.; Menegatti, M.; Peyvandi, F. Rare inherited coagulation disorders: No longer orphan and neglected. Res. Pract. Thromb. Haemost. 2024, 8, 102460. [Google Scholar] [CrossRef]
- Lewandowska, M.D.; Connors, J.M. Factor XI Deficiency. Hematol./Oncol. Clin. N. Am. 2021, 35, 1157–1169. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Pabinger, I.; Ragni, M.; Abdul-Kadir, R.; Berntorp, E.; Blanchette, V.; Bodó, I.; Casini, A.; Gresele, P.; Lassila, R.; et al. Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: An EHA Consensus Report. HemaSphere 2019, 3, e286. [Google Scholar] [CrossRef]
- Kietaibl, S.; Ahmed, A.; Afshari, A.; Albaladejo, P.; Aldecoa, C.; Barauskas, G.; De Robertis, E.; Faraoni, D.; Filipescu, D.C.; Fries, D.; et al. Management of severe peri-operative bleeding: Guidelines from the European Society of Anaesthesiology and Intensive Care: Second update 2022. Eur. J. Anaesthesiol. 2023, 40, 226–304. [Google Scholar]
- World Federation of Hemophilia. Compendium of Assessment Tools [Internet]. 2014. Available online: https://elearning.wfh.org/resource/compendium-of-assessment-tools/ (accessed on 18 January 2025).
- Tosetto, A.; Rodeghiero, F.; Castaman, G.; Goodeve, A.; Federici, A.B.; Batlle, J.; Meyer, D.; Fressinaud, E.; Mazurier, C.; Goudemand, J.; et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: Results from a multicenter European study (MCMDM-1 VWD). J. Thromb. Haemost. 2006, 4, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Riddel, J.; Rand, M.L.; Tosetto, A.; Silva, M.; James, P.D. Evaluation of the diagnostic utility for von Willebrand disease of a pediatric bleeding questionnaire. J. Thromb. Haemost. 2009, 7, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Šrámek, A. Usefulness of Patient Interview in Bleeding Disorders. Arch. Intern. Med. 1995, 155, 1409. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.C.; Arnold, D.M.; Coller, B.S.; James, P.D.; Neunert, C.E.; Lillicrap, D.; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Adler, M.; Kaufmann, J.; Alberio, L.; Nagler, M. Diagnostic utility of the ISTH bleeding assessment tool in patients with suspected platelet function disorders. J. Thromb. Haemost. 2019, 17, 1104–1112. [Google Scholar] [CrossRef]
- Gresele, P.; Falcinelli, E.; Bury, L.; Pecci, A.; Alessi, M.; Borhany, M.; Heller, P.G.; Santoro, C.; Cid, A.R.; Orsini, S.; et al. The ISTH bleeding assessment tool as predictor of bleeding events in inherited platelet disorders: Communication from the ISTH SSC Subcommittee on Platelet Physiology. J. Thromb. Haemost. 2021, 19, 1364–1371. [Google Scholar] [CrossRef]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Borhany, M.; Fatima, N.; Abid, M.; Shamsi, T.; Othman, M. Application of the ISTH bleeding score in hemophilia. Transfus. Apher. Sci. 2018, 57, 556–560. [Google Scholar] [CrossRef]
- Toret, E.; Ay, Y.; Karapinar, T.H.; Oymak, Y.; Kavakli, K.; Vergin, R.C. Evaluation of Bleeding Phenotype of Inherited Factor VII Deficiency in Children With a Bleeding Assessment Tool and Global Assays. J. Pediatr. Hematol./Oncol. 2020, 42, e527–e530. [Google Scholar] [CrossRef]
- Fasulo, M.R.; Biguzzi, E.; Abbattista, M.; Stufano, F.; Pagliari, M.T.; Mancini, I.; Gorski, M.M.; Cannavò, A.; Corgiolu, M.; Peyvandi, F.; et al. The ISTH Bleeding Assessment Tool and the risk of future bleeding. J. Thromb. Haemost. 2018, 16, 125–130. [Google Scholar] [CrossRef]
- Ambaglio, C.; Zane, F.; Russo, M.C.; Preti, P.S.; Scudeller, L.; Klersy, C.; Gamba, G.; Squizzato, A. Preoperative bleeding risk assessment with ISTH-BAT and laboratory tests in patients undergoing elective surgery: A prospective cohort study. Haemophilia 2021, 27, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Vries, M.J.; van der Meijden, P.E.; Kuiper, G.J.; Nelemans, P.J.; Wetzels, R.J.; van Oerle, R.G.; Lancé, M.D.; Cate, H.T.; Henskens, Y.M. Preoperative screening for bleeding disorders: A comprehensive laboratory assessment of clinical practice. Res. Pract. Thromb. Haemost. 2018, 2, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Stefan, M.; Tomescu, D.; Predoi, C.; Goicea, R.; Perescu, M.; Popescu, M.; Dorobanțu, D.; Droc, G.; Andrei, Ș.; Știru, O.; et al. Less (transfusion) is more-enhancing recovery through implementation of Patient Blood Management in cardiac surgery: A retrospective, single-centre study of 1174 patients. J. Cardiovasc. Dev. Dis. 2023, 10, 266. [Google Scholar] [PubMed]
- James, P.D.; Connell, N.T.; Ameer, B.; Di Paola, J.; Eikenboom, J.; Giraud, N.; Haberichter, S.; Jacobs-Pratt, V.; Konkle, B.; McLintock, C.; et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021, 5, 280–300. [Google Scholar] [CrossRef]
- Franchini, M.; Capra, F.; Targher, G.; Montagnana, M.; Lippi, G. Relationship between ABO blood group and von Willebrand factor levels: From biology to clinical implications. Thromb. J. 2007, 5, 14. [Google Scholar] [CrossRef]
- Kim, B. Diagnostic workup of inherited platelet disorders. Blood Res. 2022, 57, S11–S19. [Google Scholar] [CrossRef]
- Palma-Barqueros, V.; Revilla, N.; Sánchez, A.; Cánovas, A.Z.; Rodriguez-Alén, A.; Marín-Quílez, A.; González-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. [Google Scholar] [CrossRef]
- Chai-Adisaksopha, C.; Nevitt, S.J.; Simpson, M.L.; Janbain, M.; Konkle, B.A. Bypassing agent prophylaxis in people with hemophilia A or B with inhibitors. Cochrane Database Syst. Rev. 2017, 9, CD011441. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Gupta, N.; Guo, Y.; Bangalore, S.; Berger, J.S. Perioperative bleeding and thrombotic risks in patients with Von Willebrand disease. J. Thromb. Thrombolysis 2017, 44, 67–70. [Google Scholar] [CrossRef]
- Brignardello-Petersen, R.; El Alayli, A.; Husainat, N.; Kalot, M.; Shahid, S.; Aljabirii, Y.; Britt, A.; Alturkmani, H.; El-Khechen, H.; Motaghi, S.; et al. Surgical management of patients with von Willebrand disease: Summary of 2 systematic reviews of the literature. Blood Adv. 2022, 6, 121–128. [Google Scholar] [CrossRef]
- Connell, N.T.; Flood, V.H.; Brignardello-Petersen, R.; Abdul-Kadir, R.; Arapshian, A.; Couper, S.; Grow, J.M.; Kouides, P.; Laffan, M.; Lavin, M.; et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Adv. 2021, 5, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Hermans, C.; Apte, S.; Santagostino, E. Invasive procedures in patients with haemophilia: Review of low-dose protocols and experience with extended half-life FVIII and FIX concentrates and non-replacement therapies. Haemophilia 2021, 27, 46–52. [Google Scholar] [CrossRef]
- Lowell, A.E.; Calgi, M.P.; Caruso, J.J.; Man, L.M.; McNeil, J.S. Perioperative Management of Hemophilia Patients. Curr. Anesth. Rep. 2024, 14, 354–365. [Google Scholar] [CrossRef]
- Kwak, J.; Mazzeffi, M.; Boggio, L.N.; Simpson, M.L.; Tanaka, K.A. Hemophilia: A Review of Perioperative Management for Cardiac Surgery. J. Cardiothorac. Vasc. Anesth. 2022, 36, 246–257. [Google Scholar] [CrossRef]
- Poston, J.N.; Kruse-Jarres, R. Perioperative hemostasis for patients with hemophilia. Hematology 2022, 2022, 586–593. [Google Scholar] [CrossRef]
- Kozek-Langenecker, S.A.; Ahmed, A.B.; Afshari, A.; Albaladejo, P.; Aldecoa, C.; Barauskas, G.; De Robertis, E.; Faraoni, D.; Filipescu, D.C.; Fries, D.; et al. Management of severe perioperative bleeding: Guidelines from the European Society of Anaesthesiology: First update 2016. Eur. J. Anaesthesiol. 2017, 34, 332–395. [Google Scholar] [CrossRef]
- Kozek-Langenecker, S.A.; Afshari, A.; Albaladejo, P.; Santullano, C.A.A.; De Robertis, E.; Filipescu, D.C.; Fries, D.; Görlinger, K.; Haas, T.; Imberger, G.; et al. Management of severe perioperative bleeding: Guidelines from the European Society of Anaesthesiology. Eur. J. Anaesthesiol. 2013, 30, 270–382. [Google Scholar]
- Cattaneo, M.; Mannucci, P.M. Desmopressin (DDAVP). In Platelets; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1111–1120. Available online: https://linkinghub.elsevier.com/retrieve/pii/B978012813456600062X (accessed on 12 January 2020).
- Windyga, J.; Dolan, G.; Altisent, C.; Katsarou, O.; López Fernández, M.F.; Zülfikar, B.; EHTSB Collaborators. Practical aspects of DDAVP use in patients with von Willebrand Disease undergoing invasive procedures: A European survey. Haemophilia 2016, 22, 110–120. [Google Scholar] [CrossRef]
- Desborough, M.J.; Oakland, K.; Brierley, C.; Bennett, S.; Doree, C.; Trivella, M.; Hopewell, S.; Stanworth, S.J.; Estcourt, L.J. Desmopressin use for minimising perioperative blood transfusion. Cochrane Database Syst. Rev. 2017, 7, CD001884. [Google Scholar] [CrossRef]
- Loomans, J.I.; Kruip, M.J.; Carcao, M.; Jackson, S.; van Velzen, A.S.; Peters, M.; Santagostino, E.; Platokouki, H.; Beckers, E.; Voorberg, J.; et al. Desmopressin in moderate hemophilia A patients: A treatment worth considering. Haematologica 2018, 103, 550–557. [Google Scholar] [CrossRef]
- Furqan, F.; Sham, R.; Kouides, P. Efficacy and safety of half-dose desmopressin for bleeding prophylaxis in bleeding disorder patients undergoing predominantly low to moderate risk invasive procedures. Am. J. Hematol. 2020, 95, E285–E287. [Google Scholar] [CrossRef] [PubMed]
- Seaman, C.D. Efficacy of adjusted weight-based dosing of desmopressin (1-deamino-8-d-arginine vasopressin in type 1 von Willebrand disease. Blood Coagul. Fibrinolysis 2023, 34, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ribkoff, J.; Olson, S.; Raghunathan, V.; Al-Samkari, H.; DeLoughery, T.G.; Shatzel, J.J. The many roles of tranexamic acid: An overview of the clinical indications for TXA in medical and surgical patients. Eur. J. Haematol. 2020, 104, 79–87. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Huang, Q.; Zeng, H.J.; Ma, J.; Shen, B.; Zhou, Z.K.; Pei, F.X. Tranexamic acid may benefit patients undergoing total hip/knee arthroplasty because of haemophilia. BMC Musculoskelet. Disord. 2019, 20, 402. [Google Scholar] [CrossRef]
- Van Galen, K.P.; Engelen, E.T.; Mauser-Bunschoten, E.P.; Van Es, R.J.; Schutgens, R.E. Antifibrinolytic therapy for preventing oral bleeding in patients with haemophilia or Von Willebrand disease undergoing minor oral surgery or dental extractions. Cochrane Database Syst. Rev. 2019, CD011385. [Google Scholar] [CrossRef]
- Wilson, R.D. Preoperative diagnosis and management of inherited bleeding disorders in female adolescents and adults. Can. J. Surg. 2023, 66, E246–E263. [Google Scholar] [CrossRef]
- Shima, M. Current status and future prospects of activated recombinant coagulation factor VIIa, NovoSeven®, in the treatment of haemophilia and rare bleeding disorders. Ann. Hematol. 2023, 103, 2647–2658. [Google Scholar] [CrossRef]
- Castaman, G. The role of recombinant activated factor VII in the haematological management of elective orthopaedic surgery in haemophilia A patients with inhibitors. Blood Transfus. 2017, 15, 478–486. [Google Scholar] [CrossRef]
- Giansily-Blaizot, M.; Schved, J.F. Recombinant human factor VIIa (rFVIIa) in hemophilia: Mode of action and evidence to date. Ther. Adv. Hematol. 2017, 8, 345–352. [Google Scholar] [CrossRef]
- Recht, M.; Rajpurkar, M.; Chitlur, M.; D’OIron, R.; Zotz, R.; Di Minno, G.; Cooper, D.L.; Poon, M. Independent adjudicator assessments of platelet refractoriness and rFVIIa efficacy in bleeding episodes and surgeries from the multinational Glanzmann’s thrombasthenia registry. Am. J. Hematol. 2017, 92, 646–652. [Google Scholar] [CrossRef]
- Zotz, R.B.; Poon, M.C.; Di Minno, G.; D’Oiron, R.; Glanzmann Thrombasthenia Registry Investigators. The International Prospective Glanzmann Thrombasthenia Registry: Pediatric Treatment and Outcomes. TH Open 2019, 3, e286–e294. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Severity of Disease | Factor Activity | Clinically Relevant Bleeds |
|---|---|---|
| Mild | 5–40% | Major trauma, surgery, rarely spontaneous |
| Moderate | 1–5% | Minor trauma, surgery, sometimes spontaneous |
| Severe | <1% | Spontaneous |
| Disease | Screening Test | Specific Tests |
|---|---|---|
| vWD | None | vWF antigen Ristocetin cofactor F VIII |
| IPD | None | CBC Peripheral blood smear Functional (aggregation and secretion) tests PFA-100 |
| Haemophilia A | aPTT elevated PT normal | F VIII Inhibitor screening Genetic testing |
| Haemophilia B | aPTT elevated PT normal | F IX Inhibitor screening Genetic testing |
| Disease | Specific Treatment | Non-Specific, Adjuvant Treatment |
|---|---|---|
| vWD | vWF FVIII DDAVP in type 1 | Antifibrinolytics |
| IPD | Platelet transfusion, not prophylactically | DDAVP Antifibrinolytics rVIIa in Glanzmann thrombasthenia |
| Haemophilia A | FVIII BPAs if high-reactive inhibitors rFVIIa or aPCC | DDAVP in mild HA Antifibrinolytics |
| Haemophilia B | FIX BPAs if high-reactive inhibitors rFVIIa | Antifibrinolytics |
| Haemophilia Type | Preoperative Level | Intraoperative Level | Days 1–3 | Days 4–6 | Days 7–14 |
|---|---|---|---|---|---|
| Haemophilia A | 80–100% | 80–100% | 60–80% | 40–60% | 30–50% |
| Haemophilia B | 60–80% | 60–80% | 30–50% | 30–50% | 20–40% |
| Recommendations | 2013 | 2017 | 2023 |
|---|---|---|---|
| Referral to haematologist for assessment, planning and management | 2C for preoperative assessment and planning 1C for vWD, IPDs and haemophilia perioperative management | 2C for assessment 1C for perioperative management, in centres with expertise in IBDs | 1B for perioperative management, in centres with expertise in IBDs |
| The use of bleeding assessment tools for detecting and predicting the perioperative risk of bleeding before surgery and invasive procedures in patients with suspected or confirmed IBDs. | 1C | 1C | 2B |
| Individualised preoperative haemostatic correction depending on the specific disorder, type of surgery and individual factors (bleeding phenotype). | 2C | ||
| Replacement/substitution therapy with factor concentrates, either plasma-derived or recombinant products, for major bleeding/surgery in patients with vWD or haemophilia A and B. | 1C | 1C | 1C |
| For haemophilia patients with inhibitors—administer either rFVIIa or aPCCs. | 2C | 2C | 2C |
| Routine perioperative platelet transfusion in patients with IPDs. | Against—2C | Against—2C | Against—2C |
| There is insufficient data to recommend routine perioperative supplementation of deficient factors in patients with rare bleeding disorders. | No recommendation | No recommendation | No recommendation |
| Desmopressin as a first-line treatment for minor bleeding/surgery, after a trial testing and in the absence of contraindications. | 1C for vWD 2C for IPDs | 1C for vWD 2C for IPDs and haemophilia A | 2C for vWD and haemophilia A No recommendation for IPDs—insufficient data |
| Perioperative antifibrinolytics as adjunct therapy in haemophilia, vWD or IPDs. | 2C | 2C | 2B for haemophilia, vWD. |
| Perioperative antifibrinolytics as monotherapy | - | - | 2C—in patients with IPDs and in patients with haemophilia or vWD for minor mucosal or dental procedures |
| Recombinant factor VII activated considered in patients with Glanzmann thrombasthenia undergoing surgery | 1C | 1C | 2C |
| Recombinant factor VII activated used in perioperative bleeding due to inherited factor VII deficiency. | 2C | 2C | 2C |
| There is insufficient data to recommend the use of recombinant factor VII activated in perioperative bleeding for patients with other RBDs. | C | No recommendation | No recommendation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ștefan, M.; Filipescu, D.; Predoi, C.; Văleanu, L.; Andrei, Ș.; Tomescu, D. Normal Haemostasis, Inherited Bleeding Disorders and Surgery: What Does the Anaesthesiologist Need to Know? Medicina 2025, 61, 1087. https://doi.org/10.3390/medicina61061087
Ștefan M, Filipescu D, Predoi C, Văleanu L, Andrei Ș, Tomescu D. Normal Haemostasis, Inherited Bleeding Disorders and Surgery: What Does the Anaesthesiologist Need to Know? Medicina. 2025; 61(6):1087. https://doi.org/10.3390/medicina61061087
Chicago/Turabian StyleȘtefan, Mihai, Daniela Filipescu, Cornelia Predoi, Liana Văleanu, Ștefan Andrei, and Dana Tomescu. 2025. "Normal Haemostasis, Inherited Bleeding Disorders and Surgery: What Does the Anaesthesiologist Need to Know?" Medicina 61, no. 6: 1087. https://doi.org/10.3390/medicina61061087
APA StyleȘtefan, M., Filipescu, D., Predoi, C., Văleanu, L., Andrei, Ș., & Tomescu, D. (2025). Normal Haemostasis, Inherited Bleeding Disorders and Surgery: What Does the Anaesthesiologist Need to Know? Medicina, 61(6), 1087. https://doi.org/10.3390/medicina61061087